

Abstract
Potential metal interactions with the cleavage site of a minimal hammerhead ribozyme (mHHRz) were probed using 31P NMR-detected Cd2+ titration studies of HHRz constructs containing a phosphorothioate (PS) modification at the cleavage site. The mHHRz nucleophile position was replaced by either a 2′-F or a 2′-NH2 in order to block cleavage activity during the study. The 2′-F/PS cleavage site mHHRz construct, in which the 2′-F should closely imitate the atom size and electronegativity of a 2′OH, demonstrates low levels of metal ion association (<1 ppm 31P chemical shift changes). This observation indicates that having an atom size and electrostatic properties that are similar to the 2′-OH are not the governing factors in allowing metal interactions with the scissile phosphate of the mHHRz. With a 2′-NH2 substitution, a large upfield change in 31P NMR chemical shift of the phosphorothioate peak (Δ~3 ppm with 6 equivalents added Cd2+) indicates observable Cd2+ interactions with the substituted site. Since a 2′-NH2, but not a 2′-F, can serve as a metal ligand, these data suggest that a metal ion interaction with the HHRz cleavage site may include both the scissile phosphate and the 2′ nucleophile. Control samples in which the 2′-NH2/PS unit is placed either next to the mHHRz cleavage site (at U16.1), in a duplex, or in a amUPSU dinucleotide, show much weaker interactions with Cd2+. Results with these control samples indicate that simply the presence of a 2′-NH2/PS unit does not create a strong metal binding site, reinforcing the possibility that the 2′-NH2-moderated Cd-PS interaction is specific to the mHHRz cleavage site. Upfield chemical shifts of both 31P and H2′ 1H resonances in amUPSU are observed with addition of Cd2+, consistent with the predicted metal coordination to both 2′-NH2 and phosphorothioate ligands. These data suggest that metal ion association with the HHRz cleavage site may include an interaction with the 2′-OH nucleophile.
Keywords: Hammerhead, ribozyme, RNA, metal, NMR, phosphorothioate
The Hammerhead ribozyme (HHRz) is a self-cleaving ribozyme that catalyzes the reversible attack of a 2′-OH on the proximal 3′ phosphodiester bond, resulting in a 2′,3′ cyclic phosphate and 5′-OH leaving group. The HHRz consists of 3 base-paired stems with a 3-helix junction of conserved nucleotides forming the active site (1–3). Originally discovered in viroids and satellite viruses where the HHRz functions in genomic processing during rolling circle replication (1, 4, 5), the HHRz motif has since been found in a variety of genomes including a recent discovery of a discontinuous HHRz motif in mouse (6, 7). The function of the HHRz in higher organisms is currently unknown. One of the earliest-discovered ribozymes, the HHRz has been the subject of many mechanistic studies aimed at understanding the bases for RNA-directed catalysis.
As a negatively charged biopolymer, RNA structure and function depends strongly on cations. The precise contributions of metal ions to ribozyme catalysis are of great interest to the RNA community, and numerous studies have been performed on HHRz constructs to determine the metal requirements of this ribozyme. Many studies have been performed on minimal, or ‘truncated’ HHRz (mHHRz) domains that contain the conserved core surrounded by three base-paired stems (Figure 1). Native ‘extended’ HHRz’s (exHHRz) contain additional internal or terminal loops in Stems I and II that mediate tertiary contacts, resulting in a more active ribozyme (Reviewed in (7)). Substitution experiments have been used as one approach to discern roles for cations in ribozyme function. Both mHHRz’s and exHHRz’s have appreciable activity in very high concentrations (1–10 M) of monovalent cations, including NH4+, indicating that cation-supported RNA structure and electrostatic neutralization are large contributions to HHRz activity (8, 9). However, for both HHRz constructs it has been suggested that a more efficient divalent ion-dependent reaction channel exists in which a coordinated metal ion plays a specific catalytic role (10,11). In the mHHRz, activity in 4 M Na+ or Li+ is only 50-fold lower than Mg2+-dependent reaction rates. With the exception of two key phosphorothioate substitutions that are discussed below (11), the monovalent-dependent activity has a similar response to many active-site substitutions. Thus, the contribution of many important groups to catalysis is the same whether catalysis is supported by monovalent or divalent cations. There is, however, an added beneficial interaction that can only be satisfied with divalent ions and an inner-sphere interaction. In the case of the exHHRz, which can reach ≥100-times faster single-turnover rates in Mg2+ or other divalent cations than does the mHHRz, activity supported by monovalent cations is lower by 2–3 orders of magnitude than maximum rates with divalent cations (10, 12). Preliminary phosphorothioate studies also indicate sensitivity to direct metal coordination at specific sites in the exHHRz (13). While these data indicate that the HHRz is not an obligate metalloenzyme, it is likely that at physiological ionic strengths, HHRz catalysis occurs predominantly via divalent cation-based mechanisms as appreciable rates in their absence are only achieved in >~4 M monovalent ions. In physiological levels of monovalent cations, both types of HHRz constructs are dependent on divalent metal cations for global folding and catalysis, but the onset of activity occurs at lower divalent concentrations for the more stabilized exHHRzs (14).
Figure 1.

(A) Sequence of truncated HHRz (trHHRz) used in this study. Nucleotides in blue identify the substrate strand, and green nucleotides represent the enzyme strand. The conserved core is shown in red, and an arrow identifies the site of cleavage (cleavage site, CS) between nucleotides C17 and G1.1. (B) 2′-nucleophile substitutions used in this study, in combination with phosphorothioate substitutions. (C) X-ray crystal structure of an all-RNA mHHRz (PDB1MME) (48) showing location of the phosphodiester groups 3′ to C17 (CS) and U16.1 examined in this study using 31P NMR spectroscopy.
The most likely additional role for divalent cations in HHRz catalysis has been predicted based on thiophosphate interference (‘metal-rescue’) experiments. In these experiments, a phosphorothioate substitution replaces a non-bridging oxygen with sulfur, resulting in a lower affinity for hard metals such as Mg2+. If the metal-RNA interaction is important for catalysis, the Mg2+-dependent rate is significantly altered upon sulfur substitution of the putative ligand. The addition of a thiophilic metal such as Cd2+ would then be expected to rescue catalysis by coordinating to the sulfur substitute. Working with the mHHRz, Wang and co-workers (11) found that under single turnover conditions and a constant background of 10 mM ‘hard’ ions (Ca2+ or Mg2+), Rp phosphorothioate substitutions at both the A9 and scissile phosphate positions result in loss of activity that can be substantially rescued by the addition of Cd2+. Moreover, the Cd2+-dependence of activity in constructs containing Rp phosphorothioate substitutions at both of these sites fits best to a model in which a single metal ion bridges between them. This ‘bridging metal’ model is based on apparent affinities of rescuing metal ions in mHHRz constructs harboring single or double substitutions at the putative metal-binding sites as well as the ability of a single Cd2+ equivalent to rescue a dual PS substitution under stoichiometric conditions. Because the apparent affinity of the rescuing metal ion is sensitive to substitutions at the A9 site but not the cleavage site, the model further predicts that a single rescuing metal ion is bound to the A9 phosphate/G10.1 imino N in the ground state of the mHHRz, and that this same metal makes a bridging contact with the scissile phosphate only further along the reaction pathway in the transition state of the reaction, and potentially in an unstable intermediate that is only transiently populated before reaction.
Crystallographic and spectroscopic studies of mHHRz’s have consistently identified a metal ion coordinated in the ‘A9/G10.1 pocket’ consisting of the A9 pro-R phosphate oxygen and the N7 imino group of G10.1. In crystal structures of mHHRz’s, the active site is somewhat open and the A9 site is located >10Å from the cleavage site scissile phosphate (7). In the ‘bridging metal’ model (11), it is proposed that the mHHRz exists predominantly in this open structure and must undergo a significant conformational change to form a more compact structure representing the catalytically active, and thermodynamically unstable intermediate form of the HHRz that has important functional groups primed catalysis. In the unstable, active-form intermediate, the A9 metal ion is brought into proximity for bridging contact with the scissile phosphate. Potential roles for the critical bridging metal ion include stabilizing the active conformation and/or lowering the free energy of the negatively charged phosphorane transition state (11).
31P NMR spectroscopy in combination with phosphorothioate substitutions can be used as a tool to identify and investigate the properties of potential RNA metal ligands (15–18). A single non-bridging oxygen phosphorothioate substitution results in two ~60 ppm downshifted 31P peaks corresponding to the Rp and Sp stereoisomers of the phosphorothioate (17, 19). Coordination of Cd2+ to sulfur will cause an upfield chemical shift of the relevant stereoisomer peak, allowing for specific identification of metal-RNA interactions (15–17, 19). Such behavior is found in 31P NMR spectra of an A9 phosphorothioate in the mHHRz (reproduced in Figure 2A), which confirmed the A9/G10.1 site as a relatively high-affinity metal binding site (17). By contrast, NMR studies to date probing the cleavage site C1.1 phosphorothioate have found little evidence of this site being a metal ligand (17, 20). For such spectroscopic experiments, the activity of the mHHRz has been blocked by one of a variety of 2′ substitutions at the nucleophile. Specifically, previous unsuccessful attempts to observe metal interactions with the mHHRz scissile phosphate by 31P NMR spectroscopy have employed 2′-deoxy and 2′-OMe nucleophile substitutions (17, 20). Overall, results from these previous spectroscopic studies are consistent with the predictions of the ‘bridging metal’ model: in ground-state mHHRz structures, metal coordination is observed at the A9 site, but not at the cleavage site. In context of the model, spectroscopic evidence for the interaction of a metal ion with the scissile phosphate would be unlikely, as this coordination event is only proposed to happen in either a thermodynamically unstable intermediate or the transition state.
Figure 2.

Cd2+-induced phosphorothioate 31P chemical shift changes in the mHHRz and control samples. (A) Rp or Sp PS substitutions 5′ to A9 (blue, red) or at the cleavage site (CS) with a 2′-OMe nucleophile substitution (yellow, green), and in a control duplex (purple, gold). Data reproduced from Maderia et al. 2000 (17)). (B) Rp or Sp PS substitutions in the HHRz with a 2′-NH2 substitution (2′NH2/PS) at the cleavage site (CS, red and blue) or at U16.1 (green and pink). Also shown are data for a control duplex (2′NH2/PS) (purple and gold), and a dinucleotide amUPSU (brown and green). All RNA concentrations are ~300–500 μM.
Studies of ribozyme structure generally require repression of catalytic activity, and this is often achieved by substitution of the nucleophile. mHHRz activity can be repressed by 2′-H, -OMe, -F, or -NH2 nucleophile substitutions (21). Previous NMR studies of metal interactions with the C1.1 phosphate in the mHHRz employed 2′-H and 2′-OMe substitutions to block activity (reproduced in Figure 2A) (17, 20). These studies reported only small chemical shifts upon addition of up to several molar equivalents of Cd2+, indicating that the C1.1 phosphate is a very low-affinity binding site in these substituted constructs and consistent with the model in which a metal interaction with the scissile phosphate does not occur in the dominant ground-state structure. However, an alternative that bears consideration is that a lack of spectroscopic observation of the metal-scissile phosphate interaction could be due to changes in the characteristics of the 2′ functional group. The chemical and structural properties of these substitutions differ from those of the natural 2′OH, in particular with respect to potential metal coordination or hydrogen bonding capabilities that may be required to stabilize metal interactions. For example, although a 2′-OMe substitution is thought to have similar effects on sugar pucker as a 2′-OH (22, 23), the 2′-OMe is larger, more hydrophobic and does not participate as strongly in hydrogen bonds or metal ion coordination. Similarly, a 2′-deoxy substituent removes hydrogen-bonding and metal coordination properties at that site.
In this paper we report results of 31P NMR spectroscopy experiments that probe Cd2+ interactions with a minimal HHRz construct containing a phosphorothioate substitution at the scissile phosphate in combination with either a 2′-NH2 or 2′-F substitution in the nucleophile position to block HHRz catalysis. A 2′-F is expected to have a similar atomic radius and electrostatic properties to a 2′-OH and to favor a C3′ endo sugar pucker (23), but it is not expected to be available for metal coordination or strong hydrogen bonding (24). By contrast, a 2′-NH2 substitution is expected to be available for both hydrogen bonding and metal coordination, and favors the 2′ endo sugar conformation thought to be required for in-line attack by the nucleophile in the ribozyme reaction (22, 23). We find, surprisingly, that the 2′-NH2 substitution is permissive for metal binding to the mHHRz scissile phosphate, whereas 2′-F substitution does not result in observable metal coordination. Recognizing that close placement of amine and thioate ligands might create an ‘artificial’ chelate site with inner-sphere Cd2+ coordination that would increase apparent affinities significantly above the background level, control studies are presented that monitor metal interactions in constructs in which the 2′-NH2/PS substitution is placed in a dinucleotide, a duplex, and in the mHHRz at a site adjacent to the cleavage site. The Cd-thiophosphate interaction at the mHHRz cleavage site appears to be ≥10-fold stronger than in these control constructs containing the same 2′-NH2/PS substitution in different contexts. Preliminary results from 1H NMR spectroscopy of a U(2′-NH2)PSU dinucleotide are consistent with a model in which both the phosphate and 2′ nucleophile can coordinate a single divalent metal. Taken together, the dependence of metal interactions on 2′ nucleophile identity provokes the suggestion that metal coordination to the HHRz scissile phosphate may also involve the 2′-nucleophile position. Integration of these results with the existing ‘bridging-metal’ hypothesis is discussed. In general, the results highlight the potential sensitivity of metal ion association with ribozyme active sites upon substitutions within the putative coordination environment.
Materials and Methods
Oligonucleotides
Hammerhead ribozyme RNA oligomers shown in Figure 1 with their respective modifications were purchased from Dharmacon (CO) and deprotected according to manufacturer’s instructions. Deprotected RNA was purified by denaturing gel electrophoresis on 20%/7 M urea polyacrylamide gels and electroeluted from the gel slice. Eluted RNA was dialyzed against 5 mM triethanolamine (TEA; pH 7.0), 100 mM Na+, for at least 48 hours with several reservoir changes, concentrated (Centricon-3, Millipore), ethanol precipitated, and resuspended into buffer to form a stock solution. Substrate strand RNA with a 2′ substituent at the U16.1 nucleotide was deprotected used without further purification. Oligonucleotide concentration was determined by UV absorbance at 260 nm and extinction coefficients provided by the manufacturer. Control RNA duplexes consisting of the HHRz substrate strand with a phosphorothioate substitution indicated by an asterisk (5′ ACGCUC*GCUCGCG 3′) and its unmodified complement were also obtained from Dharmacon, as were amUPSU dinucleotides.
Separation of phosphorothioate isomers
Phosphorothioate-substituted RNA consisting of a mixture of phosphorothioate diastereomers, Rp and Sp, were separated using reverse phase HPLC (Akta Purifier, Amersham Biosciences) on a C18 column (5μ6.3/250, Amersham Biosciences) fitted with a guard column (Upchurch, Oak Harbor, WA) (17, 25). For oligomers, a stationary phase of 0.1 M ammonium acetate and a mobile phase of 80% acetonitrile/20% 0.1 ammonium acetate was used. For the separation of the dinucleotide diastereomers, a more volatile buffer system composed of a 10 mM triethylammonium acetate stationary phase and a mobile phase of 70% acetonitrile/30% triethylammonium acetate, was employed that allowed for removal of the buffer by lyophilization or rotoevaporation. All buffers were filtered using a 20 μM filter flask and degassed by bubbling through helium for 30 minutes before use. Gradients were adjusting based on the size of RNA, with larger RNAs typically requiring a more shallow gradient.
The configurations of separated phosphorothioate diastereomers were determined by digestion with phosphodiesterase I (Crotalus adamanteus venom, USB) (25). While the Rp phosphorothioate linkage is cleaved, the uncleaved Sp phosphorothiaote linkage results in a dinucleotide which can be indentified using HPLC separation. Typical digestion mixtures contained 4.0 nmoles of RNA, 0.1 M Tris (pH 8.5), 0.3 mM dithiothreitol (DTT), 0.3 mM MgCl2, 0.1 units of snake venom, and 8 units of calf alkaline phosphatase (USB). Reactions were incubated at 37 °C for 8 hours.
NMR spectroscopy
All NMR samples were placed in Shigemi D2O matched quartz tubes (Shigemi Inc., Allison Park, PA). All samples contained at least 400 μM RNA in a volume of 200 μL. 31P NMR spectra of the substrate strand containing a phosphorothioate at the cleavage site were collected first, and then a 1:1 substrate to enzyme hybrid was formed by heating the sample to 90 °C for 90 seconds followed by bench cooling. Cd2+ was then added to the sample. Experiments were performed in 5mM HEPES (pH 8.5) and 100mM Na+. This pH was chosen to ensure deprotonation of 2′-NH2 substitutions. 31P NMR experiments were performed at 15 °C unless otherwise indicated.
For metal cation titrations, a metal stock solution was made in deionized H2O or D2O, and the appropriate volume was added directly to the NMR sample tube. Control experiments indicated no change in pH in the buffer systems employed. In order to prevent nonspecific degradation of the RNA in the presence of metals, no additional heating steps were used. MgCl2 and MnCl2 metal stocks were purchased from Sigma. CdCl2 and Cd(NO3)2 were purchased from Alfa Aesar. All were 99.9%+ purity.
1H-decoupled 31P NMR spectra were recorded at 202 MHz on a Varian Unity Spectrometer with a 5 mM broadband probe. A pulse sequence of a 500 ms delay, 30° pulse, followed immediately by an acquisition of 0.4 seconds was used. At least 10,000 transients (approximately 8 hours) were collected for each sample. Unless otherwise noted, samples were suspended in a buffer of 5 mM HEPES (pH 8.5) and 100 mM NaCl. An external reference solution of trimethyl phosphate (TMP) or phosphoric acid was used. Where appropriate, the equilibrium dissociation constant (Kd) of the metal-RNA interaction and the total shift for a fully bound RNA-M2+ complex ([Δ]T) was found by fitting to a 1:1 binding model (Equation 1) (26).
| (1) |
[MC]T and M are the total concentration of model compound/RNA and added metal, respectively, and Δobs is the observed change in chemical shift for each data point.
1H NMR spectroscopy was performed on a Varian Inova 500-MHz spectrometer equipped with triple-axis gradient probes. 1H samples were run in 10 mM sodium cacodylate buffer (pH 7.4) and 100 mM NaCl. 5,5-Dimethylsilapentanesulfonate (DSS) with a chemical shift of 0 ppm was used as an internal reference. Use of cacodylate buffers results in a residual methyl proton resonance at 3.8 ppm (27). Double-quantum filtered (DQF) homonuclear 1H-1H correlated spectroscopy (COSY) and 1H-31P-COSY experiments were performed on RNA samples that had been exchanged in D2O by lyophilization or vacuum drying the sample, adding D2O, and repeating multiple times. NMR spectra were processed using NMRpipe software and VarianNMR software and visualized using nmrDraw and Inkscape (28).
Hammerhead ribozyme kinetics
Cleavage activity of the trHHRz WT and variant with a C17 2′-NH2/PS (cleavage site) substitution were measured at indicated single timepoints under the conditions of the NMR experiment (~400 μM enzyme and substrate strands, 5 mM Hepes pH 8.5, 100 mM NaCl) except at 24 °C. Kinetics of the trHHRz WT and variant with a U16.1 2′-NH2/PS substitution were measured in single-turnover conditions (10:1 enzyme:substrate) in 25 mM MOPS pH 7.0, 100 mM NaCl, 1.5 μM enzyme and 0.15 μM substrate plus trace amount of 32P-labeled substrate, and the indicated divalent metal ion concentrations at 20 °C. Data are plotted as (fraction cleaved, f) vs. time, and fit to a single-exponential rate kobs expression of [f(t) = fT (1−ekobs*t)].
Results
A 2′-NH2, but not 2′-F, is permissive for Cd2+ coordination to the HHRz scissile phosphorothioate
Previous studies have probed metal interactions with the hammerhead ribozyme cleavage site phosphodiester bond by monitoring the downshifted 31P NMR feature that results from non-bridging phosphorothioate substitution (17, 20). To inhibit activity, either a 2′-H or 2′-OMe subsitution of the nucleophile has also been included. Results from these 2′-X/PS (X=H or OMe) have yielded only small 31P chemical shift changes upon addition of Cd2+ that are inconsistent with a strong metal site interaction (reproduced in Figure 2A).
We first extended these studies with a 2′-F substitution at the nucleophile position of the HHRz (Figure 3 and 2B). Like the 2′-H and 2′-OMe substitutions, a 2′-F substitution suppresses activity, enabling NMR experiments that test metal binding to ligands in the active site. The 2′-F is expected to more closely mimic a 2′-OH in size and electrostatics than a 2′-OMe. Upon addition of Cd2+ to this 2′-F/PS doubly substituted HHRz, however, no significant changes in the 31P chemical shift were observed for the SP isomer (Figure 3 and 2B). A slight upfield chemical shift was observed when Cd2+ was added to the RP isomer, as was also observed in an earlier report using the 2′-OMe construct (17). These data indicate that the HHRz with a 2′-F/PS scissile site has only a very weak, yet possibly stereo-specific propensity for metal interactions.
Figure 3.

31P NMR spectra of HHRz with a Rp or Sp 2′-F/PS substitution at the cleavage site. Experiments were performed in 5mM HEPES (pH 8.5) and 100 mM Na+ at 15 °C.
A 2′-NH2 nucleophile substitution is also expected to suppress catalytic activity of the HHRz. Like a 2′-OH, a 2′-NH2 is expected to be able to participate in hydrogen bonding and also be an available metal ligand. Additionally, based on studies of uridine mononucleotides, a 2′-NH2 substitution is expected to favor the 2′ endo sugar pucker (22, 23). The 2′ endo conformation, which would favor in-line attack, has been observed for the C1.1 ribose in crystals of an extended HHRz construct thought to most accurately predict the active site (29–31). Studies of the pKa of a 2′-NH2 on various nucleobase analogs indicate that at a pH greater than 7, the amine should be in its unprotonated, neutral state (32–35). Single-turnover kinetic studies for both WT and C1.1 2′-NH2 substituted HHRz constructs, performed in 5 mM HEPES (pH 8.5) and 100 mM Na+, showed ~1% cleavage over 36 hours in the absence of divalent cations. With addition of divalent metal ions, the 2′-NH2 HHRz showed only 2% (in 50 mM Mg2+) to ~6% (in 20 mM Cd2+ or 20 mM Mn2+) product formation in 36 hours, while the WT HHRz cleaved more than 70% of its substrate under these metal conditions in one hour (Supplementary Figure S1). Thus, the 2′-NH2/PS HHRz retains only very weak cleavage ability that would not interfere with an NMR study.
Upon addition of Cd2+, 31P NMR spectra of a racemic mixture of HHRz constructs containing C1.1 2′-NH2/PS substitutions showed significant upfield chemical shifts (Figure 4 and 2B) of both peaks. This effect was much greater than the shifts observed for the other cleavage site PS constructs with 2′-OMe or 2′-F substitutions (Figure 2B), suggesting significant metal association with the cleavage site containing the 2′-NH2/PS substitution. In particular, the furthest downfield peak originating at 58 ppm, which can be assigned to the Rp diastereomer based on previous work (17), moves upfield and merges with the Sp peak in an estimated >3 ppm upfield shift over addition of 6 equivalents of Cd2+ (final concentration ~2.2 mM Cd2+/0.1 M NaCl). This result in combination with the previous 31P NMR studies on constructs that lacked a potential metal ligand at the 2′ position, and showed little evidence for metal binding, indicates that the 2′ position may play a role in metal association with the cleavage site in the mHHRz.
Figure 4.

31P NMR of HHRz samples with 2′-NH2/PS substitution (mixture of PS diastereomers) at the cleavage site (C17, left) or the 5′ neighboring position (U16.1, right). Data were obtained at 15°C in 5 mM HEPES (pH 8.5) and 100 mM NaCl with initial RNA concentrations of 450 μM (left) and 440 uM (right).
Control studies establish specificity in the HHRz active site Cd2+-phosphorothioate interaction
Both amines and phosphorothioate thio groups are expected to be good ligands for Cd2+, a moderately soft metal ion (36, 37). These two proximal substitutions in the 2′-NH2/PS constructs may therefore create a non-specific Cd2+ binding site that recruits metals that would not otherwise bind to the substituted position. Furthermore, the HHRz active site is located at an RNA three-helix junction, which may increase the effective concentration of metal ions through electrostatic effects (17, 38–40) and therefore enhance an otherwise weak, non-specific interaction. To control for these possibilities, we tested Cd2+ association with a dinucleotide model and with two derivatives of the mHHRz containing the NH2/PS substitutions.
In order to test the ability of this double substitution to be a general soft metal-binding pocket outside the context of the HHRz, a amUPSU dinucleotide containing the NH2/PS substituted site was tested for its ability to bind Cd2+. The substituted dinucleotide was separated into its stereoisomers by reverse-phase HPLC. A metal titration showed less than a 0.5 ppm chemical shift change for both isomers through addition of up to 10 equivalents of Cd2+ (Figure S2). Addition of 75 molar equivalents of Cd2+ still only caused upfield shifts of ~1 ppm. A fit of the titration data to a 1:1 binding model (Equation 1) indicates a Kd for Cd2+ binding to the NH2/PS site of 22 mM for the SP isomer and 47 mM for the RP isomer (0.1 M NaCl) (Figure S3). These low calculated KD’s reflect weak metal binding and reinforce the conclusion that the double 2′-NH2/PS substitution alone does not create a high-affinity ‘recruiting’ metal binding pocket.
Metal ions are known to condense around the dense negative charge of the phosphodiester backbone of an RNA helix (reviewed in (39–41)). An increase of the effective metal concentration near the double 2′-NH2/PS substitution in any RNA helix could potentially enhance the apparent affinity of this site for Cd2+. We therefore tested an RNA construct composed of the HHRz substrate strand with the cleavage site 2′NH2/PS substitution in the context of a duplex with its complement strand. This construct showed < 1 ppm upfield chemical shift with addition of up to 5 equivalents of Cd2+ for both diastereomers (Figure S4). Similar very weak Cd2+-S interactions were found previously for 2′-H/PS and 2′-OMe/PS substitutions within helices (17). Taken together, these results also reflect a relatively weak Cd2+-sulfur interaction for phosphorothioates outside of the context of the HHRz active site, regardless of the identity of the proximal 2′ position.
The increased charge density at the three-helix junction of the HHRz active site could further enhance metal concentrations in that region (12, 17, 38–40), increasing the propensity for non-specific metal interactions. For this reason, we tested an HHRz construct in which the 2′-NH2/PS substitution was placed one nucleotide 5′ to the scissile phosphate, at position U16.1 (Figure 1). The mHHRz construct also contained an inhibitory 2′-OMe at the C17 cleavage site. 31P NMR spectra of the mixed diastereomers of this U16.1 2′NH2/PS mHHRz control construct showed less than a 1ppm change of the phosphorothioate-shifted peaks upon the addition of up to 6 equivalents of Cd2+ (Figure 4, right). These data indicate that simply positioning a 2′-NH2 near a phosphorothioate, even at a three-helix junction, does not necessarily create a high affinity metal binding site in the mHHRz. To ensure that the U16.1 double substitution does not significantly alter the local architecture of the mHHRz active site, we tested the activity of a catalytically competent (scissile 2′-OH) mHHRz with the U16.1 2′-NH2/PS substitution. Previous work has established that the mHHRz activity is tolerant of U16.1 2′-deoxy and PS substitutions (42, 43). The U 16.1 2′-NH2/PS construct, as a mixture of diastereomers, demonstrated near wild-type catalytic activity (Figure S5), indicating that the U16.1 double substitution did not negatively alter the structure of the three-helix junction. These control studies confirm that the observed metal interactions in the double substituted 2′-NH2/PS HHRz are specific to the scissile phosphate.
Cd2+-NH2 coordination in the amUPSU dinucleotide model suggested by 1H DQF-COSY spectroscopy
The preceding data indicate that a proximal 2′-functional group that is able to participate in hydrogen bonding and/or metal coordination may be required for a relatively high affinity metal interaction with a phosphorothioate at the mHHRz active site. 31P NMR spectra alone, however, do not confirm that the metal bound to the phosphorothioate sulfur atom is also coordinating the 2′ functional group of its 5′ ribose. One potential probe for this interaction is the chemical shift of the ribose 2′-H, which is sensitive to the electronegativity of its vicinal 2′-functional group (23). Metal coordination to the 2′-group is expected to change the electrostatic properties of the ligand, and therefore change the chemical shift of the 2′-H (23, 44).
In an initial investigation of metal interaction with the 2′ amine in a 2′-NH2/PS binding site, we used 1H NMR to determine the change in chemical shift of the 2′-H upon the addition of Cd2+ in separated isomers of the amUPSU dinucleotide model. Assignments for the 1′-H, 2′-H, and 3′-H ribose protons were based on 1H-31P COSY and DQF-COSY experiments (Figures 5, S6 and S7). For uridine, the previously reported value for the H2′ chemical shift with a 2′-OH substituent is 4.34 ppm, and with a 2′-NH2 is 3.56 ppm (23). Similar values of 4.4 ppm and 3.6 ppm, respectively, were assigned here (Figure 5). Upon addition of 40 mM Cd2+ to either Rp or Sp dinucleotide, shifts are observed in the DQF-COSY H1′-H2′ and H2′-H3′ cross peaks of the 2′-NH2 substituted ribose (Figure 5 and supplemental material). Metal coordination to the 2′-NH2 would result in electron density delocalization away from the H2′, and indeed downfield chemical shifts are observed for 1H resonance from the H1′, H2′, and the H3′ positions upon addition of Cd2+. This result is consistent with previous observations that H2′ resonances move downfield as the electronegativity of the other 2′ substituents is increased (23). While the ribose 1H chemical shift changes could be due to electron withdrawing influence of metal coordination to the 2′-NH2, they would also be sensitive to changes in ring conformation (45). In both diastereomers of amUPSU in the absence of Cd2+, the J1′2′ coupling constant appears to be larger than the J3′4′ coupling constant, suggesting that the 2′-NH2 sugar system has a dominantly C2′-endo sugar conformation (45, 46) and consistent with previous results from NMR studies of uridine mononucleotides (23). Addition of Cd2+ does not change the relative J1′2′–J3′4′ values (Figure 5 and S7). Thus, the observed chemical shift changes in the 2′-NH2 ribose system upon the addition of Cd2+ do not appear to be due to a change in the relative sugar conformation populations. In combination with the Cd2+-dependent upfield 31P chemical shift, these 1H NMR data are consistent with Cd2+ chelation by both 2′-NH2 and PS sulfur ligands in the amUPSU dinucleotides. These data further suggest that the 31P chemical shift changes observed upon addition of Cd2+ to the mHHRz cleavage site 2′-NH2/PS construct may also indicate 2′-NH2 coordination to the metal ion.
Figure 5.

DQF-COSY spectra of amUSpU in absence (black) and presence (red) of 40 mM CdCl2. Data were obtained in D2O at 10 °C in 10 mM sodium cacodylate (pH 7.4) and 100 mM NaCl. Crosspeak assignments are shown for the 2′NH2 ribose system (solid lines) and partial assignment for the 2′OH ribose (dotted line).
Discussion
Potential bidentate 2′-OH/phosphate coordination by divalent metals in the HHRz active site
Repressing activity in ribozymes is a requirement for biophysical studies that are aimed at examining structures occurring prior to the cleavage step. Previous NMR studies of metal interactions with the mHHRz have employed either a 2′-deoxy or a 2′-OMe nucleophile substitution to block activity(17, 20). Metal rescue experiments have identified the proR non-bridging oxygen atoms of both the A9 and scissile phosphates as catalytically important metal ligands in this system, and the bridging-metal hypothesis of Wang and coworkers predicts simultaneous coordination by these ligands in the transition state of the reaction (11). While NMR experiments have confirmed the A9 site to be a high affinity metal coordination site, there has been no previous spectroscopic evidence for metal coordination to the scissile phosphate (17, 20). These data are consistent with a model in which the mHHRz maintains predominantly a ‘ground-state’, open structure with a populated high-affinity A9/G10.1 metal site, and infrequently samples an unstable active structure in which the metal ion may acquire a non-bridging oxygen ligand from the scissile phosphate (11). It is also possible, however, that the cleavage site 2′ substitutions used previously to block activity in the spectroscopic studies may also have blocked the ability for a metal to interact with this site.
Any substitution within the active site of the HHRZ may have substantial positive or negative effects on metal coordination. While some substitutions may change the geometry of potential ligands, others may have steric and/or electrostatic effects. Even more problematic would be a substitution that replaces a potential metal ligand for an atom with no metal-binding capabilities. In this study, we continued the spectroscopic investigation of mHHRz scissile phosphate metal coordination by testing the Cd2+-binding abilities of mHHRz cleavage-site phosphorothioates in combination with either a 2′-F or 2′-NH2 nucleophile substitution. Whereas the 2′-F construct failed to exhibit metal binding to the scissile phosphate, the 2′-NH2 HHRz demonstrated 31P shifts consistent with innersphere metal ion coordination.
Although a potential nucleophile, the 2′-NH2-substituted mHHRz shows a loss of divalent metal-dependent catalysis that is not rescued by the addition of the soft metal Cd2+, indicating that these RNA constructs are stable during 31P NMR-detected metal titration experiments. A 2′-NH2 with an Rp phosphorothioate at the mHHRz cleavage site, however, exhibits up to ~3 ppm upfield changes in chemical shift upon addition of 6 equivalents of Cd2+ (Figure 2B and 4). Similar chemical shift changes were previously observed to result from Cd2+-phosphorothioate interactions at the HHRz A9 position (17), a metal-binding site that has also been demonstrated biochemically (11, 47) as well as by crystallography (48) and solution EPR experiments (49). The similar magnitude of 31P chemical shift change observed with the 2′-NH2/PS substitution indicates that this construct has a similar ability to associate with metals at the scissile phosphate, and that the metal coordination and/or hydrogen-bonding properties of the 2′-NH2 are a contributing factor.
The observation that of the four 2′ nucleophile substitutions tested in spectroscopic studies, the 2′-NH2 is the only functional group that presents an available metal ligand and is the only group resulting in observable metal interactions with the scissile phosphate raises the possibility that metal coordination by the native 2′- OH, or a metal-related hydrogen bonding interaction with this position, is relevant in context of the wild type ribozyme. There are several modes by which a single divalent metal may interact with the HHRz to promote catalysis (Scheme 1), including interactions with both the nucleophile and a non-bridging phosphodiester oxygen atom as shown in model C of Scheme 1, and or with the leaving group as shown in model B. The data presented here suggest the interactions in model C, in which a metal ion coordinating the proR oxygen of the scissile phosphate also coordinates to the nucleophilic 2′-OH. Functional and crystallographic evidence for this type of interaction in a ribozyme has been established for the Group I intron (reviewed in (50)). The results of our current experiments do not preclude interaction of an additional metal ion at the mHHRz cleavage site. However, the inability in previous NMR experiments to observe significant metal-phosphorothioate interactions in the absence of a metal-coordinating 2′-nucleophile does indicate that interactions such as in A or B of Scheme 1 do not easily occur in the ground-state of the mHHRz.
Scheme 1.
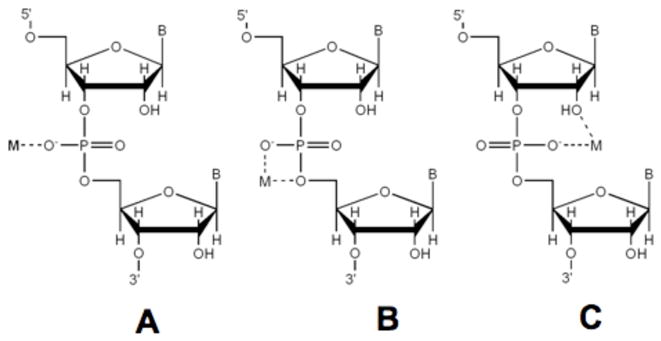
While 31P NMR-based results are suggestive of a bidentate metal ion coordination to both nucleophile and scissile phosphate in the mHHRz, further evidence for metal coordination to the 2′-nucleophile would include observation of metal-dependent shifts in signals from that position. Initial evidence for this coordination mode is found in the 1H NMR spectra of the dinucleotide amUPSU model system, obtained in the presence and absence of Cd2+. The upfield shifts of vicinal 2′H protons in Figure 5 are consistent with coordination of the 2′-NH2 by Cd2+ in both stereoisomers of the dinucleotide, supporting a bidentate metal-binding model. Similar studies probing this interaction in the HHRz are a goal for future experiments.
Cd2+ interactions with the mHHRz 2′-NH2/PS cleavage site are not due solely to ‘recruitment’
As discussed above, substitutions in the active site of a ribozyme can have drastic effects on the ability for a metal to specifically coordinate RNA ligands. These effects can either be prohibitive of a metal coordination event or stabilize a metal coordination that is not representative of the wild type system. In the present 31P NMR studies, a non-bridging phosphate oxygen is substituted with sulfur in order to differentiate the target site from all the other phosphodiester linkages in the RNA construct. This single substitution replaces the relatively hard oxygen atom with a soft metal-coordinating sulfur. For this reason, the common ‘hard’ divalent ion Mg2+ is replaced with ‘borderline-soft’ (and diamagnetic) Cd2+ in our NMR experiments to enable coordination to the substituted site. The other modification to the wild type system in these studies requires 2′ substitutions that prevent early steps of the HHRz reaction, allowing observation of an uncleaved HHRz conformation. A 2′NH2 was the only substitution that supported metal binding at the mHHRz cleavage site, and while this is an interesting result because it has hydrogen-bonding and metal coordination characteristics more similar to hydroxyl than any of the other attempted substitutions, an amine group is also considered a ‘soft’ ligand that, like sulfur, could enhance Cd2+ affinities by orders of magnitude (36, 37). The concern then, is that by placing two soft metal ligands in close proximity, the overall enhancement in metal-binding ability would recruit Cd2+ to an otherwise non-native site. This level of artificial enhancement in the Cd2+ ion affinities could mean that the metal coordination observed here is a artifact of the construct and not relevant to the wild type reaction.
The potential for metal ion ‘recruitment’ by the 2′-NH2/PS site was tested using control constructs containing the 2′-NH2/PS unit in context of a dinucleotide model, an RNA helix, and a nearby position in the mHHRz. Phosphorothioates embedded within these control constructs all demonstrate very weak interactions with Cd2+, as evidenced by <1 ppm 31P chemical shift changes in up to ~3 mM Cd2+ (Figure 2B), which is a result that mirrors previous findings for 2′-OMe/PS or 2′-H/PS substitutions embedded in single-strand or duplex RNA strands (17). The observation of >3 ppm 31P chemical shift changes induced by Cd2+ when the 2′-NH2/PS substitution is placed at the mHHRz scissile phosphate is therefore suggestive of a higher-affinity metal-binding pocket that is influenced by its local environment, and not general binding caused solely by the introduction of 2′-NH2/PS Cd2+ ligands.
Relevance of the Cd2+-NH2/PS active-site metal ion interaction to existing mHHRz activity models
Wang and co-workers established through metal-rescue experiments that a single metal ion is responsible for metal rescue of both the A9 and scissile phosphorothioates despite a measured distance of greater than 10Å between these ligands in structural studies of the mHHRz (11). These and other data lead to a model of the mHHRz reaction in which the ribozyme exists predominantly in an inactive open conformation, and samples an active but thermodynamically unstable closed conformation that brings the two putative metal ligands close enough for a single metal ion to bridge between them. The prediction is that this active conformation, and thus the metal-scissile phosphate interaction, would be difficult to observe spectroscopically due to its thermodynamic instability. Thus, in context of the bridging-metal model, the cleavage-site metal interaction observed in this study is surprising. Three potential explanations for these results are described below.
One possible scenario concerning the significance of these results is that the interaction observed in this study is adventitious and unrelated to mHHRz catalysis. As is common with structured RNAs, both solution and X-ray crystallographic studies of the mHHRz have predicted multiple divalent cation binding sites. It is possible that the interaction observed here with the mHHRz cleavage site is affected by the 2′ sugar substituent, but that it does not represent the metal-scissile phosphate interaction that is revealed in mechanistic studies. The fact that the interaction is more easily observed within the mHHRz active site than in the control samples could be ascribed to the electrostatics of the mHHRz active site, which is expected to form a relatively negative electrostatic pocket (17, 38, 40). Our attempted control for this situation, the 2′NH2/PS unit adjacent to the mHHRz cleavage site, could have slightly different geometric and electrostatic properties that influence its ability to coordinate Cd2+ relative to the cleavage site position. Even if the metal coordination event observed here is not relevant to mHHRz catalysis, its sensitivity to the 2′-substituent still provides a demonstration of the potential importance of ribose functional group changes on the metal coordination properties of neighboring phosphodiester ligands.
If the metal interaction with the mHHRz cleavage site that is observed in these experiments is indeed related to the functional, bridging metal ion, then its properties are subject to predictions based on the existing model. First, it must be postulated that the 2′-NH2/PS substitution somehow stabilizes the otherwise thermodynamically unstable ‘active’ conformation that brings the A9 and cleavage site metal ligands into proximity, allowing sufficient population of the bridging metal site to be observed by NMR. One could argue that simultaneous addition of both the cleavage site 2′ amine and the thio ligands have created a Cd2+ site with sufficient thermodynamic stability to allow partial population of this bridging state, while at the same time inhibiting the subsequent cleavage step. If these NMR studies are in fact observing the functional bridging metal ion, then the apparent affinity of the metal ion interaction with the cleavage site that is observed by NMR should be sensitive to substitutions at the A9 site. One particular expectation would be that the NMR chemical shift changes should appear at Cd2+ concentrations that are expected to fill the A9/G10.1 site. Because of the electrostatic properties of nucleic acids and presence of a charge-shielding counterion atmosphere, as well as possible cation-induced structural changes, metal ion association with RNA is best described in terms of K1/2 for the observable rather than KD for an equilibrium dissociation constant (39, 41, 51). These NMR experiments were performed by adding equivalents of Cd2+ rather than dialyzing to a known equilibrium Cd2+ concentration, further complicating interpretation in terms of quantitative affinities since there may be Cd2+ association with additional sites on the RNA. This means that the ‘free’ Cd2+ concentration is unknown but is possibly overestimated, leading to an overestimation of apparent K1/2 or Kd values. Thus, while these data indicate that a 2′-NH2 substitution promotes more facile association of Cd2+ with the PS-substituted mHHRz cleavage site than do 2′-X/PS (X=H, OMe, F) substitutions, the data of Figure 2B cannot be interpreted in terms of thermodynamic affinities for this site. With these caveats in mind, however, one can still compare behavior between similar NMR experiments. It is clear that the Cd2+-thiophosphate interaction with the 2′-NH2/PS cleavage site (Figure 2B) requires higher Cd2+ concentrations than those needed to fill the mHHRz 2′-OH/PS A9 site (Figure 2A). At similar RNA concentrations, signal changes from the 2′-NH2/PS cleavage site are > 3 ppm and not saturated by addition of 6 equivalents, or ~ 2.7 mM Cd2+ (0.1 M NaCl), whereas the A9 PS-substituted mHHRz signal is saturating at ~700 uM Cd2+ (0.1 M NaCl). A very approximate ≥4x ratio of apparent affinities can be estimated from these data, which is broadly consistent with the increase in Cd2+ affinity expected upon substitution of the A9 pro-R oxygen for sulfur as measured by Wang and coworkers.2 These are very qualitative observations, however, and further experiments exploring additional modifications of these sites would be required before a strong link could be drawn between the current experiments and population of the bridging metal site in the mHHRz.
As a final scenario, it is possible that the mHHRz samples in these NMR experiments are not sampling the thermodynamically unstable intermediate that would allow simultaneous metal ion coordination to both the A9 and the scissile phosphate, but that the results are still relevant to the active state of the ribozyme. Even if these experiments are primarily sampling the open ribozyme conformation, the fact that the addition of the 2′-NH2 ligand makes the scissile phosphorothioate more amenable to metal ion coordination (than when the site includes a 2′-H, F, or OMe) could be an indication of the same propensity in the active conformation of the mHHRz. As is observed here, in the bridging and active conformer an interaction with a 2′ ligand may strengthen metal ion association with the cleavage site phosphodiester and thereby stabilize the active conformer and tune reactivity in that state.3
While apparently weaker than the high-affinity A9 mHHRz site, the Cd2+ interaction with the 2′NH2/PS cleavage site is still significantly enhanced relative to several control samples. In 0.1 M NaCl, apparent Kd values of 20–50 mM are measured for Cd2+ binding to the 2′NH2/PS unit in the dinucleotide models (Figure S3), at least 10-fold weaker than the interaction with the mHHRz cleavage site. Interestingly, the weak but measurable Cd2+ Kd values for the dinucleotide models reflect slightly higher affinity for the Sp thiophosphate, while the Sp stereoisomer of the mHHRz construct had less significant 31P chemical shift changes upon addition of metal (Figure 2B). This result, in agreement with the stereospecificity found in metal-rescue experiments (11, 25, 52, 53), highlights the potential organization of the mHHRz active site in comparison with the free dinucleotide.
Relationship to current activity models for the extended Hammerhead Ribozyme
Recent research has focused on exHHRz constructs whose extended stems I and II include tertiary contacts that stabilize the formation of the active state. New crystal structures of extended HHRzs exhibit a more compact active site, with many interactions that satisfy predictions from kinetic data obtained with the mHHRz (31, 43). Unlike the case of the mHHRz, and consistent with the predictions of Wang and coworkers (11), in structures of the extended HHRz the A9/G10.1 metal-binding pocket is within a feasible distance to the scissile phosphate to allow a metal ion to bridge the pro-R oxygens. The bridging metal ion interaction is still not observed in these structures, however, despite a ~5 Å distance between the two potential pro-R ligands. Mn2+ occupies only the A9/G10 pocket in crystallographic studies of a pre-cleaved Schistosome exHHRz with a 2′-OMe substitution at the nucleophile (29, 30). Based on biochemical predictions and crystallographic contacts, a current acid-base model for HHRz catalysis is proposed that includes activation of the HHRz 2′-nucleophile by deprotonation by the N6 of G12, and protonation of the scissile phosphate leaving group by the 2′-OH of G8 (30, 31, 54–56). In addition to being sensitive to substitutions of G8 and G12, activities of exHHRz’s are sensitive to the nature of added divalent cation (12, 13, 57), and active-site phosphorothioate inhibition with rescue by Cd2+ has been demonstrated in the extended HHRz (13). Molecular dynamics and biochemical studies have also suggested a metal ion interaction with the 2′-OH of G8, proposed to activate it as the general acid (30, 55, 58). Taken together, these observations lead to a current model of combined nucleobase and metal ion contributors to catalysis by the HHRz, with details remaining to be developed.
The experiments reported here could indicate that the 2′-OMe substitution used in crystallographic studies of the exHHRz may inhibit potential metal interactions with the nucleophile. A recent X-ray structure of a slow-cleaving G12A sTRSV exHHRz mutant containing a native 2′-OH nucleophile shows, however, that substitutions at the nucleophile position do not cause gross rearrangements of the catalytic core. The G12A structure does exhibit slight changes in scissile phosphate and nucleophile positions in comparison with the previous Schistosoma structure (29). No localized metal ions were reported in the pre-cleaved structure of the G12A exHHRz. In a structure collected post-cleavage, however, densities for two Mg2+ ions are reported, one of which is near to the 2′ nucleophile (now the cyclic phosphate) and the other of which maintains the A9/G10.1 position. While the appearance of the new metal ion near to the 2′ nucleophile in a post-cleavage structure is intriguing, this interaction is not yet predicted from functional data and possibly is relevant only to the cleaved form of the ribozyme or to the crystallographic state.
Summary
The data presented here show that Cd2+ interactions with a cleavage-site phosphorothioate in the mHHRz are significantly enhanced by inclusion of a proximal 2′-NH2 instead of 2′-H, F, or OMe substituents. These results demonstrate an influence of substituents at the 2′ nucleophile on metal ion coordination to the scissile phosphate. In the case of the Cd2+-2′-NH2/PS interaction, the Cd2+ is proposed to directly coordinate to the nucleophile position. If present in the active site of the native HHRz, such a bidentate metal-ion coordination could allow metal ions to activate the nucleophile for deprotonation while also coordinating the phosphate, and thereby also aid in transition state stabilization. Other potential avenues by which the 2′-position might influence metal coordination in the HHRz could be via hydrogen-bonding to a metal aqua ligand, or by electrostatics upon deprotonation. Further studies will be required to assess the applicability of these and other metal ion binding modes to activity in the exHHRz.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM058096 to VJD, 5T32GM008523 to EMO, 5T32GM007759 to WLW) and the NSF (CHE0111696 to VJD)
We thank Professor Andy LiWang and Dr. Xiangming Kong (Department of Biochemistry and Biophysics, Texas A&M University) for assistance 1H DQF-COSY NMR data collection and analysis, Dr. Michael Strain for assistance with NMR experiments at the University of Oregon, and Chiharu Graybill (University of Oregon, Institute of Molecular Biology) for additional 31P NMR data collection on the mHHRz. We also appreciate receiving thoughtful comments on this work from the Reviewers.
Abreviations
- HHRz
hammerhead ribozyme
- mHHRz
minimal (or truncated) HHRz
- exHHRz
extended HHRz
- RNA
ribonucleic acid
- DNA
deoxyribonucleic acid
- WT
wild-type
- NMR
nuclear magnetic resonance
- EPR
electron paramagnetic resonance
- PS
phosphorothioate
- Rp or Sp
phosphorothioate substitutions 5′ to the indicated site with Rp or Sp configurations
- OMe
OCH3 group
- amUPSU
uridine dinucleotide with a 2′-NH2 substitution on the 5′ ribose and a phosphorothioate linkage
- TEA
triethanolamine
- HEPES
N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- TMP
trimethyl phosphate
- nt
nucleotide
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
- DTT
dithiothreitol
- MD
molecular dynamics
Footnotes
One estimate of expected affinities comes from the mechanistic studies of Wang and coworkers (11), who measure activity-based Cd2+ Kd’s for the unsubstituted mHHRz A9 site of ~100 μM-280 μM in backgrounds of 10 mM Mg2+ or Ca2+, respectively. Their values for the interaction of Cd2+ with a A9-Rp phosphorothioate substitution are ~30–70 μM, an increase of 3–4x higher affinity presumably due to the sulfur-Cd2+ interaction. Since our NMR studies are performed under different ionic strength, pH, and temperature conditions, we cannot directly compare expected values (and as described in the text, the NMR studies are not performed under conditions allowing for valid K1/2 measurements). However, a rough extrapolation might predict a similar difference of 3–4 fold between the Cd2+ affinity for the unsubstituted and thio-substituted A9 site. This value is qualitatively consistent with the differences in binding behavior of Figures 2A (PS at A9) and 2B (unsubstituted A9).
Comparison with the control constructs indicates a greater propensity towards Cd2+ interactions at the mHHRz cleavage site when both 2′NH2/PS positions are available to interact with the metal ion. It is difficult, however, to relate this observation to quantitative predictions about the ‘native’ cleavage site state containing Mg2+ and potential bidentate 2′OH/PO ligands. Though the Mg2+ ion would be presented with two ‘hard’ ligands, in the case of Mg2+ the ion would exchange a high-affinity aqua ligand for the 2′OH, an exchange potentially driven only by the chelate effect. By contrast, the Cd2+ ion has a much higher affinity for amine over aqua ligands, which might significantly favor interaction with the 2′NH2. Still, the fact that addition of a 2′NH2 was necessary to observe the Cd2+ interaction with the mHHRz cleavage site might suggest that in the native state, contribution from the 2′ position is also important for this interaction. As suggested by a Reviewer, one such contribution could be deprotonation of the 2′OH along the reaction pathway, increasing negative electrostatic potential and thereby stabilizing the bridging metal ion. Interestingly, MD simulations (58) based on the Schistosoma exHHRz crystal structure with Mg2+ placed in the A9/G10.1 site, but starting with a deprotonated 2′-O− nucleophile, have shown much more facile formation of the bridging metal state in comparison with simulations based on the fully protonated nucleophile.
SUPPORTING INFORMATION AVAILABLE
Activities of mHHRz constructs with a C1.1 2′-NH2 or a U16.1/PS substitution, Cd2+-dependent 31P NMR spectra of amUPSU diastereomers or 2′-NH2/PS substitutions within RNA helices, binding isotherms for amUPSU diastereomers, and 1H-31P COSY and 1H-1H DQF-COSY spectra of amUPSU diastereomers in the presence and absence of Cd2+. This material is available free of charge via the Internets at http://pubs.acs.org.
References
- 1.Forster AC, Symons RH. Self-cleavage of virusoid RNA is performed by the proposed 55-nucleotide active site. Cell. 1987;50:9–16. doi: 10.1016/0092-8674(87)90657-x. [DOI] [PubMed] [Google Scholar]
- 2.Haseloff J, Gerlach WL. Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature. 1988;334:585–591. doi: 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- 3.Uhlenbeck OC. A small catalytic oligoribonucleotide. Nature. 1987;328:596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 4.Hutchins CJ, Rathjen PD, Forster AC, Symons RH. Self-cleavage of plus and minus RNA transcripts of avocado sunblotch viroid. Nucleic Acids Res. 1986;14:3627–3640. doi: 10.1093/nar/14.9.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prody GA, Bakos JT, Buzayan JM, Schneider IR, Bruening G. Autolytic processing of dimeric plant-virus satellite RNA. Science. 1986;231:1577–1580. doi: 10.1126/science.231.4745.1577. [DOI] [PubMed] [Google Scholar]
- 6.Martick M, Horan LH, Noller HF, Scott WG. A discontinuous hammerhead ribozyme embedded in a mammalian messenger RNA. Nature. 2008;454:899–902. doi: 10.1038/nature07117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blount KE, Uhlenbeck OC. The structure-function dilemma of the hammer head ribozyme. Ann Rev Biophys Biomol Struct. 2005;34:415–440. doi: 10.1146/annurev.biophys.34.122004.184428. [DOI] [PubMed] [Google Scholar]
- 8.O’Rear JL, Wang SL, Feig AL, Beigelman L, Uhlenbeck OC, Herschlag D. Comparison of the hammerhead cleavage reactions stimulated by monovalent and divalent cations. RNA. 2001;7:537–545. doi: 10.1017/s1355838201002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curtis EA, Bartel DP. The hammerhead cleavage reaction in monovalent cations. RNA. 2001;7:546–552. doi: 10.1017/s1355838201002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roychowdhury-Saha M, Burke DH. Distinct reaction pathway promoted by non-divalent-metal cations in a tertiary stabilized hammerhead ribozyme. RNA. 2007;13:841–848. doi: 10.1261/rna.339207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry. 1999;38:14363–14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- 12.Boots JL, Canny MD, Azimi E, Pardi A. Metal ion specificities for folding and cleavage activity in the Schistosoma hammerhead ribozyme. RNA. 2008;14:2212–2222. doi: 10.1261/rna.1010808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osborne EM, Schaak JE, Derose VJ. Characterization of a native hammerhead ribozyme derived from schistosomes. RNA. 2005;11:187–196. doi: 10.1261/rna.7950605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blount KF, Uhlenbeck OC. The Structure-Function Dilemma of the Hammerhead Ribozyme. Annu Rev Biophys Biomol Struct. 2005;34:415–440. doi: 10.1146/annurev.biophys.34.122004.184428. [DOI] [PubMed] [Google Scholar]
- 15.Pecoraro V, Hermes J, Cleland W. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry. 1984;23:5262–5271. doi: 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- 16.Maderia M, Horton TE, DeRose VJ. Metal interactions with a GAAA RNA tetraloop characterized by P-31 NMR and phosphorothioate substitutions. Biochemistry. 2000;39:8190–8200. doi: 10.1021/bi000140l. [DOI] [PubMed] [Google Scholar]
- 17.Maderia M, Hunsicker LM, DeRose VJ. Metal-phosphate interactions in the hammerhead ribozyme observed by P-31 NMR and phosphorothioate substitutions. Biochemistry. 2000;39:12113–12120. doi: 10.1021/bi001249w. [DOI] [PubMed] [Google Scholar]
- 18.Huppler A, Nikstad L, Allmann A, Brow D, Butcher S. Metal binding and base ionization in the U6 RNA intramolecular stem-loop structure. Nat Struct Biol. 2002;9:431–435. doi: 10.1038/nsb800. [DOI] [PubMed] [Google Scholar]
- 19.Eckstein F. Nucleoside phosphorothioates. Annu Rev Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- 20.Suzumura K, Yoshinari K, Tanaka Y, Takagi Y, Kasai Y, Warashina M, Kuwabara T, Orita M, Taira K. A reapprisal, based on 31P NMR, of the direct coordination of a metal ion with the phosphoryl oxygen at the cleavage site of a hammerhead ribozyme. J Am Chem Soc. 2002;124:8230–8236. doi: 10.1021/ja0202098. [DOI] [PubMed] [Google Scholar]
- 21.Pieken W, Olsen D, Benseler F, Aurup H, Eckstein F. Kinetic characterization of ribonuclease-resistant 2′-modified hammerhead ribozymes. Science. 1991;253:314–317. doi: 10.1126/science.1857967. [DOI] [PubMed] [Google Scholar]
- 22.Hruska FE, Mak A, Singh H, Shugar D. Proton magnetic-resonance study of effect of 2′-O-methylation on ribonucleoside conformation. Can J Chem-Rev. 1973;51:1099–1106. [Google Scholar]
- 23.Guschlbauer W, Jankowski K. Nucleoside conformation is determined by the electronegativity of the sugar substituent. Nucleic Acids Res. 1980;8:1421–1433. doi: 10.1093/nar/8.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunitz J, Taylor R. Organic fluorine hardly ever accepts hydrogen bonds. Chem Eur J. 1997;3:89–98. [Google Scholar]
- 25.Slim G, Gait MJ. Configurationally defined phosphorothioate-containing oligoribonucleotides in the study of the mechanism of cleavage of hammerhead ribozymes. Nucleic Acids Res. 1991;19:1183–1188. doi: 10.1093/nar/19.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez RL, Jr, Tinoco I., Jr Solution structure and thermodynamics of a divalent metal ion binding site in an RNA pseudoknot. J Mol Biol. 1999;289:1267–1282. doi: 10.1006/jmbi.1999.2841. [DOI] [PubMed] [Google Scholar]
- 27.Roberts GCK. NMR of Macromolecules. Oxford University Press; Oxford: 1993. [Google Scholar]
- 28.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 29.Chi YI, Martick M, Lares M, Kim R, Scott WG, Kim SH. Capturing hammerhead ribozyme structures in action by modulating general base catalysis. PLoS Biol. 2008;6:2060–2068. doi: 10.1371/journal.pbio.0060234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martick M, Lee TS, York DM, Scott WG. Solvent structure and hammerhead ribozyme catalysis. Chemistry & Biology. 2008;15:332–342. doi: 10.1016/j.chembiol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martick M, Scott WG. Tertiary Contacts Distant from the Active Site Prime a Ribozyme for Catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aurup H, Tuschl T, Benseler F, Ludwig J, Eckstein F. Oligonucleotide duplexes containing 2′-amino-2′-deoxycytidines: thermal stability and chemical reactivity. Nucleic Acids Res. 1994;22:20–24. doi: 10.1093/nar/22.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobbs J, Sternbach H, Sprinzl M, Eckstein F. Polynucleotides containing 2′-amino-2′-deoxyribose and 2′-azido-2′-deoxyribose. Biochemistry. 1973;12:5138–5145. doi: 10.1021/bi00749a018. [DOI] [PubMed] [Google Scholar]
- 34.Verheyden J, Wagner D, Moffatt J. Synthesis of some pyrimidine 2′-amino-2′-deoxynucleosides. J Org Chem. 1971;37:250–254. doi: 10.1021/jo00801a002. [DOI] [PubMed] [Google Scholar]
- 35.Dai Q, Lea CR, Lu J, Piccirilli JA. Syntheses of (2 ′)3 ′-N-15-Amino-(2 ′)3 ′-deoxyguanosine and determination of their pK(a) values by N-15 NMR spectroscopy. Org Lett. 2007;9:3057–3060. doi: 10.1021/ol071129h. [DOI] [PubMed] [Google Scholar]
- 36.Martell AE, Smith RE. Critical Stability Constants. 1–4. Plenum; New York: 1971–1974. [Google Scholar]
- 37.Martell AE, Hancock RD. Metal Complexes in Aqueous Solutions. Plenum; New York: 1996. [Google Scholar]
- 38.Mohanty U, Spasic A, Kim HD, Chu S. Ion atmosphere of three-way junction nucleic acid. J Phys Chem B. 2005;109:21369–21374. doi: 10.1021/jp050005o. [DOI] [PubMed] [Google Scholar]
- 39.Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu VB, Herschlag D. Unwinding RNA’s secrets: advances in the biology, physics, and modeling of complex RNAs. Curr Opin Struct Biol. 2008;18:305–314. doi: 10.1016/j.sbi.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeRose VJ. Characterization of nucleic acid metal ion binding by spectroscopic techniques. In: Hud NV, editor. Nucleic Acid-Metal Ion Interactions. Royal Society of Chemistry; Cambridge: 2008. pp. 154–175. [Google Scholar]
- 42.Kore AR, Eckstein F. Hammerhead Ribozyme Mechanism: A Ribonucleotide 5′ to the Substrate Cleavage Site Is Not Essential. Biochemistry. 1999;38:10915–10918. doi: 10.1021/bi991332n. [DOI] [PubMed] [Google Scholar]
- 43.Nelson J, Uhlenbeck O. Hammerhead redux: Does the new structure fit the old biochemical data? RNA. 2008;14:605–615. doi: 10.1261/rna.912608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uesugi S, Miki H, Ikehara M, Iwahashi H, Kyogoku Y. Linear relationship between electronegativity of 2′-substituents and conformation of adenine nucleosides. Tet Lett. 1979:4073–4076. [Google Scholar]
- 45.Haasnoot CAG, Deleeuw FAAM, Altona C. The Relationship between Proton-Proton Nmr Coupling-Constants and Substituent Electronegativities. 1 An Empirical Generalization of the Karplus Equation. Tetrahedron. 1980;36:2783–2792. [Google Scholar]
- 46.Kim SG, Lin LJ, Reid BR. Determination of Nucleic-Acid Backbone Conformation by H-1-NMR. Biochemistry. 1992;31:3564–3574. doi: 10.1021/bi00129a003. [DOI] [PubMed] [Google Scholar]
- 47.Peracchi A, Beigelman L, Scott EC, Uhlenbeck OC, Herschlag D. Involvement of a specific metal ion in the transition of the hammerhead ribozyme to its catalytic conformation. J Biol Chem. 1997;272:26822–26826. doi: 10.1074/jbc.272.43.26822. [DOI] [PubMed] [Google Scholar]
- 48.Scott WG, Finch JT, Klug A. The crystal structure of an all-RNA hammerhead ribozyme: a proposed mechanism for RNA catalytic cleavage. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 49.Vogt M, Lahiri S, Hoogstraten CG, Britt RD, DeRose VJ. Coordination environment of a site-bound metal ion in the hammerhead ribozyme determined by 15N and 2H ESEEM spectroscopy. J Am Chem Soc. 2006;128:16764–16770. doi: 10.1021/ja057035p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stahley MR, Adams PL, Wang JM, Strobel SA. Structural metals in the group I intron: A ribozyme with a multiple metal ion core. J Mol Biol. 2007;372:89–102. doi: 10.1016/j.jmb.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das R, Travers KJ, Bai Y, Herschlag D. Determining the Mg2+ stoichiometry for folding an RNA metal ion core. J Am Chem Soc. 2005;127:8272–8273. doi: 10.1021/ja051422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knoll R, Bald R, Furste JP. Complete identification of nonbridging phosphate oxygens involved in hammerhead cleavage. RNA. 1997;3:132–140. [PMC free article] [PubMed] [Google Scholar]
- 53.Ruffner DE, Uhlenbeck OC. Thiophosphate interference experiments locate phosphates important for the hammerhead RNA self-cleavage reaction. Nucleic Acids Res. 1990;18:6025–6029. doi: 10.1093/nar/18.20.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han J, Burke JM. Model for general acid-base catalysis by the hammerhead ribozyme: pH-activity relationships of G8 and G12 variants at the putative active site. Biochemistry. 2005;44:7864–7870. doi: 10.1021/bi047941z. [DOI] [PubMed] [Google Scholar]
- 55.JMT, Perrin DM. Probing general acid catalysis in the hammerhead ribozyme. J Amer Chem Soc. 2009;131:1520–5126. doi: 10.1021/ja807790e. [DOI] [PubMed] [Google Scholar]
- 56.Thomas JM, Perrin DM. Probing General Base Catalysis in the Hammerhead Ribozyme. J Amer Chem Soc. 2008;130:15467–15475. doi: 10.1021/ja804496z. [DOI] [PubMed] [Google Scholar]
- 57.Roychowdhury-Saha M, Burke DH. Extraordinary rates of transition metal ion-mediated ribozyme catalysis. RNA. 2006;12:1846–1852. doi: 10.1261/rna.128906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee TS, Lopez CS, Giambasu GM, Martick M, Scott WG, York DM. Role of Mg2+ in hammerhead ribozyme catalysis from molecular simulation. J Amer Chem Soc. 2008;130:3053–3064. doi: 10.1021/ja076529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.