

Abstract
Cells are constantly exposed to genotoxic events that can damage DNA. To counter this, cells have evolved a series of highly conserved DNA repair pathways to maintain genomic integrity. The ATM protein kinase is a master regulator of the DNA double-strand break (DSB) repair pathway. DSBs activate ATM's kinase activity, promoting the phosphorylation of proteins involved in both checkpoint activation and DNA repair. Recent work has revealed that 2 DNA damage response proteins, the Tip60 acetyltransferase and the mre11-rad50-nbs1 (MRN) complex, co-operate in the activation of ATM in response to DSBs. MRN functions to target ATM and the Tip60 acetyltransferase to DSBs. Tip60's chromodomain then interacts with histone H3 trimethylated on lysine 9, activating Tip60's acetyltransferase activity and stimulating the subsequent acetylation and activation of ATM's kinase activity. These results underscore the importance of chromatin structure in regulating DNA damage signaling and emphasize how histone modifications co-ordinate DNA repair. In addition, human tumors frequently exhibit altered patterns of histone methylation. This rewriting of the histone methylation code in tumor cells may impact the efficiency of DSB repair, increasing genomic instability and contributing to the initiation and progression of cancer.
Keywords: Tip60, ATM, histone methylation, H3K9me3, chromodomain, DNA repair, lysine demethylase
Tip60 (KAT5) is a ubiquitously expressed acetyltransferase which is an essential player in multiple signaling pathways, including transcriptional regulation, steroid receptor function, chromatin remodeling, histone acetylation, DNA repair and maintaining stem cell function 1-3. Tip60 acetylates the ε-amino groups of lysine residues on both histone and non-histone proteins 3, 4, including such diverse targets as histone H2A and H4 5-8, the androgen receptor 9, p53 10, 11, enzymes involved in glucose metabolism 12, the ATM kinase 13-16 and others 1, 12. Acetylation of these proteins by Tip60 is linked to changes in functional activity, including, for example, activation of the ATM kinase 16 and regulation of p53 function 10, 11. Tip60, like most acetyltransferases, exhibits limited sequence specificity for protein acetylation. Instead, Tip60 interacts with multiple protein partners that target Tip60 to specific substrates to promote their acetylation 1, 3. The complexity of Tip60 interactions is highlighted by the observation that Tip60 is a highly connected “hub” protein, interacting with multiple proteins in complex regulatory pathways 17. Consequently, inactivation of Tip60 is embryonic lethal in mice 18, underscoring the essential role of Tip60 in development. The Tip60 acetyltransferase therefore controls the acetylation of wide range of cellular proteins which are required to maintain cell viability. This review will focus on the role of Tip60 in regulating DNA double-strand break (DSB) repair.
Tip60 and DNA repair
DNA damage can arise through errors which occur during DNA replication or by exogenous agents which directly damage the DNA. DNA double-strand breaks (DSBs), which are produced by exposure to ionizing radiation, are particularly difficult for the cell to repair. Consequently, mammalian cells have evolved a highly complex mechanism to detect and repair these types of DNA lesions 19. Tip60 plays a key role in DSB repair, and is required to maintain genomic integrity and regulate the repair of DNA damage 7, 16, 20. Inactivation of Tip60 leads to increased sensitivity to ionizing radiation 7, 13, 14, 16, 21 and increased levels of chromosomal aberrations 13. Tip60 is also required for the activation of the ATM kinase 16, which phosphorylates multiple DNA damage response proteins, including nbs1, p53, chk2 and SMC1 (reviewed in 22) in response to DSB detection. Aberrant expression of Tip60 has been detected in prostate, breast and colorectal cancer 23-26. Further, Tip60 is a haplo-insufficient tumor suppressor, and loss of heterozygosity at the Tip60 locus and reduced levels of nuclear Tip60 have been detected in breast cancer 27. These observations demonstrate that a key function of Tip60 is to protect cells from genomic instability and to suppress potentially transforming events which can lead to cancer. Work from many laboratories has contributed to our current understanding of how Tip60 regulates genomic stability. This work indicates that Tip60 participates in 2 major pathways involved in DSB repair – chromatin remodeling at DSBs by the NuA4-Tip60 complex and acetylation and activation of the ATM kinase. The ability of Tip60 to regulate chromatin structure at DSBs is mediated through its interaction with the NuA4 complex 28, a multifunctional remodeling complex which is recruited to DSBs 21, 29. The NuA4-Tip60 complex acetylates histones H2AX and H4 at DSBs 6, 21, 29-31, facilitating both turnover of H2AX and modifying chromatin architecture to facilitate DSB repair (reviewed in 32, 33). Although this function for Tip60 is critical in DSB repair, this review will focus on the emerging role of Tip60 in the acetylation and activation of the DNA damage responsive ATM kinase.
Tip60 and ATM activation
The ATM kinase is the product of the ataxia telangiectasia gene 34. Patients with A-T have impaired antibody production, neurodegeneration, increased cancer risk and are extremely sensitive to IR-induced DNA damage, indicating an underlying defect in the repair of DSBs 34. Cells derived from A-T patients which lack functional ATM protein exhibit defects in DSB repair and loss of DNA-damage activated cell cycle checkpoints 22, 35, resulting in increased sensitivity to ionizing radiation. Cloning of the A-T gene identified ATM as a member of the PIK kinase family of DNA-damage activated kinase, which includes the DNA-PKcs and ATR kinases 36. Subsequent work from many laboratories identified 100s of proteins 37 phosphorylated by ATM in response to DNA damage, including key components of the DSB repair pathway such as p53, nbs1, chk2, brca1 and H2AX (reviewed in 19, 34). The phosphorylation of the c-terminal of the histone variant H2AX, (termed γH2AX 38) by ATM plays a pivotal role in DSB repair. H2AX is rapidly phosphorylated on chromatin domains surrounding the DSB, extending up to 1Mb on either side of the break 39. The mdc1 scaffold protein then binds directly to γH2AX, providing a platform to recruit and retain other DNA repair proteins, including 53BP1, RNF8 and brca1, at the DSB 40-44. The end result is the accumulation of DNA repair proteins on large (megabase) chromatin domains on either side of the DSB, which can be visualized by immunofluorescence techniques using antibodies against components of these complexes 42-44. Thus activation of ATM's kinase activity initiates a signal transduction pathway which leads to recruitment of DNA repair complexes to DSBs and the activation of cell cycle checkpoints. ATM activation is therefore a crucial step in the detection and repair of DSBs.
Much work has focused on understanding how DSBs upregulate the kinase activity of ATM. DNA damage leads to increased autophosphorylation of ATM at multiple sites 45, including serine 1981 46. This autophosphorylation of ATM was proposed to initiate conversion of inactive ATM dimers to active ATM monomers 46, and this was supported by the observation by several groups that mutation of ATM autophosphorylation sites blocks ATM activation 45, 46 and dimer-monomer transition 14. However, recent biochemical studies have shown that in vitro activation of ATM kinase activity can be achieved in the absence of significant serine 1981 autophosphorylation 47, 48. In addition, mouse models in which 1 or more of the ATM autophosphorylation sites were mutated lacked any detectable defect in ATM function 49, 50. It is possible that these different results are explained by differences the mechanism of ATM activation between mouse and human cells, or that the murine model system employed has influenced the outcome 49, 50. Although more work is required to resolve these issues, the data indicates that autophosphorylation of ATM is not the primary mechanism for ATM activation, since autophosphorylation is dispensable for ATM function under some conditions 47, 48, 50.
An additional contributor to ATM activation is the MRN DNA binding complex 51-55. MRN consists of the mre11 nuclease, which contains a DNA binding domain, the rad50 ATPase, which functions as a structural component, and the nbs1 protein, which contains several phosphorylation sites and functions as a regulatory factor for MRN 51. ATM interacts with the MRN complex 55, 56, and studies have clearly shown that mutation or deletion of the mre11, rad50 or nbs1 components of MRN significantly reduce the activation of ATM's kinase activity by DNA damage in vivo 52-55. In addition, biochemical studies demonstrate that purified MRN is sufficient to activate ATM's kinase activity in vitro 47, 48, 57. These 2 lines of evidence indicate that MRN is upstream of ATM and is essential for the full activation of ATM's kinase activity. In addition to MRN, recent work identified the Tip60 acetyltransferase as an essential factor required for ATM activation.
Tip60's acetyltransferase activity is rapidly activated by ionizing radiation, leading to the acetylation and activation of the ATM kinase 16. Loss of Tip60 activity prevents ATM acetylation and blocks the activation of ATM's kinase activity, indicating a crucial role for Tip60 in ATM activation. Tip60 and ATM form a complex in which Tip60 associates with the highly conserved FATC domain at the extreme c-terminal of ATM (figure 1) 16, 58. This interaction promotes the acetylation of lysine 3016 of ATM by Tip60 14, 16, 58. Mutation of lysine 3016 blocks the activation of ATM's kinase activity by DNA damage, indicating that Tip60-dependent acetylation of ATM is a key step in the activation of ATM's kinase activity. Subsequently, other groups confirmed a role for Tip60 and acetylation of ATM in regulating ATM activity 59-61. The ATM acetylation site is located in the PIKK Regulatory Domain (PRD) of ATM, which is wedged between the c-terminal FATC domain and the kinase domain of ATM (figure 1). The ATM acetylation site is highly conserved among higher eukaryotes 14, whereas the autophosphorylation sites are not 46, 49, 50, suggesting that acetylation is an evolutionarily conserved event in ATM regulation. Further, the c-terminal PRD-FATC domain structure is critical for the kinase activity of several PIK protein family members, including mTor 62, DNA-PKcs 63, 64 and ATM 58. Studies on ATR demonstrate that binding of TopBP1 (a regulator of ATR function) to the PRD of ATR activates ATR's kinase activity 65. Based on the results presented here and on studies on related PIKs 36, 66, it is proposed that acetylation of lysine 3016 by Tip60, which is located in the PRD between the FATC and kinase domains, alters the conformation of the FATC domain. This altered conformation of the FATC domain could allow substrate proteins access to the kinase domain, as well as positively regulating the intrinsic kinase activity of the kinase domain. One additional factor to be considered is the nature of the interaction between Tip60 and the FATC domain of ATM. The original studies demonstrated that ATM and Tip60 copurify from cells 16, 58, and that mutation or deletion of the FATC domain abolished this interaction. However, detailed in vitro studies from our laboratory (B. Price, unpublished studies) have failed to detect a direct interaction between ATM and Tip60, indicating that additional protein factors are required for the formation of the ATM-Tip60 complex (figure 1). Identifying these factors will provide additional insights into the complexity of Tip60 regulation of ATM.
Figure 1. Acetylation and activation of ATM by Tip60.

Domain structure of ATM, including the PIK Regulatory Domain (PRD) and the conserved FAT and FATC domains. ? Indicates unidentified protein factor(s) hypothesized to be required for recruiting Tip60 to the ATM complex. IR = Ionizing Radiation, Ac = acetylation.
The final step in understanding Tip60's role in the DNA damage response is to determine how DSBs increase the acetyltransferase activity of Tip60 and mediate its ability to acetylate ATM. Clues as to the potential mechanism are provided by recent work examining the chromodomain at the N-terminal of Tip60 13. Chromodomains are specialized binding modules containing conserved hydrophobic amino-acids which interact with methyl groups on methylated lysine residues 67. Lysine methylation is a common post-translational modification of histones in which the ε-amino group of lysine is either mono-, di- or trimethylated 68, 69, with each methylation state encoding specific functional information. For example, methylation of histone H3 on lysines 9 or 27 and histone H4 on lysine 20 is associated with the inactive heterochromatin 69-71, whereas methylation of histone H3 on lysines 4 and 36 is associated with transcriptionally active genes 72-74. Histone methylation is a dynamic signal transduction process, controlled by histone methyltransferases 75, 76 and histone demethylases 77, 78. Methylated lysine residues on histones provide docking sites for a variety of specialized methyl-lysine binding domains, including tudor domains, PHD fingers, and chromodomains 67, 79, 80. For example, the chromodomain of HP1α interacts with H3K9me3 81, 82. Histone methylation therefore represents a signaling system which creates specific sites for recruitment of chromatin modifying complexes to the chromatin.
There is now strong evidence that Tip60's chromodomain interacts specifically with H3K9me3 13. This interaction between Tip60 and H3K9me3 functions as an allosteric regulator, increasing the catalytic activity of Tip60. Further, mutations in the conserved hydrophobic domains of the chromodomain block both the interaction between Tip60 and H3K9me3 and the upregulation of Tip60's acetyltransferase activity by DNA damage. As a result, inactivating mutations in Tip60's chromodomain inhibits the subsequent acetylation and activation of ATM's kinase activity by Tip60 13. Further, when global H3K9me3 levels were reduced, either by overexpressing KDM4D, a H3K9me3 demethylase 78 or by genetic inactivation of the major H3K9 methyltransferases, Suv39h1 and Suv39h2 83, Tip60 activation following DNA damage was significantly decreased 13. Further, cells with reduced levels of H3K9 methylation displayed increased sensitivity to ionizing radiation and increased genomic instability 13. The chromodomain of Tip60 therefore functions as the sensor for activation of Tip60 by DNA damage, indicating that the recruitment of Tip60 to DSBs leads to interactions between Tip60's chromodomain and methylated lysine residues on histones, and that this interaction activates Tip60's HAT activity.
Although these results demonstrate a key role for acetylation of the PRD domain of ATM by Tip60 in activating ATM's kinase activity, it is critical to incorporate the published data on the role of the MRN complex into this model. As discussed earlier, there is significant data, both from cell based 52-55 and biochemical systems 47, 48, 57, that the MRN complex makes a crucial contribution to activating ATM's kinase activity. Tip60 is stably associated with ATM in cells, so that ATM and Tip60 are recruited together to DSBs 16, 58. In a recent paper 13, it was demonstrated that when the MRN complex was inactivated, the acetylation and the activation of ATM by Tip60 were defective. Further, loss of functional MRN delayed both the recruitment of Tip60 to DSBs and significantly reduced the activation of Tip60's acetyltransferase activity by DNA damage 13. These results are consistent with a model (figure 2) in which the inactive ATM-Tip60 complex is recruited to MRN at DSBs. This recruitment of ATM-Tip60 to DSBs then allows the chromodomain of Tip60 to interact with nearby histone H3 which is trimethylated on lysine 9 (H3K9me3), activating Tip60's acetyltransferase activity through allosteric regulation of the acetyltransferase domain. Tip60 then acetylates ATM, which in turn activates ATM's kinase activity. However, a key area which remains to be addressed are the relative contributions of the MRN complex and Tip60 to ATM activation. MRN could simply serve to recruit and concentrate the inactive ATM-Tip60 complex at DSBs, and therefore stabilize the interaction between Tip60 and H3K9me3 at DSBs. This would lead to activation of Tip60's acetyltransferase activity and acetylation and activation of ATM kinase activity. However, purified MRN can activate ATM in an in vitro biochemical system which appears to lack Tip60 48, 57, indicating that MRN plays an active role in upregulating ATM kinase activity. For example, interaction between MRN and ATM may alter the structure of the ATM dimer, activating ATM's kinase activity and increasing autophosphorylation of ATM. This, in turn, would lead to conversion of inactive ATM dimers to active ATM monomers. Acetylation of the PRD domain of ATM by Tip60 would then serve to “fix” ATM in this active conformation, allowing ATM to maintain kinase activity even after dissociation from MRN. In purified systems, MRN may still activate ATM, but the absence of the normal mechanism for ATM inactivation (such as phosphatases and histone deacetylases: 84) may reduce or eliminate the normal in vivo requirement for acetylation of ATM by Tip60. The requirement for both MRN and Tip60 for ATM activation in vivo, as well as the potential contributions of other proteins 85, will give continuing insights into the regulation of this critical enzyme.
Figure 2. A mechanism for ATM activation.
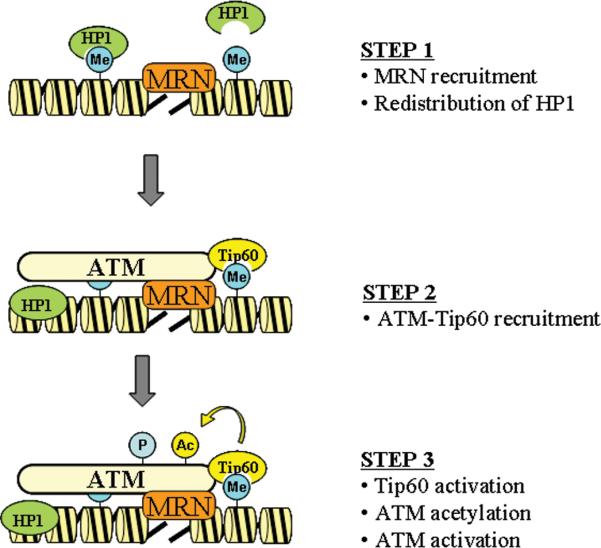
STEP 1: Following DSB production, MRN is recruited to DSB. In parallel, HP1 proteins are released from H3K9me3, and either retained on the chromatin, or released to the nucleoplasm by a process involving phosphorylation of HP1 by the CK2 kinase 91. It is not know if MRN participates in this process. STEP 2: Inactive ATM-Tip60 complex is recruited to the DSB by MRN, facilitating interactions between Tip60's chromodomain and H3K9me3. STEP 3: Interaction between MRN and ATM, in combination with acetylation of ATM by Tip60, activates ATM's kinase activity. Me = methylation, Ac = acetylation, P = phosphorylation.
Tip60 activation
The ability of Tip60 to utilize H3K9me3 for activation at DSBs raises several intriguing questions about the role of H3K9me3 in the DNA damage response. The first question relates to how Tip60 can locate available H3K9me3 on the chromatin adjacent to a DSB (figure 2). One possibility is that the chromatin undergoes de novo methylation of H3K9 on chromatin domains surrounding DSBs, implying recruitment of H3K9me3 methyltransferases (such as suv39h1 and suv39h2 83) to the DSBs. Since ATM and Tip60 activation is essentially maximal within minutes after DNA damage 13, 46, this would require an exceptionally rapid signaling system to recruit H3K9 methyltransferases and then methylate H3 on lysine 9 at DSBs. In fact, published studies indicate that the global levels of H3K9me3 are not significantly altered after DNA damage 13, 86, implying that methylation of H3K9 is not regulated by DNA damage. However, detecting small changes in H3K9 methylation localized to the chromatin at DSBs (which may involve < 1% of the total chromatin) against the normal background of H3K9me3 will likely require more sensitive techniques than e.g. western blot analysis to detect small changes in H3K9me3 at specific chromosomal sites. More sophisticated techniques, such as ChIP based assays, will be required to address this issue.
An alternative mechanism for Tip60 activation at DSBs could involve utilization of pre-existing H3K9me3 for activation of Tip60. This would require that H3K9me3 be evenly distributed across the entire chromatin to ensure correct Tip60 activation. However, H3K9me3 is predominantly located in the compacted, gene poor heterochromatic regions of the chromatin 70, 71, 87, where it is associated with members of the HP1 protein family 88. This suggests that Tip60 may be preferentially activated by DSBs generated within heterochromatin, which would be consistent with reports that the repair of DSBs in heterochromatin requires ATM 89. However, H3K9me3 is also located in non-heterochromatic regions 90, indicating that Tip60 function is unlikely to be entirely restricted to heterochromatic regions during DNA repair. However, it is unlikely that H3K9me3 is evenly distributed across the chromatin, and there are likely to be large chromatin domains lacking H3K9me3. Although it is possible that these regions are transiently methylated during the DNA damage response, a clearer understanding of the distribution of H3K9me3 across the entire chromatin is needed to fully understand how this modification regulates Tip60 activity after DSB production.
A further item is that the majority of the cellular H3K9me3 is bound by the HP1 family of proteins 81, 82, potentially making H3K9me3 unavailable for interaction with Tip60 (figure 2). Recent work demonstrates that, following DNA damage, the chromodomain of HP1β is phosphorylated by casein kinase 2, and this phosphorylation leads to rapid release of HP1β from H3K9me3 13, 91, followed by re-association at later times 91. This suggests a simple mechanism in which the DNA-damage induced release of HP1β 13, 91 generates domains of H3K9me3 for interaction with Tip60's chromodomain. However, this simple idea is more complex than originally thought. A recent study reported that HP1 proteins are actively recruited to sites of DNA damage, and that this recruitment was independent of their ability to associate with H3K9me3 92, 93. Instead, HP1 proteins were localized to the damaged chromatin via their chromoshadow domain. Further, DSB repair within heterochromatin requires the ATM-dependent phosphorylation of the heterochromatin protein kap-1 89. Kap-1 phosphorylation then promotes relaxation of the heterochromatin and facilitates DSB repair 94. Although these studies agree that HP1 plays an important role in DSB repair, it is unclear exactly how HP1 is mobilized in response to DNA damage. A potential explanation is that phosphorylation of the chromodomain of HP1 initially releases HP1 from H3K9me3 91. However, this pool of HP1 is not released into the nucleoplasm, but is instead retained at DSBs through interaction between the chromoshadow domain of HP1 and the chromatin 92. This would be consistent with the observation that the heterochromatin binding protein kap-1 remains associated with the heterochromatin even after phosphorylation by ATM in response to DNA damage 94. Reorganization of the chromatin, and in particular altering the strength of the interactions between HP1, H3K9me3 and kap-1 may therefore be important for introducing both flexibility into the chromatin and to increase access of Tip60 to previously buried H3K9me3 sites.
Conclusion and implications
The recruitment of the ATM-Tip60 complex to DSBs leads to interaction with H3K9me3 on the chromatin. This interaction activates Tip60 acetyltransferase activity, leading to the acetylation and activation of the ATM kinase. Tip60 is therefore activated by a novel mechanism involving direct interaction between methylated histones and Tip60's chromodomain, and indicates that chromatin structure plays a critical role in DSB repair. The tumor suppressor functions of Tip60 are therefore mediated by interactions between Tip60's chromodomain and methylated histones at DSBs. This implies that alterations in global histone methylation patterns would affect Tip60 activity and therefore its ability to regulate DNA repair. In fact, studies have demonstrated aberrant histone methylation patterns in breast and other cancers 95. In particular, altered patterns of H3K9 methylation have been reported in both gastric adenocarcinoma 96, and medullablastoma 97. Further, genetic inactivation of histone methyltransferases, including Dot1, Suv4-20h and Suv39h1/h2, leads to aberrant histone methylation, genomic instability, checkpoint activation and aberrant DNA repair 83, 98-101. These results imply that altered histone methylation patterns, including changes in H3K9me3 levels, may contribute to the etiology and progression of cancer through influencing the activation of the ATM-Tip60 pathway in response to DNA damage. DNA damage occurring in regions of low/absent H3K9me3 may be repaired with lower efficiency than in regions of high H3K9me3 density. Further, changes in the distribution and density of H3K9me3 across the chromatin during tumor progression may significantly impact genomic stability and influence the sensitivity of tumors to chemo- and radio-therapy. Epigenetic therapies aimed at inhibiting H3K9 methyltransferases (to decrease H3K9me3) or H3K9me3 demethylases (to increase H3K9 methylation) may therefore be effective at sensitizing tumor cells to radiation therapy and/or protecting normal tissue from radiation damage.
Acknowledgements
We thank A. D'Andrea and members of the Price laboratory for critical discussions and reading of the manuscript. This work was supported by grants from the NCI (CA64585 and CA93602) and the DOD Breast Cancer Program to BDP, and by an NCI training grant to YS (T32 CA09078).
Abbreviations
- A-T
ataxia telangiectasia
- H3K9me3
histone H3 trimethylated on lysine 9
- HP1α
heterochromatin protein 1a
- KDM4A
lysine demethylase 4A
- MRN
mre11-rad50-nbs1 complex
- PIK
PI 3-kinase
- PRD
PIK Regulatory Domain
References
- 1.Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006 doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Fazzio TG, Huff JT, Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162–74. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrozza MJ, Utley RT, Workman JL, Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19:321–9. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- 4.Marmorstein R. Structure of histone acetyltransferases. J Mol Biol. 2001;311:433–44. doi: 10.1006/jmbi.2001.4859. [DOI] [PubMed] [Google Scholar]
- 5.Kimura A, Horikoshi M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells. 1998;3:789–800. doi: 10.1046/j.1365-2443.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- 6.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates Iii JR, et al. Acetylation by Tip60 Is Required for Selective Histone Variant Exchange at DNA Lesions. Science. 2004 doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 7.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 8.Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–96. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem. 2002;277:25904–13. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 10.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–39. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–51. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, et al. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–84. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009 doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. 2007;27:8502–9. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y, Jiang X, Chen S, Price BD. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006;580:4353–6. doi: 10.1016/j.febslet.2006.06.092. [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–7. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 18.Hu Y, Fisher JB, Koprowski S, McAllister D, Kim MS, Lough J. Homozygous disruption of the Tip60 gene causes early embryonic lethality. Dev Dyn. 2009;238:2912–21. doi: 10.1002/dvdy.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–73. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 21.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 22.Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 2005;569:123–32. doi: 10.1016/j.mrfmmm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 23.ME LL, Vidal F, Gallardo D, Diaz-Fuertes M, Rojo F, Cuatrecasas M, et al. New p53 related genes in human tumors: significant downregulation in colon and lung carcinomas. Oncol Rep. 2006;16:603–8. doi: 10.3892/or.16.3.603. [DOI] [PubMed] [Google Scholar]
- 24.Halkidou K, Gnanapragasam VJ, Mehta PB, Logan IR, Brady ME, Cook S, et al. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene. 2003;22:2466–77. doi: 10.1038/sj.onc.1206342. [DOI] [PubMed] [Google Scholar]
- 25.Mattera L, Escaffit F, Pillaire MJ, Selves J, Tyteca S, Hoffmann JS, et al. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene. 2009;28:1506–17. doi: 10.1038/onc.2008.499. [DOI] [PubMed] [Google Scholar]
- 26.Sakuraba K, Yasuda T, Sakata M, Kitamura YH, Shirahata A, Goto T, et al. Down-regulation of Tip60 gene as a potential marker for the malignancy of colorectal cancer. Anticancer Res. 2009;29:3953–5. [PubMed] [Google Scholar]
- 27.Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:1063–7. doi: 10.1038/nature06055. [DOI] [PubMed] [Google Scholar]
- 28.Doyon Y, Cote J. The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev. 2004;14:147–54. doi: 10.1016/j.gde.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, et al. Binding of Chromatin-Modifying Activities to Phosphorylated Histone H2A at DNA Damage Sites. Mol Cell. 2004;16:979–90. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol. 2007;27:7028–40. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jha S, Shibata E, Dutta A. Human Rvb1/Tip49 is required for the histone acetyltransferase activity of Tip60/NuA4 and for the downregulation of phosphorylation on H2AX after DNA damage. Mol Cell Biol. 2008;28:2690–700. doi: 10.1128/MCB.01983-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–17. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Altaf M, Saksouk N, Cote J. Histone modifications in response to DNA damage. Mutat Res. 2007;618:81–90. doi: 10.1016/j.mrfmmm.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 34.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 35.Meyn MS. Ataxia-telangiectasia, cancer and the pathobiology of the ATM gene. Clin Genet. 1999;55:289–304. doi: 10.1034/j.1399-0004.1999.550501.x. [DOI] [PubMed] [Google Scholar]
- 36.Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. Embo J. 2009;28:3067–73. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 38.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 41.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 42.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. Embo J. 2006;25:3504–14. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 47.Dupre A, Boyer-Chatenet L, Gautier J. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat Struct Mol Biol. 2006;13:451–7. doi: 10.1038/nsmb1090. [DOI] [PubMed] [Google Scholar]
- 48.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 49.Daniel JA, Pellegrini M, Lee JH, Paull TT, Feigenbaum L, Nussenzweig A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J Cell Biol. 2008;183:777–83. doi: 10.1083/jcb.200805154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–5. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- 51.Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–8. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- 52.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003;22:5612–21. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina San Martin B, Van Laethem F, et al. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol. 2005;7:675–85. doi: 10.1038/ncb1270. [DOI] [PubMed] [Google Scholar]
- 54.Cerosaletti K, Wright J, Concannon P. Active role for nibrin in the kinetics of atm activation. Mol Cell Biol. 2006;26:1691–9. doi: 10.1128/MCB.26.5.1691-1699.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 56.You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–79. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–6. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 58.Jiang X, Sun Y, Chen S, Roy K, Price BD. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem. 2006;281:15741–6. doi: 10.1074/jbc.M513172200. [DOI] [PubMed] [Google Scholar]
- 59.Kim YC, Gerlitz G, Furusawa T, Catez F, Nussenzweig A, Oh KS, et al. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat Cell Biol. 2009;11:92–6. doi: 10.1038/ncb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Balajee AS, Su T, Cen B, Hei TK, Weinstein IB. The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage. J Cell Biol. 2008;183:253–65. doi: 10.1083/jcb.200711150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eymin B, Claverie P, Salon C, Leduc C, Col E, Brambilla E, et al. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol. 2006;26:4339–50. doi: 10.1128/MCB.02240-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi T, Hara K, Inoue H, Kawa Y, Tokunaga C, Hidayat S, et al. Carboxyl-terminal region conserved among phosphoinositide-kinase-related kinases is indispensable for mTOR function in vivo and in vitro. Genes Cells. 2000;5:765–75. doi: 10.1046/j.1365-2443.2000.00365.x. [DOI] [PubMed] [Google Scholar]
- 63.Beamish HJ, Jessberger R, Riballo E, Priestley A, Blunt T, Kysela B, et al. The C-terminal conserved domain of DNA-PKcs, missing in the SCID mouse, is required for kinase activity. Nucleic Acids Res. 2000;28:1506–13. doi: 10.1093/nar/28.7.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Priestley A, Beamish HJ, Gell D, Amatucci AG, Muhlmann-Diaz MC, Singleton BK, et al. Molecular and biochemical characterisation of DNA-dependent protein kinase-defective rodent mutant irs-20. Nucleic Acids Res. 1998;26:1965–73. doi: 10.1093/nar/26.8.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008;22:1478–89. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mordes DA, Cortez D. Activation of ATR and related PIKKs. Cell Cycle. 2008;7:2809–12. doi: 10.4161/cc.7.18.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–40. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–81. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 69.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 70.Regha K, Sloane MA, Huang R, Pauler FM, Warczok KE, Melikant B, et al. Active and repressive chromatin are interspersed without spreading in an imprinted gene cluster in the mammalian genome. Mol Cell. 2007;27:353–66. doi: 10.1016/j.molcel.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 72.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 73.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–16. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 76.Trievel RC. Structure and function of histone methyltransferases. Crit Rev Eukaryot Gene Expr. 2004;14:147–69. doi: 10.1615/critreveukaryotgeneexpr.v14.i3.10. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 78.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–81. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 79.Bienz M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem Sci. 2006;31:35–40. doi: 10.1016/j.tibs.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 80.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–7. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 82.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–3. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 83.Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–37. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 84.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, et al. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. Embo J. 2004;23:4451–61. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanu N, Behrens A. ATMINistrating ATM signalling: regulation of ATM by ATMIN. Cell Cycle. 2008;7:3483–6. doi: 10.4161/cc.7.22.7044. [DOI] [PubMed] [Google Scholar]
- 86.Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. Embo J. 2009;28:1878–89. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peng JC, Karpen GH. Epigenetic regulation of heterochromatic DNA stability. Curr Opin Genet Dev. 2008;18:204–11. doi: 10.1016/j.gde.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–5. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- 89.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 90.Vakoc CR, Sachdeva MM, Wang H, Blobel GA. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol. 2006;26:9185–95. doi: 10.1128/MCB.01529-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–6. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 92.Luijsterburg MS, Dinant C, Lans H, Stap J, Wiernasz E, Lagerwerf S, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185:577–86. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ball AR, Jr., Yokomori K. Revisiting the role of heterochromatin protein 1 in DNA repair. J Cell Biol. 2009;185:573–5. doi: 10.1083/jcb.200904033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 95.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 96.Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol. 2008;15:1968–76. doi: 10.1245/s10434-008-9927-9. [DOI] [PubMed] [Google Scholar]
- 97.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–72. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Giannattasio M, Lazzaro F, Plevani P, Muzi-Falconi M. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem. 2005;280:9879–86. doi: 10.1074/jbc.M414453200. [DOI] [PubMed] [Google Scholar]
- 99.Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25:8430–43. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tardat M, Murr R, Herceg Z, Sardet C, Julien E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol. 2007;179:1413–26. doi: 10.1083/jcb.200706179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sakaguchi A, Steward R. Aberrant monomethylation of histone H4 lysine 20 activates the DNA damage checkpoint in Drosophila melanogaster. J Cell Biol. 2007;176:155–62. doi: 10.1083/jcb.200607178. [DOI] [PMC free article] [PubMed] [Google Scholar]