

Abstract
Cyclin-dependent protein kinases (CDKs) are key regulators of the eukaryotic cell cycle and of the eukaryotic transcription machinery. Here we report the characterization of Pfcrk-3 (Plasmodium falciparum CDK-related kinase 3; PlasmoDB identifier PFD0740w), an unusually large CDK-related protein whose kinase domain displays maximal homology to those CDKs which, in other eukaryotes, are involved in the control of transcription. The closest enzyme in Saccharomyces cerevisiae is BUR1 (bypass upstream activating sequence requirement 1), known to control gene expression through interaction with chromatin modification enzymes. Consistent with this, immunofluorescence data show that Pfcrk-3 colocalizes with histones. We show that recombinant Pfcrk-3 associates with histone H1 kinase activity in parasite extracts and that this association is detectable even if the catalytic domain of Pfcrk-3 is rendered inactive by site-directed mutagenesis, indicating that Pfcrk-3 is part of a complex that includes other protein kinases. Immunoprecipitates obtained from extracts of transgenic parasites expressing hemagglutinin (HA)-tagged Pfcrk-3 by using an anti-HA antibody displayed both protein kinase and histone deacetylase activities. Reverse genetics data show that the pfcrk-3 locus can be targeted only if the genetic modification does not cause a loss of function. Taken together, our data strongly suggest that Pfcrk-3 fulfils a crucial role in the intraerythrocytic development of P. falciparum, presumably through chromatin modification-dependent regulation of gene expression.
Plasmodium falciparum, the protozoan parasite responsible for the most virulent form of human malaria, causes 1 to 3 millions deaths annually, mostly among children in sub-Saharan Africa. This mortality is expected to rise with the global emergence and spread of drug-resistant parasites, making the discovery of alternative control agents an urgent task (43). The identification of potential targets is now greatly facilitated by the availability of genomic databases for several species of the Plasmodium genus (www.plasmoDB.org) (49). Plasmodium cell cycle regulators represent attractive candidate targets for intervention, because (i) their activities are most probably essential to parasite survival, and (ii) the overall organization of the cell cycle in malaria parasites differs considerably from that in mammalian cells; this is reflected by atypical properties of the enzymatic machinery controlling cell cycle progression, suggesting that specific inhibition is achievable (12, 15).
The progression of the eukaryotic cell cycle is tightly controlled by a family of protein kinases, the cyclin-dependent kinases (CDKs), whose active forms are composed of a catalytic subunit (CDK) and a regulatory subunit (cyclin) (39). While several mammalian CDKs (CDK1, -2, -3, -4, -6, and -7) function in cell cycle control, others (CDK8, -9, -10, and -11) are part of the transcription machinery. CDK7 is a regulator both of cell cycle progression (through its activity as a CDK-activating kinase [CAK]) and of transcription (through its activity as a component of the general transcription factor TFIIH) (26). CDK8 and -9 regulate transcription by phosphorylating the C-terminal domain of the large subunit of RNA polymerase II (2, 18); BUR1, the Saccharomyces cerevisiae CDK9 homologue previously known as SGV1, has been shown to regulate transcription through selective control of histone modifications (7, 17, 30). CDK10 regulates transcription and cell cycle progression by modulating the activity of the Ets2 transcription factor, a regulator of CDK1 expression (27). CDK11 interacts with the general precursor mRNA splicing factors and with RNA polymerase II, thereby playing a role in transcript production and the regulation of RNA processing (37). Finally, CDK5 has neuron-specific functions (11).
Among the 85 (or 99, depending on the criteria used for inclusion) eukaryotic protein kinase (ePK) sequences that were identified in the P. falciparum kinome (3, 52), 18 clustered within the CMGC group (CDKs, MAPKs [mitogen-activated protein kinases], GSK3 [glycogen synthase kinase 3], and CDK-like), with 6 sequences more closely related to CDKs than to other CMGC subfamilies. By analogy with their functions in other eukaryotes, and despite the unique characteristics of the Plasmodium cell cycle (13, 31, 41) and transcription machineries (1, 6, 8–10), it is likely that the Plasmodium CDK-related kinases play key roles in cell cycle progression and transcription in the parasite. Among those gene products, PfPK5 (22, 32, 47), Pfcrk-1 (14), Pfmrk (34, 35, 53), and PfPK6 (5) have been the subjects of biochemical or structural investigations. However, the only reverse genetics-based information published so far regarding the function of CDKs in the parasite life cycle is that for Pbcrk-1, the orthologue of Pfcrk-1 in Plasmodium berghei, which is essential for erythrocytic schizogony (46).
Here we report the functional characterization of pfcrk-3 (PlasmoDB identifier PFD0740w), a gene encoding an unusually large CDK-related protein (1,339 amino acids) whose kinase domain displays maximal homology to those CDKs which, in other eukaryotes, are involved in the control of transcription. The enzyme associates with a kinase activity present in parasite extracts, and this association is detectable even if the catalytic domain of Pfcrk-3 is rendered inactive by site-directed mutagenesis, suggesting that Pfcrk-3 is part of a complex containing other protein kinases. We demonstrate that Pfcrk-3 interacts with a histone deacetylase (HDAC) in parasite extracts, and we provide reverse genetics evidence strongly suggesting that the pfcrk-3 gene plays a crucial role in parasite proliferation during the asexual erythrocytic cycle.
MATERIALS AND METHODS
GST-Pfcrk-3 expression plasmid and site-directed mutagenesis.
The Pfcrk-3 catalytic domain was amplified from the 3D7 cDNA clone by using oligonucleotides carrying a BamHI (forward primer, CGGGGATCCGATAAAAGAATGTAAGTTACACA) or a SalI (reverse primer, GGGGTCGACTTATCCTTTTTGATTACTCTGT) site (underlined). The PCR product was inserted into the pGEX4T3 plasmid (Amersham Biosciences) at the BamHI and SalI sites. GST-Pfcrk-3-K445M, a plasmid encoding a mutant glutathione S-transferase (GST)-Pfcrk-3 fusion protein with an alteration from lysine to methionine at residue 445, was obtained by site-specific mutagenesis using the overlap extension PCR technique (21). The plasmids were electroporated into Escherichia coli strain BL21, and the inserts were verified by DNA sequencing prior to protein expression.
Expression and purification of recombinant proteins.
GST, GST-Pfcrk-3, and GST-Pfcrk-3-K445M were induced in Escherichia coli (strain BL21 codon+) with 0.5 mM isopropyl-β-thiogalactopyranoside at 30°C for 4 h. Cells were harvested and resuspended in ice-cold sonication buffer (phosphate-buffered saline [PBS] [pH 7.5], 0.1% Triton, 1 mM EDTA, 1 mM dithiothreitol [DTT]) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride and Complete mixture inhibitor tablet from Roche) and 100 μg/ml lysozyme. After 10 min on ice, the suspension was sonicated and clarified by centrifugation at 11,000 × g for 30 min at 4°C. The resulting supernatant was incubated with glutathione Sepharose resin (Sigma) for 1 h. The resin was washed four times with sonication buffer and once with a buffer containing 50 mM Tris-HCl (pH 8.7)-75 mM NaCl. The protein concentration was determined using the Bio-Rad dye reagent according to the manufacturer's recommendations with bovine serum albumin as a standard. Aliquots of purified proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining.
Parasite culture and preparation of parasite extracts.
The P. falciparum clone 3D7 was cultured in vitro by standard methods (25). The parasites were grown in human erythrocytes at 5% hematocrit in complete RPMI 1640 medium in 25-cm2 ventilated flasks. The flasks were kept in a 37°C incubator under 5% CO2. To remove serum and leukocytes, the human blood was washed three times in RPMI 1640 and the buffy coat removed. The medium was changed daily. Parasitemia was measured daily by examining Giemsa-stained blood smears and was kept between 0.5% and 10%.
Fresh or frozen saponin-lysed P. falciparum (3D7) pellets were sonicated in radioimmunoprecipitation assay (RIPA) buffer (30 mM Tris [pH 8.0], 150 mM NaCl, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT, 10 μM ATP, 0.5% Triton X-100, 1% NP-40, 10 mM β-glycerophosphate, 10 mM NaF, 0.1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM benzamidine, and Complete protease inhibitors). Lysates were cleared by centrifugation at 10,000 × g for 15 min at 4°C, and the total amount of protein in the supernatant was measured by the Bio-Rad protein assay.
Pulldown experiments.
Glutathione-agarose beads coated with GST, GST-Pfcrk-3, or GST-Pfcrk-3-K445M were incubated in parasite extracts or in RIPA buffer alone at 4°C under mild agitation for 1 h (100 μg of total parasite proteins for 10 μg of recombinant proteins on beads). The beads were then washed three times in RIPA buffer, once in RIPA buffer with 0.1% SDS, and once in a standard kinase buffer containing 10 mM sodium fluoride, 10 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, and Complete mixture protease inhibitors. Beads were resuspended in a volume of kinase buffer equal to the volume of beads. A standard kinase assay was then performed in a final volume of 30 μl, and the samples were analyzed by SDS-PAGE and autoradiography.
Kinase assays.
Kinase assays were performed as described previously (32). Briefly, reactions (30 μl) were performed in a standard kinase buffer containing 20 mM Tris-HCl (pH 7.5), 20 mM MgCl2, 2 mM MnCl2, 10 mM ATP, and 5 μCi [γ-32P]ATP, using 0.5 μg of recombinant kinase (or immunoprecipitated material [see below]) and 5 μg of substrate (myelin basic protein [MBP] or histone H1). After 30 min at 30°C, the reaction was stopped by the addition of Laemmli buffer, and the reaction product was loaded onto a 12% SDS-polyacrylamide gel. Following Coomassie blue staining, the gels were dried and exposed for autoradiography.
Antibody production, immunoprecipitation, and immunofluorescence analyses of wild-type Pfcrk-3.
Chicken immunoglobulin (IgY) antibodies against peptides H2N-CKNRRTLNEDMLSVVD-CONH2 (named VVD) and H2N-PNERDIKTLRNLPCTN-CONH2 (named PNG), derived from protein sequences (residues 539 to 553 and 840 to 855, respectively, encoded by the PFD0740w gene), were synthesized by Auspep, coupled to rabbit albumin carrier, and inoculated into chickens (the PNG peptide was derived from an early version of PlasmoDB, and the sequence was subsequently changed to PNERDIKYLRNLPCWN; the two substitutions appear not to have prevented recognition of Pfcrk-3 by the antibodies [see Results]). The antibodies were isolated and affinity purified on a peptide-affinity matrix as described previously (19). The IgYs were used in immunoprecipitation as described previously (38). Briefly, an anti-Pfcrk-3 antibody bound to protein A Sepharose beads was incubated with an extract from a parasite at a specific stage (late trophozoite or schizont). Because of the low affinity of chicken antibodies for protein A Sepharose beads, the antibody was first incubated with a rabbit anti-chicken antibody before being coupled to protein A Sepharose beads. After incubation, protein A Sepharose bead-bound complexes were washed and assayed for kinase activity.
For immunofluorescence analysis, P. falciparum-infected erythrocytes were fixed with 4% paraformaldehyde and 0.0075% glutaraldehyde as described previously (51). The cells were probed with chicken anti-Pfcrk-3 (1:400) and mouse anti-histone (1:500; Chemicon International) and were subsequently incubated with Alexa Fluor 488-conjugated goat anti-chicken IgY (Molecular Probes) and Alexa Fluor 555-conjugated goat anti-mouse IgG (Molecular Probes). Confocal images were acquired using a laser-scanning microscope (LSM 510; Carl Zeiss).
Immunoprecipitation of HA-tagged Pfcrk-3.
Parasites expressing hemagglutinin (HA)-tagged Pfcrk-3 or wild-type 3D7 parasites were obtained by saponin lysis and were then solubilized in M-PER protein extraction reagent (Pierce) containing 25 U/500 μl Benzonase and a protease inhibitor cocktail (Roche) for 20 min at 4°C. The lysates were cleared at 9,000 × g and 4°C for 5 min prior to immunoprecipitation (IP). For each IP, 500 μg protein lysate (in a 100-μl total volume) was incubated with 10 μl anti-HA agarose slurry (Pierce) overnight at 4°C and was then washed three times, for 5 min each time, with 500 μl of cold TBS (25 mM Tris, 0.15 M NaCl [pH 7.2]) buffer.
Histone deacetylase assay.
To detect HDAC enzyme activity in the Pfcrk-3 complex, duplicate samples were immunoprecipitated as described above, followed by a final wash in HDAC assay buffer (Millipore Corporation). The beads were further incubated for 16 h at 37°C in 60 μl HDAC assay buffer containing 100 μM acetylated fluorogenic peptide and 500 μM NAD cofactor with or without HDAC inhibitors (10 mM nicotinamide and/or 2 mM trichostatin A). To test for the presence of NAD-independent HDAC activities in the Pfcrk-3 immunoprecipitates, duplicate experiments were also performed in the absence of the cofactor. The beads were pelleted at 9,000 × g for 30 s, and 40 μl of the supernatant was transferred to each well of a half-volume plate. Twenty microliters of activator solution was added for 15 min, and the resulting fluorescence was recorded using a plate reader (excitation filter, 360/40 nm; emission filter, 460/40 nm).
Transfection plasmids. (i) pCAM-BSD-crk-3.
A 1,092-bp DNA fragment (nucleotides 1277 to 2368 of the pfcrk-3 open reading frame [ORF]) was amplified by PCR from P. falciparum genomic DNA, using primers (forward, GGGGGGATCCGCATATGGAGATGTTTGGATGGC; reverse, GGGGCGGCCGCTGGTGGTCTATACCATAATGTAATAACTC) containing BamHI or NotI sites (underlined). The amplicon was digested with BamHI and NotI and was inserted at these sites into the pCAM-BSD vector (48).
(ii) pCAM-BSD-crk-3-HA.
The pCAM-BSD-HA vector was generated by introducing a sequence encoding a single HA tag and the 3′ untranslated region (3′ UTR) of the P. berghei dhfr-ts gene into the multiple cloning site of pCAM-BSD (see Fig. 5A). The 3′ end of the Pfcrk-3 coding region (652 bp, omitting the stop codon) was amplified by PCR from genomic DNA, using primers with PstI or BamHI restriction sites, which allowed insertion of the amplified product into the pCAM-BSD-HA vector.
Fig. 5.

HA tagging of the pfcrk-3 locus. (A) Strategy for C-terminal tagging of pfcrk-3. The locations of PCR primers used for genotyping are indicated by arrows, restriction sites by vertical lines, and DNA probes by horizontal bars. See the text for details. (B) PCR analysis of HA-tagged locus. Total DNA isolated from blasticidin-resistant parasites transfected with pCAM-BSD-crk-3-HA and from wild-type 3D7 parasites was subjected to PCR using the primers indicated. (Left) Primers OL-3 and -4 (diagnostic for pCAM-BSD-crk-3 episome or concatemeric inserts); (right) primers OL-5 and -4 (diagnostic for 3′ integration). (C) Southern blot analysis. Total DNA was extracted from blasticidin-resistant parasites transformed with pCAM-BSD-crk-3 and from wild-type 3D7 parasites, and 3 μg was digested with PstI and SwaI, run on a 0.8% agarose gel, transferred to a Hybond membrane, and probed with the blasticidin resistance cassette (see panel A). The membrane was stripped and probed with a pfcrk-3 fragment that is not present in the pCAM-BSD-crk-3-HA plasmid (see panel A for the location of the probes, which are indicated by horizontal bars underneath the loci). The band corresponding to the linearized episome or concatemeric insert is detected in the transfected parasites (left panel, lower band). The band corresponding to the wild-type locus (right panel, right lane) is replaced by a larger band of the size expected from the recombination of the plasmid into the locus (both panels, left lanes).
Parasite transfection and genotype characterization.
Ring-stage parasites were electroporated with 60 μg of plasmid DNA (pCAM-BSD-crk-3 or pCAM-BSD-crk-3-HA) as described previously (16). Blasticidin was added to a final concentration of 2.5 μg/ml 48 h after transfection. Resistant parasites appeared 4 to 5 weeks posttransfection and were cloned by limiting dilution.
For PCR detection of (i) the integration of the pCAM-BSD-crk-3-HA plasmid at the 3′ flank of the insert, (ii) the integration of the pCAM-BSD-crk-3 knockout plasmid, and (iii) the episome, the following primers were used to amplify products from total DNA isolated from parasite lines (see below): OL-1 (GGTTCATCAAGTTGGACAAGGAGG), OL-2 (CACAACTCCACATATCAACCGATGC), OL-3 (TATTCCTAATCATGTAAATCTTAAA), OL-4 (CAATTAACCCTCACTAAAG), and OL-5 (GAAAGGCTTATCTTCGAAGTA). Primers OL-1, OL-2, and OL-5 correspond to pfcrk-3 sequences, while primers OL-3 and OL-4 correspond to pCAM-BSD vector sequences flanking the insertion site.
For Southern blot analysis, total DNA was obtained as follows. Parasite pellets obtained by saponin lysis were resuspended in PBS and were treated with 150 μg/ml proteinase K and 2% SDS at 55°C for 2 h. The DNA was precipitated with ethanol and 0.3 M sodium acetate after phenol-chloroform-isoamyl alcohol (25:24:1) extraction. The DNA was digested with PstI and SwaI, transferred to a Hybond N membrane, and hybridized to the pCAM-BSD or pfcrk-3 probe.
RESULTS
Bioinformatic analysis of Pfcrk-3.
Phylogenetic analysis of the P. falciparum kinome (3, 52) identified 18 protein kinases belonging to the CMGC group, including Pfcrk-3. Among the different families that constitute the CMGC group, Pfcrk-3 clearly clusters within the CDK family, and more precisely with the CDK8 to -11 group, which comprises CDKs involved in transcriptional control (Fig. 1). BLASTP analysis confirmed the relatedness of Pfcrk-3 to transcriptional CDKs, with the BUR1 enzyme from yeast giving the highest score (see Discussion). The 11 “invariant” residues that are conserved in most protein kinases (20, 29) are present in Pfcrk-3, as are the two signatures that define membership in the protein kinase family (4) (see Fig. S1 in the supplemental material). The polypeptide conforms with a high score to the Pfam protein kinase domain (Pfam entry PF00069; E value, 3.6e−77).
Fig. 1.

Three-species phylogenetic tree of CDKs. Sequences in red, blue, and black characters are from P. falciparum, S. cerevisiae, and Homo sapiens, respectively. Branches with bootstrap values of >70 are shown in red, and those with bootstrap values of >40 are shown in blue. (Adapted from reference 52.)
The predicted Pfcrk-3 polypeptide (1,339 amino acids) is unusually large for a CDK, because the putative catalytic domain contains two insertions (of 198 and 20 amino acids), and there are large N-terminal (378 residues) and C-terminal (335 residues) extensions (see Fig. S1 in the supplemental material). The extensions and insertions are rich in low-complexity regions and do not display homology to any characterized motif. The cyclin-binding motif (PSTAIRE in CDK1, PITALRE in CDK9, and PITQLRE in yeast BUR1) is replaced by AKTYIRE in Pfcrk-3. The Thr14 and Tyr15 residues (human CDK2 numbering) are the targets of negative regulation by phosphorylation in several mammalian CDKs; only the Tyr15 residue is present in Pfcrk-3, since Thr14 is replaced by an Ala residue. Thr160, which is the target of activating phosphorylation by CDK-activating kinases (CAKs), is conserved in Pfcrk-3.
Expression of pfcrk-3 mRNA and protein in blood stages.
Reverse transcription-PCR (RT-PCR) using primers flanking the catalytic domain showed that (i) Pfcrk-3 mRNA is present in asexual and sexual blood stages, (ii) the gene structure (2 exons separated by a 100-bp intron) proposed in PlasmoDB is correct, and (iii) transcribed mRNA includes the predicted N- and C-terminal extensions (see Fig. S2 in the supplemental material).
Microarray data available on PlasmoDB (33) indicate that pfcrk-3 mRNA is detectable throughout the asexual cycle (as well as in sporozoites and gametocytes). Northern blot analysis allowed the detection of a single 4-kb pfcrk-3 mRNA species in trophozoites, although the signal was very weak (Fig. 2A). Probing of the same membrane with pfrhoph2 (a gene expressed in schizonts) yielded a 7.5-kb mRNA species only in the schizont stage, as expected (36); equal loading was ascertained by ethidium bromide staining, which yielded rRNA bands of similar intensities in all lanes (data not shown).
Fig. 2.

Expression of Pfcrk-3 in asexual blood stages. (A) Northern blot analysis. Total RNA was extracted from synchronized 3D7 parasites (R, rings; T, trophozoites; S, schizonts) and subjected to Northern blot analysis. (Left) pfcrk-3 probe (catalytic domain); (right) pfrhoph2 probe. (B) Western blot analysis. (Left) The immunopurified IgY antibody against VVD (a peptide derived from the Pfcrk-3 catalytic domain) was used to probe extracts from rings, trophozoites, and schizonts. (Right) An extract from unsynchronized asexual parasites (U) was probed with immunopurified IgY directed against PNG, the large insertion within the Pfcrk-3 catalytic domain.
IgY antibodies against two peptides derived from Pfcrk-3 (one in the large insertion and the other in the catalytic domain [see Fig. S1 in the supplemental material]) recognized recombinant Pfcrk-3 in Western blots (data not shown) (see below for a description of the recombinant protein). Western blotting performed with extracts from synchronous parasites using the anti-VVD antibody (directed against the catalytic domain) showed that although Pfcrk-3 mRNA was detectable predominantly during early stages, the protein was detectable throughout the asexual cycle (Fig. 2B). Pfcrk-3 appears to be proteolytically processed from a large precursor in rings, whose size approximates the expected molecular size of the full-length protein (160 kDa), to a protein of around 120 kDa at later stages, with lower-intensity bands (including one at 70 kDa) also detectable (Fig. 2B, left). Determination of the exact processing events would require additional studies using antibodies directed against the various parts of the protein. The antibody directed against the largest insertion in the catalytic domain (anti-PNG antibody) (Fig. 2B, right) yielded a similar profile, suggesting that processing consists of removal of the extensions.
In view of the clean Western blot pattern obtained with the anti-VVD antibody, we next performed immunofluorescence analysis. Consistent with the Western blot data, the Pfcrk-3 signal was detectable in all intraerythrocytic stages of the parasite and largely colocalized with the parasite histone proteins (Fig. 3). In some ring-stage parasites (e.g., the top row in Fig. 3), Pfcrk-3 appears to localize at the periphery of the nucleus, a pattern similar to the “horseshoe” pattern observed by Issar et al. with antibodies against specific histone modifications (24).
Fig. 3.

Pfcrk-3 colocalizes with histones. Images captured by laser scanning confocal microscopy show substantial colocalization of Pfcrk-3 (green) with nuclearly localized histones (red). P. falciparum 3D7 parasites were fixed with 4% paraformaldehyde-0.0075% glutaraldehyde and were probed with chicken anti-Pfcrk-3 IgY and mouse anti-histone IgG antibodies. The data in each row represent the fluorescence profile in ring-stage parasites, early trophozoites, late trophozoites, or schizonts, as indicated. DIC, differential interference contrast. Bars, 5 μm in the top row and 2 μm in all other rows.
Pfcrk-3 is crucial for asexual proliferation.
In an attempt to determine whether or not Pfcrk-3 is essential for erythrocytic development, P. falciparum 3D7 parasites were transfected with a pCAM-BSD-based construct containing the central portion of the pfcrk-3 gene in order to disrupt the locus (Fig. 4A). The resulting blasticidin-resistant parasites were then monitored for integration-specific PCR products. No integration-specific products were observed even after prolonged maintenance of the culture (up to 5 months); the only PCR products obtained corresponded to the unintegrated episome (Fig. 4B). Southern blot analysis confirmed the integrity of the wild-type locus in the transfected cells (Fig. 4C). Failure to disrupt the pfcrk-3 gene may signify either that the gene is essential for parasite asexual proliferation or that the vector was unable to recombine with the locus. To verify that the pfcrk-3 locus was indeed accessible to recombination, we attempted to modify the locus without causing loss of function of the gene product. For this purpose, we transfected wild-type parasites with the pCAM-BSD-Pfcrk-3-HA plasmid containing the 3′ end of the Pfcrk-3 coding region fused to a hemagglutinin (HA) epitope followed by the 3′ untranslated region (3′ UTR) from the P. berghei dhfr-ts gene (Fig. 5A). Following single-crossover recombination, we expect an HA-tagged, functional Pfcrk-3 protein to be expressed, but we expect no expression from the wild-type enzyme. PCR analysis of the uncloned blasticidin-resistant population performed 14 weeks after transfection readily detected integration of the construct into the pfcrk-3 locus (Fig. 5B, primers OL-5 and -4), in addition to the episome (Fig. 5B, primers OL-3 and -4). Cloned lines were obtained by limiting dilution, and PCR examination of the genotypes of individual clones at the pfcrk-3 locus demonstrated that several clones had lost the wild-type locus (data not shown). The disappearance of the wild-type locus in these clones and the presence of a modified locus of the expected size were verified by Southern blot analysis. Figure 5C shows the data for one such clone, which displays a modified locus and has either retained the episome or integrated concatemers of the plasmid.
Fig. 4.

Attempt at disrupting the pfcrk-3 gene. (A) Strategy for disruption of the pfcrk-3 gene. The transfection plasmid contains a PCR fragment spanning positions 1277 to 2368 of the 4.2-kb pfcrk-3 coding sequence. This fragment excludes two kinase subdomains essential for activity, labeled GXGXXG (a glycine-rich region required for correct orientation of ATP) and PE (a proline-glutamate motif in which the latter residue is required for the structural stability of the enzyme). Single-crossover homologous recombination results in a pseudodiploid configuration with two truncated copies, each of which lacks one of these essential motifs. Oligonucleotides are indicated by horizontal arrows, restriction sites by vertical lines, and DNA probes by horizontal bars. BSD, blasticidine deaminase cassette. (B) PCR analysis of the disrupted locus. Total DNA isolated from cloned, blasticidin-resistant parasites transfected with pCAM-BSD-crk-3 (transfectant) or from wild-type 3D7 parasites (3D7) was subjected to PCR using the primers indicated (see panel A for their locations). (Left) Primers OL-3 and -4 (diagnostic for the pCAM-BSD-crk-3 episome or concatemeric inserts); (center) primers OL-1 and -4 (diagnostic for 5′ integration); (right) primers OL-3 and -2 (diagnostic for 3′ integration). (C) Southern blot analysis. Total DNA was extracted from cloned blasticidin-resistant parasites transformed with pCAM-BSD-crk-3 (transfectant) and from wild-type 3D7 parasites (3D7) and was digested with PstI and SwaI. (Left) After transfer to a Hybond membrane, the digested DNA was probed with the blasticidin resistance cassette. (Right) The membrane was stripped, and the digested DNA was probed with a pfcrk-3 amplicon located upstream of the fragment used as an insert in the pCAM-BSD-crk-3 plasmid. The sizes of comigrating markers are given to the left. The band corresponding to the linearized episome was detected in the transfected parasites (left panel, left lane). The band corresponding to the intact wild-type locus was detected in both the transfected and the untransfected parasites (right panel, both lanes).
Association of Pfcrk-3 with protein kinase and histone deacetylase activities in parasite extracts.
In view of (i) the demonstrated role of BUR1 (the enzyme of the yeast kinome that is the closest relative to Pfcrk-3) in the regulation of chromatin modification (including through histone acetylation) (7) and (ii) the emerging importance of histone deacetylases in the regulation of gene expression in Plasmodium (see reference 50 for a recent contribution to the field), we set out to investigate whether histone deacetylase activity could be copurified with Pfcrk-3 from P. falciparum extracts. Unfortunately, the anti-Pfcrk-3 IgYs did not perform satisfactorily in immunoprecipitation assays (data not shown). We therefore resorted to the parasite line expressing an HA-tagged version of Pfcrk-3 (described in the preceding section). We first verified that an immunoprecipitate obtained with anti-HA antibodies from transgenic, but not from wild-type parasites, contained a protein kinase activity, using mammalian histone H1 (a classical substrate for assaying CDK activity) as a phosphate receiver. Indeed, kinase activity was much higher in the HA immunoprecipitate obtained from parasites expressing HA-tagged Pfcrk-3 than from untransfected 3D7 parasites (Fig. 6A), indicating that Pfcrk-3 is associated, directly or indirectly, with kinase activity (see below). The autoradiogram displays many high-molecular-weight bands in addition to the histone H1 added as a substrate; these may represent copurifying P. falciparum proteins (including Pfcrk-3 itself) acting as substrates. We then subjected the HA-immunoprecipitated material to a histone deacetylase activity assay in the presence or absence of NAD as a cofactor. As shown in Fig. 6B, no activity was obtained after immunoprecipitation from extracts of wild-type parasites (bar 1); only samples from parasites expressing the HA-tagged protein (bars 2 to 6) exhibited histone deacetylase activity. Addition of the cofactor resulted in an increase in enzyme activity (compare bars 2 and 3), suggesting the presence of both NAD-dependent and NAD-independent HDAC activities in the Pfcrk-3-associated complexes. This finding is consistent with the activity of recombinant PfSir2 as reported previously (45). Interestingly, the Pfcrk-3-associated HDAC activities were sensitive to the sirtuin inhibitor nicotinamide and the class I/II HDAC inhibitor trichostatin A. Together these data indicate that Pfcrk-3 is part of one or more complexes whose components are capable of protein phosphorylation and histone deacetylation and that it may play a role in chromatin modifications by associating with various HDAC enzymes in P. falciparum.
Fig. 6.

Immunoprecipitated HA-Pfcrk-3 associates with protein kinase and histone deacetylase activities. (A) Association of HA-Pfcrk-3 with kinase activity. Immunoprecipitates obtained with anti-HA antibodies from untransfected wild-type 3D7 parasites (lane 1) or from parasites expressing HA-tagged Pfcrk-3 (lane 2) were subjected to a kinase activity assay in a standard 30-μl reaction mixture containing 5 μg histone H1 as a substrate. The kinase reactions were then run on a 12% polyacrylamide gel, which was dried and subjected to autoradiography. (B) Histone deacetylase assay. Immunoprecipitates obtained with anti-HA antibodies from untransfected wild-type 3D7 parasites (bar 1) or from the HA-tagged Pfcrk3-expressing cell line (bars 2 to 6) were subjected to HDAC assays (Millipore). With the exception of the experiment for which results are shown in bar 2, the immunoprecipitates were incubated for 16 h at 37°C in a substrate buffer containing the sirtuin cofactor NAD. HDAC inhibitors (10 μM nicotinamide [Nmide] or 2 μM trichostatin A [TrichoA]) were used separately (bars 4 and 6) or together (bar 5). Reactions were terminated with activator solution followed by fluorescence measurements (excitation wavelength, 360 nm; emission wavelength, 460 nm). Error bars represent standard deviations from duplicate experiments.
Kinase activity and pulldown assays with recombinant GST-Pfcrk-3.
In an attempt to demonstrate that Pfcrk-3 itself possesses kinase activity, a polypeptide containing the catalytic domain plus the C-terminal extension was expressed in E. coli as a 100-kDa GST fusion (GST-Pfcrk-3). No kinase activity of the purified recombinant enzyme was observed under our experimental conditions, whereas other recombinant P. falciparum kinases were active (data not shown). We reasoned that activity of GST-Pfcrk-3 might require interaction with a cyclin-like activator. Four plasmodial cyclin-like proteins have been cloned in our laboratory (Pfcyc-1, Pfcyc-2, Pfcyc-3, and Pfcyc-4), two of which (Pfcyc-1 and Pfcyc-3) activate recombinant PfPK5 (another CDK-related enzyme) in vitro (38). Incubation of Pfcrk-3 with these four different recombinant cyclins did not cause activation of the recombinant enzyme, nor did addition of RINGO, a potent activator of some CDKs, including PfPK5 (32, 42) (data not shown).
The absence of activity of the recombinant Pfcrk-3 catalytic domain even in the presence of cyclins may result from the fact that the fusion protein lacks the large N-terminal extension, or from the absence of additional activator mechanisms, such as phosphorylation by other kinases. To address the latter issue, we repeated the kinase assay following incubation of recombinant Pfcrk-3 in parasite extracts. Pulldown experiments, in which glutathione-agarose beads loaded with GST-Pfcrk-3 were incubated in extracts from asynchronous parasites, washed, and subjected to in vitro kinase activity assays, allowed us to detect histone H1 kinase activity associated with the recombinant enzyme (Fig. 7 A, lane 2). Furthermore, under these conditions, we also observed a weak signal at approximately 100 kDa, which is likely to be GST-Pfcrk-3 itself. A much weaker signal was detected when the pulldown from the parasite extract was performed with the GST moiety alone (Fig. 7A, lane 1), and no signal was observed when GST-Pfcrk-3 was incubated in parasite lysis buffer that did not contain parasite proteins (lane 4). The activity observed in Fig. 7A, lane 2 (phosphorylation of histone H1 and of GST-Pfcrk-3), might result either from activation of Pfcrk-3 itself by a component of the pulled complex (e.g., through binding of a cyclin-like regulator or through a phosphorylation event) or from another protein kinase that had been pulled down by GST-Pfcrk-3. To distinguish between these possibilities, we repeated the pulldown experiment using a kinase-dead mutant of GST-Pfcrk-3 (GST-Pfcrk-3-K445M) in which a conserved lysine residue involved in ATP orientation was replaced by a methionine residue (Fig. 7B). This yielded a signal of an intensity similar to that obtained when the pulldown was performed with the wild-type enzyme (Fig. 7B, lanes 2 and 3), suggesting that another plasmodial protein kinase that interacts with Pfcrk-3 was responsible for at least a fraction of the detected kinase activity identified in Fig. 7A.
Fig. 7.
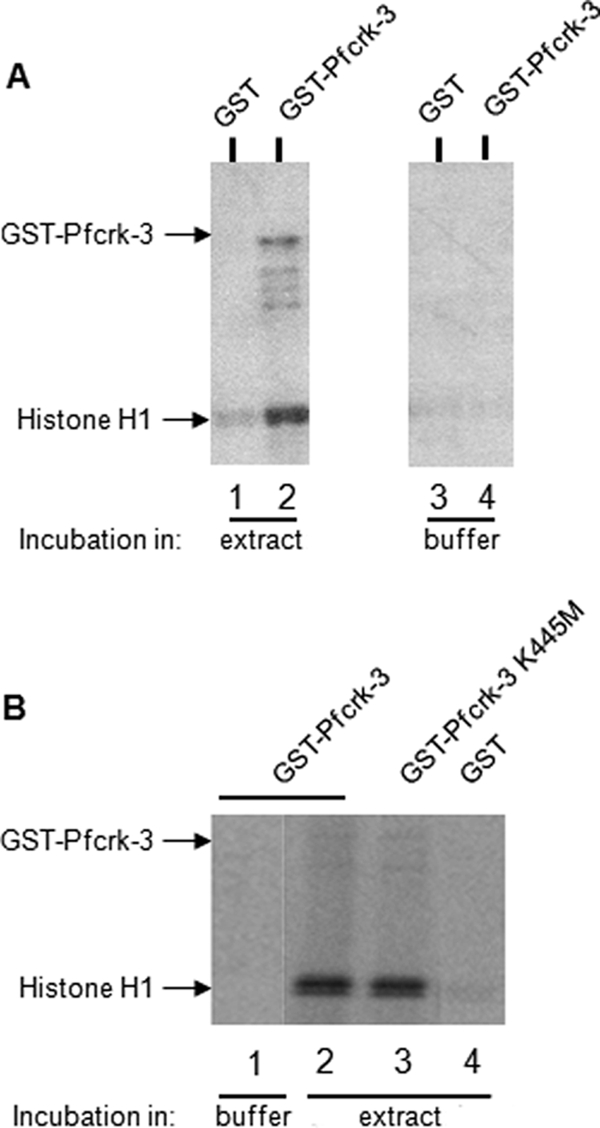
Association of recombinant GST-Pfcrk-3 with histone H1 kinase activity. (A) Pulldown experiments using wild-type GST-Pfcrk-3. One microgram of GST-Pfcrk-3 (lanes 2 and 4) or GST alone (lanes 1 and 3) bound to glutathione-agarose beads was incubated in parasite extract (lanes 1 and 2) or in parasite lysis buffer as a negative control (lanes 3 and 4). The beads were washed and resuspended in kinase assay buffer, and in vitro kinase assays were performed by addition of a reaction mixture containing radiolabeled ATP and histone H1. (B) Pulldown experiments using wild-type and kinase-dead GST-Pfcrk-3, followed by kinase assays. Portions (0.5 μg) of GST-Pfcrk-3 (lanes 1 and 2), GST-Pfcrk-3-K445M (lane 3), or GST alone (lane 4) bound to glutathione-agarose beads were incubated in parasite extracts (lanes 2 to 4) or in parasite lysis buffer as a negative control (lane 1). The beads were washed, and in vitro kinase assays were performed by addition of a reaction mixture containing the radiolabeled ATP and histone H1.
Pulldown experiments thus demonstrate that Pfcrk-3 is associated with kinase activity in parasite extracts and that at least part of this activity is due to another kinase that interacts with Pfcrk-3; we favor the hypothesis that Pfcrk-3 itself also possesses activity in vivo (see Discussion).
DISCUSSION
Bioinformatics considerations.
BLASTP analysis showed that the Pfcrk-3 kinase domain displays maximal homology to BUR1 (BLASTP score, 164; E-value, 1e−40), a yeast transcriptional CDK-related kinase previously described as SGV1 and required for recovery from mating pheromone-induced cell cycle arrest (23). When complexed to its cyclin partner, BUR2, BUR1/SGV1 phosphorylates the carboxy-terminal end of RNA polymerase II (40, 54, 55) and other transcription-related substrates (28) and regulates trimethylation (30). Thus, the BLASTP results are fully consistent with the phylogenetic analysis in which Pfcrk-3 clustered with the transcriptional CDKs, which include SGV1 and human CDK9 (Fig. 1). Another feature shared by Pfcrk-3 and BUR1 is a long C-terminal extension that is usually not found in other CDKs (including mammalian CDK9); remarkably, this extension has exactly the same size (335 amino acids) in both enzymes. Even though the Pfcrk-3 protein appears to be processed during parasite development (Fig. 2B), the C-terminal extension is presumably maintained, since the C-terminally HA-tagged enzyme can be recovered via the tag (Fig. 6).
Many CDKs are negatively regulated by phosphorylation of Thr14 and Tyr15 (human CDK2 numbering), of which only Tyr15 is conserved in Pfcrk-3. Interestingly, the inverse configuration is observed in BUR1 and CDK9, where only Thr14 is conserved; this may indicate different modes of regulation between Pfrck-3 and transcriptional CDKs in Opisthokhonts (the phylum including yeast and metazoans). In contrast, the conservation in Pfcrk-3, yeast BUR1, and human CDK9 of Thr160, the target of activating phosphorylation by CDK-activating kinases (CAKs), is consistent with the observation that BUR1 is activated by CAKs (55) and suggests that a similar mechanism may regulate Pfcrk-3 activity.
Pfcrk-3 function.
Successful in situ 3′ HA tagging (Fig. 5) establishes that the pfcrk-3 locus is recombinogenic, strongly suggesting that the lack of success in obtaining parasites with a disrupted locus is due to the fact that the gene is crucial for asexual proliferation. The possibility remains that Pfcrk-3 inactivation is not strictly lethal but causes a growth rate defect that renders parasites unable to compete with parasites that retain a wild-type locus in the transfected population. In this context, it is noteworthy that in addition to the inability of BUR1 mutants to recover from mating pheromone-dependent cell cycle arrest (23), normal growth under various conditions is affected in BUR1 and BUR2 mutants (54).
Thus, bioinformatics analyses, subcellular localization, and association with histone deacetylase activity all concur to assign Pfcrk-3 as a chromatin-associated CDK involved in the regulation of transcription. It would be of great interest, in order to gain further insight into the precise function of Pfcrk-3, to identify which of the several putative HDACs (one class I HDAC, two class II HDACs, and two class III sirtuins [44]) encoded by the P. falciparum genome is responsible for the activity we observed to be associated with HA-Pfcrk-3. Sensitivity to nicotinamide suggests that a class III HDAC, such as Pfsir2, is involved (45), but caution must be exercised until the enzyme is experimentally identified. The parasites expressing HA-tagged Pfcrk-3 represent a tool that can now be used for affinity chromatography/mass spectrometry-based identification of this and other components of the protein complex that includes Pfcrk-3. Further information on the role of Pfcrk-3 in gene expression could be gained by performing high-resolution colocalization studies to determine whether the spatial distribution of Pfcrk-3 correlates with that of specific histone modifications, as hinted by the “horseshoe” appearance of the protein distribution pattern in ring-stage parasites (Fig. 3) (24).
A recombinant protein containing the catalytic domain and the C-terminal extension, but lacking the N-terminal extension, displayed no enzymatic activity. This is expected for a CDK homologue; for example, no activity was observed with the PfPK5 CDK in the absence of a cyclin (or equivalent) activator (32). However, in contrast to what we observed with PfPK5, addition of recombinant cyclins to the reaction mixture did not result in Pfcrk-1 activation. We showed that a histone H1 kinase activity can be pulled down from parasite extracts using recombinant Pfcrk-3; kinase activity was also recovered when a kinase-dead mutant was used in the pulldown assay, suggesting that another protein kinase is present in a complex that includes Pfcrk-3. Taken together, our data are consistent with the proposition that Pfcrk-3 functions in a large complex regulating transcription. In other systems, it is well established that transcriptional complexes contain several PKs, including CDKs. If, as we suspect in view of the conservation of all residues that are important for activity, Pfcrk-3 is indeed an active CDK, it will of course be crucial for our understanding of its function to identify substrates of the enzyme. The present study generated the tools necessary to proceed with investigations in these areas and constitutes a solid basis for further work aimed at understanding the control of gene expression in malaria parasites.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by INSERM, the European Commission (FP6 ANTIMAL project and BioMalPar Network of Excellence, and FP7 MALSIG project and EviMalar Network of Excellence), and a grant from the Novartis Institute for Tropical Diseases (Singapore). J.H., L.E., and K.L.R. were supported by studentships from the French Ministère de la Défense (Délégation Générale pour l'Armement [DGA]); D.G.'s work on generating IgYs was funded by the South African National Research Foundation and Medical Research Council; and work in the D.C. laboratory is funded by a grant from the U.S. National Institutes of Health (AI73795).
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
Published ahead of print on 19 March 2010.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1. Abe M. K., Saelzler M. P., Espinosa R., III, Kahle K. T., Hershenson M. B., Le Beau M. M., Rosner M. R. 2002. ERK8, a new member of the mitogen-activated protein kinase family. J. Biol. Chem. 277:16733–16743 [DOI] [PubMed] [Google Scholar]
- 2. Akoulitchev S., Chuikov S., Reinberg D. 2000. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 407:102–106 [DOI] [PubMed] [Google Scholar]
- 3. Anamika, Srinivasan N., Krupa A. 2005. A genomic perspective of protein kinases in Plasmodium falciparum. Proteins 58:180–189 [DOI] [PubMed] [Google Scholar]
- 4. Bairoch A., Claverie J. M. 1988. Sequence patterns in protein kinases. Nature 331:22. [DOI] [PubMed] [Google Scholar]
- 5. Bracchi-Ricard V., Barik S., Delvecchio C., Doerig C., Chakrabarti R., Chakrabarti D. 2000. PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochem. J. 347(Pt. 1):255–263 [PMC free article] [PubMed] [Google Scholar]
- 6. Callebaut I., Prat K., Meurice E., Mornon J. P., Tomavo S. 2005. Prediction of the general transcription factors associated with RNA polymerase II in Plasmodium falciparum: conserved features and differences relative to other eukaryotes. BMC Genomics 6:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chu Y., Simic R., Warner M. H., Arndt K. M., Prelich G. 2007. Regulation of histone modification and cryptic transcription by the Bur1 and Paf1 complexes. EMBO J. 26:4646–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coulson R. M., Hall N., Ouzounis C. A. 2004. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res. 14:1548–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deitsch K., Duraisingh M., Dzikowski R., Gunasekera A., Khan S., Le Roch K., Llinas M., Mair G., McGovern V., Roos D., Shock J., Sims J., Wiegand R., Winzeler E. 2007. Mechanisms of gene regulation in Plasmodium. Am. J. Trop. Med. Hyg. 77:201–208 [PubMed] [Google Scholar]
- 10. De Silva E. K., Gehrke A. R., Olszewski K., Leon I., Chahal J. S., Bulyk M. L., Llinas M. 2008. Specific DNA-binding by apicomplexan AP2 transcription factors. Proc. Natl. Acad. Sci. U. S. A. 105:8393–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dhavan R., Tsai L. H. 2001. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2:749–759 [DOI] [PubMed] [Google Scholar]
- 12. Doerig C. 2005. Protein kinases regulating Plasmodium development, p. 290–310In Sherman I. W. (ed.), Molecular approaches to malaria. ASM Press, Washington, DC [Google Scholar]
- 13. Doerig C., Chakrabarti D. 2004. Cell cycle control in Plasmodium falciparum: a genomics perspective, p. 249–287 In Waters A. P., Janse C. J. (ed.), Malaria parasites, genomes, and molecular biology. Caister Academic Press, Wymondham, United Kingdom [Google Scholar]
- 14. Doerig C., Horrocks P., Coyle J., Carlton J., Sultan A., Arnot D., Carter R. 1995. Pfcrk-1, a developmentally regulated cdc2-related protein kinase of Plasmodium falciparum. Mol. Biochem. Parasitol. 70:167–174 [DOI] [PubMed] [Google Scholar]
- 15. Doerig C., Meijer L. 2007. Antimalarial drug discovery: targeting protein kinases. Expert Opin. Ther. Targets 11:279–290 [DOI] [PubMed] [Google Scholar]
- 16. Dorin-Semblat D., Quashie N., Halbert J., Sicard A., Doerig C., Peat E., Ranford-Cartwright L., Doerig C. 2007. Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Mol. Microbiol. 65:1170–1180 [DOI] [PubMed] [Google Scholar]
- 17. Finn R. D., Mistry J., Schuster-Bockler B., Griffiths-Jones S., Hollich V., Lassmann T., Moxon S., Marshall M., Khanna A., Durbin R., Eddy S. R., Sonnhammer E. L., Bateman A. 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34:D247–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garriga J., Grana X. 2004. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene 337:15–23 [DOI] [PubMed] [Google Scholar]
- 19. Goldring J. P., Thobakgale C., Hiltunen T., Coetzer T. H. 2005. Raising antibodies in chickens against primaquine, pyrimethamine, dapsone, tetracycline, and doxycycline. Immunol. Invest. 34:101–114 [PubMed] [Google Scholar]
- 20. Hanks S. K., Quinn A. M. 1991. Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 200:38–62 [DOI] [PubMed] [Google Scholar]
- 21. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 22. Holton S., Merckx A., Burgess D., Doerig C., Noble M., Endicott J. 2003. Structures of P. falciparum PfPK5 test the CDK regulation paradigm and suggest mechanisms of small molecule inhibition. Structure (Camb.) 11:1329–1337 [DOI] [PubMed] [Google Scholar]
- 23. Irie K., Nomoto S., Miyajima I., Matsumoto K. 1991. SGV1 encodes a CDC28/cdc2-related kinase required for a G alpha subunit-mediated adaptive response to pheromone in S. cerevisiae. Cell 65:785–795 [DOI] [PubMed] [Google Scholar]
- 24. Issar N., Ralph S. A., Mancio-Silva L., Keeling C., Scherf A. 2009. Differential sub-nuclear localisation of repressive and activating histone methyl modifications in P. falciparum. Microbes Infect. 11:403–407 [DOI] [PubMed] [Google Scholar]
- 25. Jensen J. B., Trager W. 1978. Some recent advances in the cultivation of Plasmodium falciparum. Isr. J. Med. Sci. 14:563–570 [PubMed] [Google Scholar]
- 26. Kaldis P. 1999. The cdk-activating kinase (CAK): from yeast to mammals. Cell. Mol. Life Sci. 55:284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kasten M., Giordano A. 2001. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene 20:1832–1838 [DOI] [PubMed] [Google Scholar]
- 28. Keogh M. C., Podolny V., Buratowski S. 2003. Bur1 kinase is required for efficient transcription elongation by RNA polymerase II. Mol. Cell. Biol. 23:7005–7018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knighton D. R., Zheng J. H., Ten Eyck L. F., Xuong N. H., Taylor S. S., Sowadski J. M. 1991. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253:414–420 [DOI] [PubMed] [Google Scholar]
- 30. Laribee R. N., Krogan N. J., Xiao T., Shibata Y., Hughes T. R., Greenblatt J. F., Strahl B. D. 2005. BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr. Biol. 15:1487–1493 [DOI] [PubMed] [Google Scholar]
- 31. Leete T., Rubin H. 1996. Malaria and the cell cycle. Parasitol. Today 12:442–444 [DOI] [PubMed] [Google Scholar]
- 32. Le Roch K., Sestier C., Dorin D., Waters N., Kappes B., Chakrabarti D., Meijer L., Doerig C. 2000. Activation of a Plasmodium falciparum cdc2-related kinase by heterologous p25 and cyclin H. Functional characterization of a P. falciparum cyclin homologue. J. Biol. Chem. 275:8952–8958 [DOI] [PubMed] [Google Scholar]
- 33. Le Roch K. G., Zhou Y., Blair P. L., Grainger M., Moch J. K., Haynes J. D., De La Vega P., Holder A. A., Batalov S., Carucci D. J., Winzeler E. A. 2003. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301:1503–1508 [DOI] [PubMed] [Google Scholar]
- 34. Li J. L., Robson K. J., Chen J. L., Targett G. A., Baker D. A. 1996. Pfmrk, a MO15-related protein kinase from Plasmodium falciparum. Gene cloning, sequence, stage-specific expression and chromosome localization. Eur. J. Biochem. 241:805–813 [DOI] [PubMed] [Google Scholar]
- 35. Li Z., Le Roch K., Geyer J. A., Woodard C. L., Prigge S. T., Koh J., Doerig C., Waters N. C. 2001. Influence of human p16(INK4) and p21(CIP1) on the in vitro activity of recombinant Plasmodium falciparum cyclin-dependent protein kinases. Biochem. Biophys. Res. Commun. 288:1207–1211 [DOI] [PubMed] [Google Scholar]
- 36. Ling I. T., Kaneko O., Narum D. L., Tsuboi T., Howell S., Taylor H. M., Scott-Finnigan T. J., Torii M., Holder A. A. 2003. Characterisation of the rhoph2 gene of Plasmodium falciparum and Plasmodium yoelii. Mol. Biochem. Parasitol. 127:47–57 [DOI] [PubMed] [Google Scholar]
- 37. Loyer P., Trembley J. H., Katona R., Kidd V. J., Lahti J. M. 2005. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell. Signal. 17:1033–1051 [DOI] [PubMed] [Google Scholar]
- 38. Merckx A., Le Roch K., Nivez M. P., Dorin D., Alano P., Gutierrez G. J., Nebreda A. R., Goldring D., Whittle C., Patterson S., Chakrabarti D., Doerig C. 2003. Identification and initial characterization of three novel cyclin-related proteins of the human malaria parasite Plasmodium falciparum. J. Biol. Chem. 278:39839–39850 [DOI] [PubMed] [Google Scholar]
- 39. Morgan D. O. 1997. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13:261–291 [DOI] [PubMed] [Google Scholar]
- 40. Murray S., Udupa R., Yao S., Hartzog G., Prelich G. 2001. Phosphorylation of the RNA polymerase II carboxy-terminal domain by the Bur1 cyclin-dependent kinase. Mol. Cell. Biol. 21:4089–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Naughton J. A., Bell A. 2007. Studies on cell-cycle synchronization in the asexual erythrocytic stages of Plasmodium falciparum. Parasitology 134:331–337 [DOI] [PubMed] [Google Scholar]
- 42. Nebreda A. R. 2006. CDK activation by non-cyclin proteins. Curr. Opin. Cell Biol. 18:192–198 [DOI] [PubMed] [Google Scholar]
- 43. Olliaro P. 2005. Drug resistance hampers our capacity to roll back malaria. Clin. Infect. Dis. 41(Suppl. 4):S247–S257 [DOI] [PubMed] [Google Scholar]
- 44. Patel V., Mazitschek R., Coleman B., Nguyen C., Urgaonkar S., Cortese J., Barker R. H., Greenberg E., Tang W., Bradner J. E., Schreiber S. L., Duraisingh M. T., Wirth D. F., Clardy J. 2009. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 52:2185–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prusty D., Mehra P., Srivastava S., Shivange A. V., Gupta A., Roy N., Dhar S. K. 2008. Nicotinamide inhibits Plasmodium falciparum Sir2 activity in vitro and parasite growth. FEMS Microbiol. Lett. 282:266–272 [DOI] [PubMed] [Google Scholar]
- 46. Rangarajan R., Bei A., Henry N., Madamet M., Parzy D., Nivez M. P., Doerig C., Sultan A. 2006. Pbcrk-1, the Plasmodium berghei orthologue of P. falciparum cdc-2 related kinase-1 (Pfcrk-1), is essential for completion of the intraerythrocytic asexual cycle. Exp. Parasitol. 112:202–207 [DOI] [PubMed] [Google Scholar]
- 47. Ross-Macdonald P. B., Graeser R., Kappes B., Franklin R., Williamson D. H. 1994. Isolation and expression of a gene specifying a cdc2-like protein kinase from the human malaria parasite Plasmodium falciparum. Eur. J. Biochem. 220:693–701 [DOI] [PubMed] [Google Scholar]
- 48. Sidhu A. B., Valderramos S. G., Fidock D. A. 2005. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol. Microbiol. 57:913–926 [DOI] [PubMed] [Google Scholar]
- 49. Stoeckert C. J., Jr., Fischer S., Kissinger J. C., Heiges M., Aurrecoechea C., Gajria B., Roos D. S. 2006. PlasmoDB v5: new looks, new genomes. Trends Parasitol. 22:543–546 [DOI] [PubMed] [Google Scholar]
- 50. Tonkin C. J., Carret C. K., Duraisingh M. T., Voss T. S., Ralph S. A., Hommel M., Duffy M. F., Silva L. M., Scherf A., Ivens A., Speed T. P., Beeson J. G., Cowman A. F. 2009. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 7:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tonkin C. J., van Dooren G. G., Spurck T. P., Struck N. S., Good R. T., Handman E., Cowman A. F., McFadden G. I. 2004. Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol. Biochem. Parasitol. 137:13–21 [DOI] [PubMed] [Google Scholar]
- 52. Ward P., Equinet L., Packer J., Doerig C. 2004. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics 5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao Z., Waters N. C., Woodard C. L., Li Z., Li P. K. 2001. Design and synthesis of Pfmrk inhibitors as potential antimalarial agents. Bioorg. Med. Chem. Lett. 11:2875–2878 [DOI] [PubMed] [Google Scholar]
- 54. Yao S., Neiman A., Prelich G. 2000. BUR1 and BUR2 encode a divergent cyclin-dependent kinase-cyclin complex important for transcription in vivo. Mol. Cell. Biol. 20:7080–7087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yao S., Prelich G. 2002. Activation of the Bur1-Bur2 cyclin-dependent kinase complex by Cak1. Mol. Cell. Biol. 22:6750–6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.