

Abstract
Background
A low level of plasma high-density lipoprotein cholesterol (HDL-C) is a risk factor for cardiovascular disease. HDL particles are modulated by a variety of lipases, including endothelial lipase (EL), a phospholipase present on vascular endothelial cells. The proprotein convertase subtilisin/kexin type 5 (PCSK5) gene product is known to directly inactivate EL, and, indirectly, cleave and activate angiopoetin-like protein 3, a natural inhibitor of EL. We therefore investigated the effect of human PCSK5 genetic variants on plasma HDL-C levels.
Methods and Results
Haplotypes at the PCSK5 locus were examined in nine multi-generational families that included 60 individuals with HDL-C<10th percentile. Segregation with low HDL-C in one family was found. Sequencing of the PCSK5 gene in 12 probands with HDL-C<5th percentile identified seven novel variants. Using a two-stage design, we first genotyped these single-nucleotide polymorphisms (SNPs) along with 163 tagSNPs and 12 additional SNPs (n=182 total) in 457 individuals with documented coronary artery disease. We identified nine SNPs associated with HDL-C (P<0.05), with the strongest results for rs11144782 and rs11144766 (P=0.002 and P=0.005 respectively). The SNP rs11144782 was also associated with very low-density lipoprotein (P=0.039), triglycerides (P=0.049) and total apolipoprotein B levels (P=0.022). In stage 2, we replicated the association of rs11144766 with HDL-C (P=0.014) in an independent sample of Finnish low HDL-C families. In a combined analysis of both stages (n=883), region-wide significance of rs11144766 and low HDL-C was observed (unadjusted P=1.86×10−4 and Bonferroni adjusted P=0.031).
Conclusions
We conclude that variability at the PCSK5 locus influences HDL-C levels, possibly through the inactivation of EL activity and consequently, atherosclerotic cardiovascular disease risk.
Keywords: cholesterol, coronary disease, genetics, lipids, lipoproteins
INTRODUCTION
Low levels of high-density lipoprotein cholesterol (HDL-C) constitute a major risk factor for coronary artery disease (CAD)1. Genetic factors account for a large component of the plasma variability of HDL-C levels with heritability being approximately 0.502. Numerous human genetic studies have identified loci contributing to HDL-C, including both linkage analyses and association studies, explaining some these variations. Despite these, the genetic and metabolic factors that regulate HDL metabolism remain incompletely understood.
The metabolism of HDL is complex and involves a carefully orchestrated interplay between the biogenesis of nascent HDL particles3, the continual exchange of lipid and protein moieties of HDL in plasma and the modulation of HDL particles by a variety of enzymes, especially lipases4. Endothelial lipase (EL), discovered by Rader and colleagues5, is a phospholipase present on vascular endothelial cells. It can be inactivated by angiopoetin-like protein 3 (ANGPTL3)6 or by secretory proprotein convertases of the subtilisin/kexin type, such as Furin, PC5/6, and PACE47. The mammalian proprotein convertases comprise a family of nine members related to bacterial subtilisin and yeast kexin-like serine proteinases, critically involved in the activation/inactivation of various physiological and pathological processes, including those implicated in regulation of vascular events. These include PC1/3, PC2, Furin, PC4, PC5/6, PACE4, PC7, SKI-1/S1P and PCSK9, encoded by the genes PCSK1 to PCSK9.
While PCSK9 has been found to play a critical role in regulating lipid levels by enhancing low-density lipoprotein (LDL) receptor degradation9, proof of in vivo functions of the proprotein convertase subtilisin/kexin type 5 (PCSK5) (OMIM:600488) and its protein, PC5/6, in dyslipidemia and cardiovascular pathologies, has yet to be established. Murine Pcsk5 is localized on chromosome 19 and encodes two alternatively spliced isoforms, soluble PC5A (915 amino acids (aa); 21 exons) and membrane bound PC5B (1877 aa; 38 exons)10. Although devoid of a transmembrane domain, PC5A can exert its proteolytic action at the cell surface, as it is retained at the plasma membrane as a complex with tissue inhibitors of metalloproteases (TIMPs) and heparin sulfate proteoglycans (HSPG)11. The essential role of Pcsk5 has been highlighted by Essalmani et al. who observed death at birth in the knock-out (KO) mice, while heterozygotes were healthy and fertile12. Except in the liver where both isoforms are equally expressed, PC5A is the major isoform in most tissues (87–100%), and only the intestine and kidney show a predominance of PC5B (74–92%)12,13. In humans, PCSK5 is located on chromosome 9q21.13, and while two alternatively spliced transcripts are described for this gene14,15, only one, generating a 21-exon isoform and typically referred to as PCSK5, has its full length nature known (NM_006200.3).
There is strong biological plausibility for the involvement of PCSK5 in lipoprotein metabolism. Jin et al7 showed that PC5A inactivates ex vivo EL and lipoprotein lipase (LPL), with both lipases being present at the vascular endothelial surface. High expression of PC5/6 in enterocytes also suggests a possible role in processing protein substrates that could regulate food and/or sterol/lipid absorption10. Additionally, recent data from a genome-wide scan reports a region close to PCSK5 on chromosome 9, implicated in lipid regulation16, but its link to HDL deficiency remains to be explored. In light of this, there is compelling evidence that suggests that PC5/6 may be a good candidate proteinase implicated in the control of circulating HDL-C levels and mutations affecting its function may influence lipoprotein metabolism and HDL subspecies more specifically. Therefore, we undertook a study to determine whether genetic variation within the PCSK5 gene contributes to variation in HDL-C levels, which consequently, could influence atherosclerotic cardiovascular disease risk.
METHODS
French Canadian family subjects
A total of nine multigenerational French Canadian families consisting of 175 genotyped members were examined and sampled in the Preventive Cardiology/Lipid Clinic of the McGill University Health Centre (MUHC). The selection criterion for probands was HDL-C<5th percentile (age and gender-matched), based on the Lipid Research Clinics Population Studies Data Book17. All subjects provided informed consent for plasma and DNA sampling, isolation, and storage. The research protocol was reviewed and approved by the ResearchEthics Board of the MUHC.
Stage 1 and 2 association study samples
French Canadian subjects
For stage 1 analysis, unrelated patients (n=457) of French Canadian descent were selected from the Cardiology Clinic of the Clinical Research Institute of Montreal that were <60 years of age and had angiographically documented CAD18. Individuals had HDL-C values ranging from HDL-C<5th percentile to HDL-C>95th percentile (age and gender-matched)17. We excluded patients with known causes of HDL deficiency (severe hypertriglyceridemia defined as plasma triglycerides (TG)>10 mmol/L, cellular phospholipid or cholesterol efflux defect or previously identified mutations in genes associated with HDL deficiency)19,20. Demographic and clinical information, medications, and lipoprotein profiles were determined on all participating subjects as previously described20. The research protocol was approved by the Research Ethics Board of the MUHC.
Finnish family subjects
The stage 2 study sample consisted of 39 Finnish low HDL-C families (426 genotyped individuals) recruited at the Helsinki and Turku central hospitals as previously described21. Each subject involved in this study provided written informed consent. The study design was approved by the ethics committees of the participating institutions. Inclusion criteria for the probands were age 30–60 years, at least 50% stenosis in one or more coronary arteries, HDL-C level below the Finnish age- and sex-specific 10th population percentile (subjects coded as affected), total cholesterol (TC)<6.3 mmol/L for males and <6.0 mmol/L for females, and TG<2.3 mmol/L for both genders21. Serum lipid and glucose parameters were measured as previously22. Exclusion criteria for the probands were type 1 and 2 diabetes mellitus, severe hepatic or renal disease, or body mass index >3021.
Haplotyping
Microsatellite genotypes were determined by deCODE Genetics (Reykjavik, Iceland) at markers D9S1777, D91876, D9S175, D9S1843 spanning 11.3 Mb and flanking the PCSK5 gene on chromosome 9q21.13. Haplotypes were constructed using Cyrillic version 2.1.3 (Cherwell Scientific Publishing Ltd) to examine the segregation of the PCSK5 locus with the low HDL-C trait in nine families with HDL-C deficiency.
Sequencing
Sequencing of the 21 exons and exon-intron boundries of the PCSK5 gene was performed in 12 unrelated individuals with HDL-C<5th percentile. Exon-specific oligonucleotides were synthesized (Integrated DNA Technologies, Coralville, Iowa, USA) and designed using the Primer3 software (http://frodo.wi.mit.edu/)23 to include at least 22 bp of intronic sequence at each intro-exon boundary. Polymerase chain reaction (PCR)-amplified fragments were purified using the Millipore purification plate (Multiscreen PCR) and directly sequenced at the McGill University and Genome Québec Innovation Centre Sequencing Platform using the Applied Biosystems 3730-xl DNA Analyzer system. The data were analyzed by Sequencing Analysis version 5.2 and Mutation Surveyor v2.41 (SoftGenetics, USA).
Single nucleotide polymorphisms (SNPs) selection
To select the most informative SNPs for the first-stage genotyping of PCSK5, we utilized a tagSNP strategy using HapMap Utah Residents with Northern and Western European Ancestry (CEU) spanning 304714bp of genomic DNA and including 2kb upstream of PCSK5 (HapMap Rel27 PhaseII+III; Haploview v.4.024). We used a minor allele frequency (MAF)>0.05 and r2 threshold of 0.80. In addition, we included novel variants identified through sequencing (n=7), as well as SNPs selected from public genetic databases [NCBI, UCSC, SeattleSNPs, and Human SNP (Broad Institute)] (n=12), for a total of 182 SNPs to be genotyped in stage 1.
In the second stage, out of 9 SNPs that provided significant evidence of association with P<0.05, we selected 2 SNPs with P≤0.01 for stage 2 genotyping.
Genotyping
Stage 1 genotyping of 182 SNPs was performed using the Sequenom iPLEX Gold Assay (Sequenom, Cambridge, MA). Locus-specific PCR primers and allele-specificdetection primers were designed using Mass ARRAY Assay Design 3.1 software. DNA was amplified in a multiplex PCR and labeled using a single base extension reaction. The products were desalted and transferred to a 384-element SpectroCHIP array. Allele detection was performed using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Mass spectrograms and clusters were analyzed by the TYPER 3.4 software. Ehrich et al.25 have previously provided details of the procedure. Prior to association analysis, quality control-check was performed by assessing integrity of genotypic data. We obtained a 96% success rate for the SNPs and 99% of subjects (451 individuals) were successfully genotyped. For the remaining SNPs, a genotyping call rate >98% was obtained. After frequency and genotype pruning, there were 169 SNPs that were analyzed.
Genotyping of the 2 second-stage replication SNPs was performed in the Finnish low HDL families using the pyrosequencing technique on the PSQHS96A platform with a >94% genotyping call rate. Both SNPs were in Hardy-Weinberg Equilibrium in the unrelated founders (P>0.5). The Pedcheck program was used to detect Mendelian errors in the families26.
Statistical analyses
Statistical analyses for the French Canadian association study were performed with PLINK v1.04 software (http://pngu.mgh.harvard.edu/purcell/plink/)27 and the SAS package v9.1.3 (SAS Institute Inc., NC, USA). Quantitative association analysis for HDL-C was performed using linear regression, after adjustments for age and sex. The additive model was tested in the stage 1 analysis as it has been shown to be robust for detecting association even when the true genetic model is not additive28. We estimated the effect of significant SNPs on the basis of the linear regression coefficient (β). Conditional analyses were performed using stepwise linear regression. Significance was set at P<0.05.
Association analysis in the Finnish family cohort was performed using the quantitative transmission disequilibrium test (QTDT)29 implemented in the genetic analysis package SOLAR30. The QTDT approach is robust to population stratification30 and has been recognized as a powerful method that utilizes data from all available relatives. The orthogonal model of association within a variance component framework that included age and sex as covariates29 was used in our analyses, where the total association was partitioned into orthogonal within and between family components.
We also performed a combined analysis of both stages for rs11144766 and rs11144782, using the Z-method to combine statistics. Test statistics from the French Canadian cohort and the Finish family-based study were weighted by the square-root of the sample size to calculate the corresponding combined P value31. To correct for multiple testing, we adjusted for 169 SNPs tested in stage 1 as well as for the 2 SNPs tested in stage 2, resulting in a Bonferroni correction for 171 independent tests in the overall combined analysis.
RESULTS
Familial Segregation Analyses
We first investigated whether genetic variability at the PCSK5 locus was associated with HDL-C in a Mendelian fashion. We examined the segregation of PCSK5 haplotypes with a severe HDL-C deficiency trait (HDL-C<10th percentile) in nine unrelated multigenerational families of French Canadian descent (175 subjects, mean number per kindred 17). Our study included 71 men and 104 women, of which 29 and 31 were affected (HDL-C<10th percentile), respectively. Mean age for males was 50±16 years and 48±19 years for women; the mean HDL-C level in affected men was 0.64±0.08 mmol/L, compared with 1.12±0.22 mmol/L in non-affected men. Similarly, HDL-C levels in affected women were 0.87±0.15 mmol/L versus 1.39±0.26 mmol/L in non-affected. Four microsatellites, D9S1777, D9S1876, D9S175 and D9S1843, located upstream and downstream of the PCSK5 locus and spanning 11.3 Mb within the 9q21.13 region, were used to construct haplotypes at the PCSK5 gene. Only one small (n=7 subjects) kindred was observed to display perfect segregation with HDL-C levels (data not shown). Thus, cumulatively, we did not find unambiguous segregation of this locus with the low HDL-C trait using a dominant model of inheritance, suggesting that PCSK5 does not exert a Mendelian monogenic effect on HDL-C in these families.
Sequencing
We undertook a thorough examination of the PCSK5 gene locus for coding and noncoding variants. Sequencing at the PCSK5 locus was performed on all 21 exons and exon-intron boundaries using 22 pairs of primers for a total of 7455 bps in 12 unrelated individuals with low HDL-C levels (<5th percentile). We identified a total of 19 polymorphisms, 7 of which were novel non-coding variants (Table 1, Figure 1). Of the 12 previously characterized SNPs, we observed 4 synonymous polymorphisms (rs7040769, rs7020560, rs2297342, rs10521468), one SNP in the 5′ untranslated region (rs12005073), and 7 intronic. Two of the novel variants were insertions: one in intron 19 (IVS19-71insTAAAA) and the other in the 5′ untranslated region (c.385insGAGCTGCGGCGGCCCGGGGCTGC). We also found a deletion in intron 20 (IVS20-50delTACTTTCAGGACTAAT), a variant in intron 4 (IVS4-3016T>A), and three polymorphisms in the 5′ and 3′ untranslated regions (c.125C>A, c.72C>T; c.323G>A respectively) (Table 1).
Table 1.
PCSK5 Polymorphisms Identified by Sequencing
| Functional Class | Location | Validated SNPs* | Novel Variants | |
|---|---|---|---|---|
| Exonic | Synonymous | Exon 1 | rs7040769 | |
| Exon 1 | rs7020560 | |||
| Exon 12 | rs2297342 | _ | ||
| Exon 18 | rs10521468 | |||
| 5′UTR | rs12005073 | c.125C>A | ||
| c.72C>T | ||||
| c.385insGAGCTGCGGCGGCCCGGGGCTGC | ||||
| 3′UTR | c.323G>A | |||
| Intronic | Int.4 | IVS4-3016T>A | ||
| Int.7 | rs2297344 | |||
| Int.8 | rs1416547 | |||
| Int.11 | rs3824474 | |||
| Int.16 | rs2270570 | |||
| Int.17 | rs1537183 | |||
| Int.19 | rs3830384 | IVS19-71insTAAAA | ||
| Int.20 | rs10869726 | IVS20-50delTACTTTCAGGACTAAT | ||
Validated SNPs correspond to variants identifying by sequencing, but previously characterized.
UTR, untranslated region; Int, intron.
Figure 1. SNP locations in the PCSK5 gene.

Schematic representation of the human PCSK5 gene locus showing the exon structure and the location of the 19 polymorphisms (bottom panel) discovered through sequencing and the 9 genetic variants associated with HDL-C (upper panel) identified by genotyping. SNPs in bold are associated with HDL-C with P<0.01. Locations are based on RefSeq NM_006200.3.
Association studies
To investigate whether common genetic variants at the PCSK5 locus influence HDL-C levels and therefore explain some of the interindividual variation of HDL-C plasma concentrations, we conducted a quantitative association analysis utilizing a two-stage approach. In stage 1, we genotyped 169 tagSNPs in 457 unrelated subjects of French Canadian descent to screen for associations. In stage 2, we genotyped significant signals (P<0.01) in Finnish low HDL families, and subsequently performed a combined analysis of the two stages to identify variants of region-wide significance. Skol et al.31 originally introduced this approach to reduce the cost of genotyping in stage 1 while maintaining the overall power of the study.
In stage 1, a total of 169 SNPs (Figure 2) were tested for association in 457 French Canadian individuals18. Using an additive model and adjusting for age and sex as covariates, we identified 9 SNPs significantly associated with HDL-C (P<0.05), with the strongest results being rs11144782 and rs11144766 (MAF 0.164, β=−0.076 mmol/L, P=0.002; MAF 0.197, β=−0.063 mmol/L, P=0.005 respectively; Table 2, Figure 1). The rare G-allele of rs11144782 decreased HDL-C levels by 0.076 mmol/L on average per allele, while the A risk allele of rs11144766 decreased plasma HDL-C levels by 0.063 mmol/L. The effect of these minor alleles on HDL-C are displayed in Table 3. In addition, of the 9 polymorphisms identified, three other SNPs were shown to be significantly associated with decreased plasma HDL-C levels (rs11144688, rs11144690, and rs1338746) while four others were associated with an increase in HDL-C (rs1339246, rs1331384, rs4745522, rs2050833) (Table 2). These 9 variants were all found in non-coding regions and were not in linkage disequilibrium (LD).
Figure 2. LD map of SNPs investigated in the PCSK5 gene.
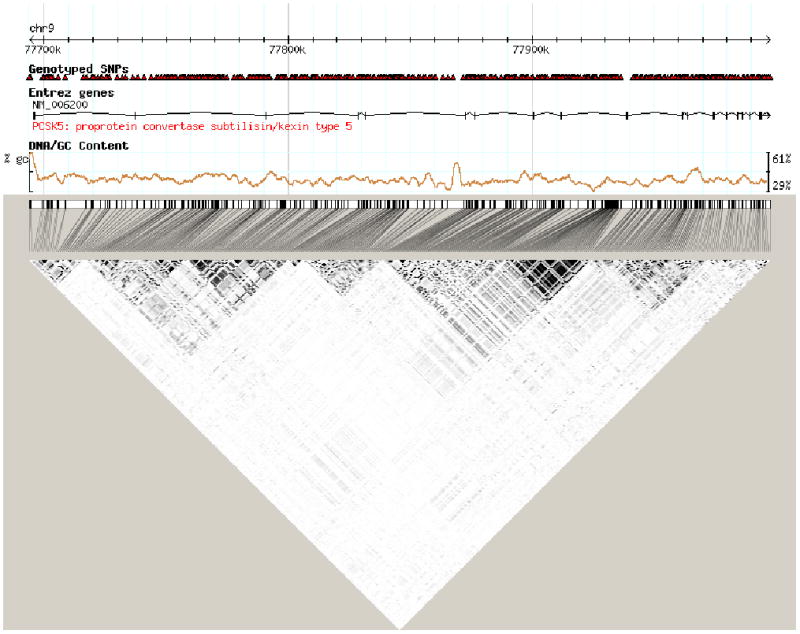
The LD map was generated using Haploview24. Numbers and white to black shading indicate r2 values (black=high, white=low).
Table 2.
Association Results of Stage 1 Analysis for the HDL-C Trait
| SNP | Effect (β) | P | MAF |
|---|---|---|---|
| rs11144782 | −0.076 | 0.002 | 0.164 |
| rs11144766 | −0.063 | 0.005 | 0.197 |
| rs1339246 | 0.056 | 0.018 | 0.176 |
| rs1331384 | 0.037 | 0.038 | 0.485 |
| rs11144688 | −0.053 | 0.039 | 0.137 |
| rs11144690 | −0.093 | 0.040 | 0.040 |
| rs1338746 | −0.036 | 0.044 | 0.424 |
| rs4745522 | 0.051 | 0.045 | 0.143 |
| rs2050833 | 0.045 | 0.045 | 0.199 |
Quantitative association analysis for HDL-C in the French Canadians was performed using linear regression, after adjustments for age and sex, under an additive model. β is the linear regression coefficient corresponding to the effect size per copy of the minor allele. Bolded SNPs represent significant variants (P≤0.01) selected for Stage 2 analysis. MAF, minor allele frequency.
Table 3.
Distribution of Genotypic Classes for rs11144782 and rs11144766 in the Stage 1 French Canadian Subjects
| rs11144782 |
rs11144766 |
|||||
|---|---|---|---|---|---|---|
| Genotypes (n) | CC (n=301) | GC (n=123) | GG (n=10) | GG (n=275) | AG (n=128) | AA (n=19) |
| Frequency | 0.69 | 0.28 | 0.02 | 0.65 | 0.30 | 0.04 |
| Mean HDL-C* (mmol/L) | 0.98±0.29 | 0.89±0.24 | 0.8±0.20 | 0.97±0.30 | 0.91±0.23 | 0.85±0.18 |
Analyses were performed using PLINK v1.04 with age and sex as covariates, under an additive model. HDL-C, high-density lipoprotein cholesterol.
The mean±SD of HDL-C are stratified by genotype, and weighted by the frequencies of their genotypic class.
We next tested our two most significant polymorphisms (P<0.01), rs11144782 and rs11144766, for association with other lipoprotein traits and, adjusting for age and sex under an additive model, observed a significant positive effect on plasma TG (P=0.049), very low-density lipoprotein cholesterol (VLDL-C; P=0.039), and apolipoprotein B (apoB P=0.022) levels (Table 4) for rs11144782, suggesting that it modulates several aspects of lipid metabolism. We also conducted a stepwise conditional regression analysis in the presence of rs11144782 and age and sex as covariates. Using this approach, we re-identified rs11144766 (P<0.001) and two other non-redundant SNPs (rs2050833, P<0.036; rs4745488, P<0.038) that contributed to the variability of HDL-C (Table 5), independently of one another, providing further evidence for the role of PCSK5 in HDL-C metabolism.
Table 4.
Lipid Traits Associated with rs11144782
| SNP | Trait | Effect (β) | P |
|---|---|---|---|
| HDL (mmol/L) | −0.076 | 0.002 | |
| rs11144782 | TG (mmol/L) | 0.502 | 0.049 |
| VLDL (mmol/L) | 0.169 | 0.039 | |
| apoBtot (mg/dL) | 10.720 | 0.022 |
Analyses were performed using PLINK v1.04 with age and sex as covariates, under an additive model. β is the linear regression coefficient, corresponding to the effect size per copy of the minor allele. HDL-C, high-density lipoprotein cholesterol; TG, triglycerides; VLDL, very low-density lipoprotein; apoBtot, total apolipoprotein B.
Table 5.
Independent Signals Found at the PCSK5 Locus for the HDL-C Trait
| SNP | Effect (β) | P |
|---|---|---|
| rs11144782 | −0.076 | 0.002 |
| rs11144766 | −0.072 | 0.001 |
| rs2050833 | 0.047 | 0.036 |
| rs4745488 | −0.039 | 0.038 |
Conditional analyses were performed using stepwise linear regression adjusting for age, sex, rs11144782, rs11144766 and rs2050833, respectively. β is the linear regression coefficient, corresponding to the effect size per copy of the minor allele.
In stage 2, we followed up rs11144782 and rs11144766 which provided the most significant associations (P≤0.01) in the French Canadian cohort, for replication in an independent sample of 39 low HDL-C Finnish dyslipidemic families (n=426)21. We tested for association between these variants and low HDL-C using QTDT with age and sex as covariates. While we did not observe an association with rs11144782, rs11144766 was found to be significantly associated with HDL-C in the same direction as in the French Canadians (P=0.014).
Next, we performed a combined analysis of the stage 1 and 2 unrelated and family samples (n=883) for the two SNPs by combining the Z statistics, as described in Materials and Methods. We observed a strong association between rs11144766 and low HDL-C (P=1.86×10−4) for the additive model and the same A risk allele. This result is region-wide significant: it surpasses the Bonferroni correction for all 171 tests performed [169 SNPs tested in stage 1 and 2 SNPs in stage 2 (Bonferroni adjusted P=0.031)]. Thus, the association between rs11144766 and HDL-C in these Finnish dyslipidemic families is consistent with the results from the French Canadian unrelated individuals, and provides strong evidence for the influence of rs11144766 on HDL-C levels.
DISCUSSION
The investigation of the molecular genetics and pathophysiology of HDL-C deficiency has been an area of fertile research. Despite a large body of information identifying HDL-C as a potent anti-atherosclerosis lipoprotein, the fundamental mechanisms underlying the genetic regulation of the HDL-C metabolic pathway remain complex and poorly understood.
In the present study, we have demonstrated that genetic variability at the PCSK5 gene modulates HDL-C levels. By sequencing the gene, we identified seven novel non-coding variants in patients with HDL-C deficiency (Figure 1). Although none of these newly identified variants represent a missense, frameshift or non-sense polymorphism with obvious functional consequences, the possibility of their regulatory role cannot be excluded and further studies to delineate their mechanistic effects are needed. We next performed an association study of PCSK5 SNPs with HDL-C through a two-stage study design. This approach was an efficient way to optimize the power to detect true associations, while minimizing the overall amount of genotypes required for sufficient regional coverage31. In stage 1, we investigated the association of PCSK5 SNPs with HDL-C levels in 457 unrelated subjects of French Canadian descent, using quantitative analyses. We identified nine significant SNPs (P<0.05, Figure 1, Table 1), five of them being associated with a decrease and four with an increase in HDL-C plasma concentration. The strongest signals, rs11144782 and rs11144766 (MAF 0.164, β=−0.076, P=0.002; MAF 0.197, β=−0.63, P=0.005 respectively), were found to be negative modulators of HDL-C levels, displaying an allele dosage-effect (Table 3): the minor allele (G) of rs11144782 was observed to contribute to a decrease of 8% in plasma HDL-C levels, while the minor allele (A) for rs11144766 lowered HDL-C serum concentration by 6% (Table 2 and 3). Interestingly, the MAF of rs11144782 and rs11144766 in the HapMap-CEU samples (MAF 0.156 and 0.142) was in concordance with both the French Canadian (MAF 0.164 and 0.197) and the Finnish study cohorts (MAF 0.168 and 0.197). In stage 2, we followed up significant markers in the previously mentioned independent study sample consisting of low HDL Finnish families to confirm the observed associations. By means of a combined analysis of both stages (n=883), region-wide significance between rs11144766 and low HDL-C was observed (unadjusted P=1.86×10−4). Using the Bonferroni correction, rs11144766 remained significant (P=0.031) after adjusting for multiple testing (n=171), providing sound and consistent evidence for its role in HDL-C metabolism.
Furthermore, in conditional regression analysis, we observed two additional SNPs putatively contributing to HDL-C (P<0.05), independent of the effects of rs11144782 and rs11144766 (Table 5). As a result, in the present study, we identified four signals at the PCSK5 locus, independent of one another. Interestingly, in stage 1 analysis, the rs11144782 variant was also associated with other lipid traits including VLDL-C (P=0.039), TG (P=0.049) and total apoB (P=0.022) levels (Table 4), translating in an absolute increase in VLDL-C (0.169 mmol/L), TG (0.502 mmol/L), and apoB (10.72 g/L) per allele respectively, suggesting that PCSK5 genetic variability may influence other aspects of lipoprotein metabolism and not solely HDL-C.
The two polymorphisms identified in this study, rs11144766 and rs11144782, are both located in introns of PCSK5. rs11144766 is found in the 10th intron, between exons coding for a region of the catalytic domain of PC5/6, while rs11144782 occurs in the 15th intron, between exons encoding the cysteine-rich motif (CRM) of PC5/6 (Figure 1). Despite their intronic localization, we show here that both of these SNPs are important regulators of HDL-C levels, potentially mediating PC5/6 activity.
There are several possible explanations for such effects. These SNPs may be involved in regulating splicing of the PCSK5 transcript, to either enhance or suppress proper intron-removal. To explore this possibility, we used the ACESCAN2 Web Server (http://genes.mit.edu/acescan2/index.html) to scan for possible sites that may affect splicing. We identified a specific GTGTGG sequence present in the rare A-allele of rs11144766 as an Intronic Splicing Enhancer (ISE). This suggests that individuals carrying this variant may have modified splicing of the PCSK5 gene which could impact PC5/6 activity. The catalytic domain of PCSK5 is crucial for its proteolytic convertase function, necessary for EL cleavage. Indeed, deleting exon 4 resulted in embryonic lethality12,13, stressing the importance of this domain. In contrast, while no ISEs were found to be associated with rs11144782, it may have other effects on splicing which could alter PC5/6 function. The CRM of the latter is involved in regulating PC5/6A localization, which, in combination with TIMPs, binds to HSPG at the cell surface32. Data reveals that the CRM confers protein-protein interactions, cell surface tethering and is essential for the efficacious processing of the human proEL precursor, likely due to a proximity effect resulting from close juxtaposition of the convertase and EL through interactions with cell-surface HSPGs8,11,33. Therefore, altering either the CRM or the catalytic domain would likely impair PC5/6 activity and, indirectly, overexpress EL. Subsequently, this might explain the observed decrease in HDL-C levels in individuals carrying these SNPs. Beyond the possibility of modulating splicing, these variants may be located in important binding sites for unknown factors, such as microRNAs. Furthermore, it is also possible that they are in LD with other un-genotyped SNPs.
Though we have highlighted the importance of rs11144782 and rs11144766 in this study, much work remains to be done to better elucidate their functional mechanisms and effects on PC5/6 activity. Despite this, our findings suggest an in vivo conceptual mechanism for how rs11144766 and rs11144782 could potentially affect HDL-C metabolism (Figure 3). We propose that by altering PC5/6 function, these variants could prevent the internal cleavage of the HSPG-bound EL by PC5/6 or its inhibition by activated ANGPTL3. As a result, EL will be fully active and its effect on HDL unopposed, resulting in a pronounced phospholipase activity and consequently, hydrolysis of HDL phospholipids that will produce smaller HDL particles. This will reduce plasma HDL-C levels, in concordance with studies in which mice overexpressing human EL revealed a marked depletion in HDL-C levels5. The EL-mediated reduction of HDL-phospholipids can also alter the lipid composition and physical properties of HDL, resulting in a diminished ability of HDL to mediate scavenger receptor class B type I-dependent cholesterol efflux34. Additionally, association of rs11144782 with increased VLDL-C, TG and apoB levels implicates the effects of other lipases, such as LPL and hepatic lipase. Further studies are however needed to elucidate their exact physiologic role on plasma lipoprotein metabolism in the presence of PCSK5 variants affecting PC5/6 function. Indeed, a PC5/6 loss of activity could allow the activation of the heparin-like glycosaminoglycans-bound LPL and its triglyceride hydrolase function on chylomicrons and VLDL, subsequently decreasing them. Similarly, ANGPTL3, a liver-derived member of the vascular endothelial growth factor family, shown to be an endogenous inhibitor of EL6 in cell-free systems, plays a potential role in regulating HDL levels35. In line with the study done by Shimizugawa et al.36 in which overexpression of ANGPTL3 in KK/San mice resulted in a marked increase of TG-enriched VLDL levels through the inhibition of LPL activity, loss of hepatic PC5/6 activity under pathophysiological, genetic or diseased conditions could increase EL and LPL activities, resulting in reduction of plasma HDL-C, VLDL-C and TG concentrations. Thus, there are several venues by which PCSK5 variants could impact lipoprotein metabolism. The relative contribution of these pathways upon PC5/6 inactivation is still unknown, and further work is required to determine the overall importance of each component in the system.
Figure 3. Conceptual model of PCSK5 variants’ effect.
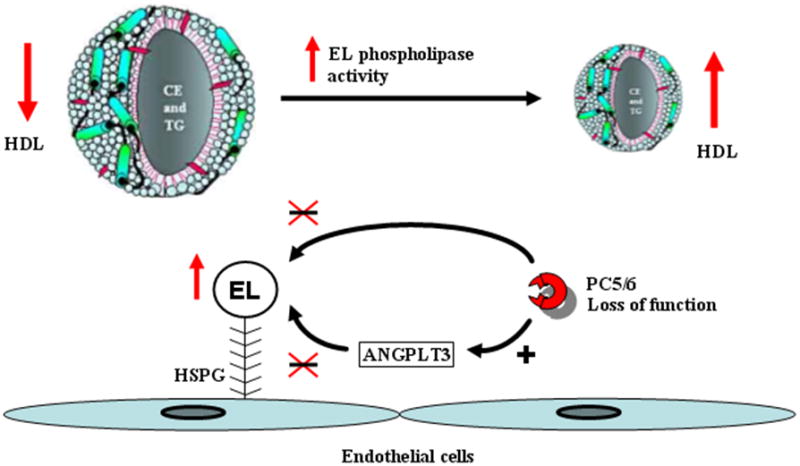
Genetic variation at PCSK5, resulting in complete or partial PC5/6 loss of function, may be responsible for the inability of PC5/6 to activate ANGPLT3 or directly inactivate EL. This will overexpress EL, increasing phospholipase activity on HDL particles, resulting in smaller HDL with decreased cholesterol content.
Given the significance of our results, a clearer understanding of the molecular interactions between the PC5/6-EL system and HDL structure, as well as the direct impact of HDL remodeling by PC5/6-EL on the reverse cholesterol transport process and endothelial function, should be the focus of future scientific studies. It would also be essential to replicate our findings in larger and more diverse study samples37 and functionally characterize these variants. Accordingly, unravelling the in vivo and in vitro effects of the PC5/6-EL system could refine our comprehension of the complex HDL metabolic pathway and provide novel insights into the human atheroprotective system in health and disease.
In conclusion, we observed an association of region-wide significance between rs11144766 and HDL-C under an additive model in an unrelated and family-based sample (n=883). These results support the contribution of PCSK5 to HDL-C levels and its pivotal role in HDL-C metabolism. While previous work by Cao et al.38 identified 2 silent SNPs in PCSK5 varying in frequency among ethnic groups, no other studies thus far have analyzed the genetic variability at the PCSK5 gene locus and its contribution to HDL-C levels. This report is therefore the first comprehensive examination of such genetic variation, implicating PCSK5 as an important and influential modulator of HDL-C serum levels in humans. These findings can thus firmly place PCSK5 on the list of genes associated with HDL-C and emphasize the need to investigate PC5/6 and its related substrates for identification of specific therapeutic targets for treatment of cardiovascular disease.
Acknowledgments
We thank the French Canadian and Finnish families who participated in this study. Leena Peltonen is thanked for sample collection, as well as E. Nikkola and M. Lupsakko for their laboratory technical assistance. We gratefully acknowledge the Montreal Genome Centre staff for their sequencing and genotyping services.
Sources of Funding
This work was supported by the Canadian Institutes of Health Research [MOP 62834, MOP 44363 and CTP 82946]; and the Heart and Stroke Foundation of Quebec. J.G. holds the McGill Novartis Chair in Cardiology at McGill University. I.I. is a recipient of a Doctoral Research Award from the Fonds de la Recherche en Santé du Québec and a Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award from the Canadian Institutes of Health Research. Z.D. is a recipient of a Doctoral Research Award from the Heart and Stroke Foundation of Canada and R.D. holds a Canada Graduate Scholarship Doctoral Award from Canadian Institutes of Health Research. This study was also funded by NIH [HL-28481 and HL082762 to P. P.]; the Clinical Research Institute Helsinki University Central Hospital Ltd [to M.-R.T.]; and the Finnish Heart Foundation [to M-R.T.]. D.W.-V. and A.H.-V. are supported by the National Human Genome Research Institute [T32 HG02536] and the American Heart Association [0725232Y] respectively.
Footnotes
DISCLOSURES
No conflict of interest to be declared.
References
- 1.Iatan I, Alrasadi K, Ruel I, Alwaili K, Genest J. Effect of ABCA1 mutations on risk for myocardial infarction. Curr Atheroscler Rep. 2008;10:413–426. doi: 10.1007/s11883-008-0064-5. [DOI] [PubMed] [Google Scholar]
- 2.Dastani Z, Engert JC, Genest J, Marcil M. Genetics of high-density lipoproteins. Curr Opin Cardiol. 2006;21:329–335. doi: 10.1097/01.hco.0000231403.94856.cd. [DOI] [PubMed] [Google Scholar]
- 3.Hassan HH, Bailey D, Lee DY, Iatan I, Hafiane A, Ruel I, Krimbou L, Genest J. Quantitative analysis of ABCA1-dependent compartmentalization and trafficking of apolipoprotein A-I: implications for determining cellular kinetics of nascent high density lipoprotein biogenesis. J Biol Chem. 2008;283:11164–11175. doi: 10.1074/jbc.M707720200. [DOI] [PubMed] [Google Scholar]
- 4.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 5.Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet. 1999;21:424–428. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- 6.Shimamura M, Matsuda M, Yasumo H, Okazaki M, Fujimoto K, Kono K, Shimizugawa T, Ando Y, Koishi R, Kohama T, Sakai N, Kotani K, Komuro R, Ishida T, Hirata K, Yamashita S, Furukawa H, Shimomura I. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27:366–372. doi: 10.1161/01.ATV.0000252827.51626.89. [DOI] [PubMed] [Google Scholar]
- 7.Jin W, Wang X, Millar JS, Quertermous T, Rothblat GH, Glick JM, Rader DJ. Hepatic proprotein convertases modulate HDL metabolism. Cell Metab. 2007;6:129–136. doi: 10.1016/j.cmet.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seidah NG, Khatib AM, Prat A. The proprotein convertases and their implication in sterol and/or lipid metabolism. Biol Chem. 2006;387:871–877. doi: 10.1515/BC.2006.110. [DOI] [PubMed] [Google Scholar]
- 9.Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lusson J, Vieau D, Hamelin J, Day R, Chretien M, Seidah NG. cDNA structure of the mouse and rat subtilisin/kexin-like PC5: a candidate proprotein convertase expressed in endocrine and nonendocrine cells. Proc Natl Acad Sci U S A. 1993;90:6691–6695. doi: 10.1073/pnas.90.14.6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nour N, Mayer G, Mort JS, Salvas A, Mbikay M, Morrison CJ, Overall CM, Seidah NG. The cysteine-rich domain of the secreted proprotein convertases PC5A and PACE4 functions as a cell surface anchor and interacts with tissue inhibitors of metalloproteinases. Mol Biol Cell. 2005;16:5215–5226. doi: 10.1091/mbc.E05-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essalmani R, Zaid A, Marcinkiewicz J, Chamberland A, Pasquato A, Seidah NG, Prat A. In vivo functions of the proprotein convertase PC5/6 during mouse development: Gdf11 is a likely substrate. Proc Natl Acad Sci U S A. 2008;105:5750–5755. doi: 10.1073/pnas.0709428105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Essalmani R, Hamelin J, Marcinkiewicz J, Chamberland A, Mbikay M, Chretien M, Seidah NG, Prat A. Deletion of the gene encoding proprotein convertase 5/6 causes early embryonic lethality in the mouse. Mol Cell Biol. 2006;26:354–361. doi: 10.1128/MCB.26.1.354-361.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miranda L, Wolf J, Pichuantes S, Duke R, Franzusoff A. Isolation of the human PC6 gene encoding the putative host protease for HIV-1 gp160 processing in CD4+ T lymphocytes. Proc Natl Acad Sci U S A. 1996;93:7695–7700. doi: 10.1073/pnas.93.15.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakagawa T, Hosaka M, Torii S, Watanabe T, Murakami K, Nakayama K. Identification and functional expression of a new member of the mammalian Kex2-like processing endoprotease family: its striking structural similarity to PACE4. J Biochem. 1993;113:132–135. doi: 10.1093/oxfordjournals.jbchem.a124015. [DOI] [PubMed] [Google Scholar]
- 16.Falchi M, Andrew T, Snieder H, Swaminathan R, Surdulescu GL, Spector TD. Identification of QTLs for serum lipid levels in a female sib-pair cohort: a novel application to improve the power of two-locus linkage analysis. Hum Mol Genet. 2005;14:2971–2979. doi: 10.1093/hmg/ddi327. [DOI] [PubMed] [Google Scholar]
- 17.NIH Washington DC. Lipid Research Clinics Population Studies Databook. Department of health and human services, Public Health Service; Washington DC: 1980. NIH publication 80–1527. [Google Scholar]
- 18.Weber M, McNicoll S, Marcil M, Connelly P, Lussier-Cacan S, Davignon J, Latour Y, Genest J., Jr Metabolic factors clustering, lipoprotein cholesterol, apolipoprotein B, lipoprotein (a) and apolipoprotein E phenotypes in premature coronary artery disease in French Canadians. Can J Cardiol. 1997;13:253–260. [PubMed] [Google Scholar]
- 19.Alrasadi K, Ruel IL, Marcil M, Genest J. Functional mutations of the ABCA1 gene in subjects of French-Canadian descent with HDL deficiency. Atherosclerosis. 2006;188:281–291. doi: 10.1016/j.atherosclerosis.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 20.Marcil M, Yu L, Krimbou L, Boucher B, Oram JF, Cohn JS, Genest J., Jr Cellular cholesterol transport and efflux in fibroblasts are abnormal in subjects with familial HDL deficiency. Arterioscler Thromb Vasc Biol. 1999;19:159–169. doi: 10.1161/01.atv.19.1.159. [DOI] [PubMed] [Google Scholar]
- 21.Pajukanta P, Allayee H, Krass KL, Kuraishy A, Soro A, Lilja HE, Mar R, Taskinen MR, Nuotio I, Laakso M, Rotter JI, de Bruin TW, Cantor RM, Lusis AJ, Peltonen L. Combined analysis of genome scans of dutch and finnish families reveals a susceptibility locus for high-density lipoprotein cholesterol on chromosome 16q. Am J Hum Genet. 2003;72:903–917. doi: 10.1086/374177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pajukanta P, Nuotio I, Terwilliger JD, Porkka KV, Ylitalo K, Pihlajamaki J, Suomalainen AJ, Syvanen AC, Lehtimaki T, Viikari JS, Laakso M, Taskinen MR, Ehnholm C, Peltonen L. Linkage of familial combined hyperlipidaemia to chromosome 1q21-q23. Nat Genet. 1998;18:369–373. doi: 10.1038/ng0498-369. [DOI] [PubMed] [Google Scholar]
- 23.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 24.Barrett JC FBMJDMJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 21:263–265. doi: 10.1093/bioinformatics/bth457. 15 A.D. [DOI] [PubMed] [Google Scholar]
- 25.Ehrich M, Bocker S, van den BD. Multiplexed discovery of sequence polymorphisms using base-specific cleavage and MALDI-TOF MS. Nucleic Acids Res. 2005;33:e38. doi: 10.1093/nar/gni038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu IP, Balise RR, Whittemore AS. Detection of disease genes by use of family data. II. Application to nuclear families. Am J Hum Genet. 2000;66:1341–1350. doi: 10.1086/302852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abecasis GR, Cardon LR, Cookson WO. A general test of association for quantitative traits in nuclear families. Am J Hum Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Havill LM, Dyer TD, Richardson DK, Mahaney MC, Blangero J. The quantitative trait linkage disequilibrium test: a more powerful alternative to the quantitative transmission disequilibrium test for use in the absence of population stratification. BMC Genet. 2005;6 (Suppl 1):S91. doi: 10.1186/1471-2156-6-S1-S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 32.Seidah NG, Prat A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J Mol Med. 2007;85:685–696. doi: 10.1007/s00109-007-0172-7. [DOI] [PubMed] [Google Scholar]
- 33.Broedl UC, Jin W, Rader DJ. Endothelial lipase: a modulator of lipoprotein metabolism upregulated by inflammation. Trends Cardiovasc Med. 2004;14:202–206. doi: 10.1016/j.tcm.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Yancey PG, Kawashiri MA, Moore R, Glick JM, Williams DL, Connelly MA, Rader DJ, Rothblat GH. In vivo modulation of HDL phospholipid has opposing effects on SR-BI-and ABCA1-mediated cholesterol efflux. J Lipid Res. 2004;45:337–346. doi: 10.1194/jlr.M300231-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, Horikoshi H, Furukawa H. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30:151–157. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- 36.Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, Ueda K, Inaba T, Minekura H, Kohama T, Furukawa H. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277:33742–33748. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
- 37.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton-Cheh C, Orho-Melander M. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 38.Cao H, Mok A, Miskie B, Hegele RA. Single-nucleotide polymorphisms of the proprotein convertase subtilisin/kexin type 5 (PCSK5) gene. J Hum Genet. 2001;46:730–732. doi: 10.1007/s100380170008. [DOI] [PubMed] [Google Scholar]