

Abstract
The Human Protein Atlas is a rich source of location proteomics data. In this work, we present an automated approach for processing and classifying major subcellular patterns in the Atlas images. We demonstrate that two different classification frameworks (support vector machine and random forest) are effective at determining subcellular locations; we can analyze over 3500 Atlas images with a high degree of accuracy, up to 87.5% for all of the samples and 98.5% when only considering samples in whose classification assignments we are most confident. Moreover, the features obtained in both of these frameworks are observed to be highly consistent and generalizable. Additionally, we observe that the features relating the proteins to cell markers are especially important in automated learning approaches.
Index Terms: Image classification, microscopy, location proteomics, machine learning, feature selection
1. Introduction
The Human Protein Atlas is a rich source of location proteomic data [1]. As it grows in size, automated tools are needed to annotate and characterize the many proteins and their conditions across cell types. Supervised learning methods, in which classifiers are trained to recognize different protein patterns, have been proven effective at analyzing subcellular patterns [2]. More recent work shows that classification can be scaled to analyze patterns across a proteome [3].
One feature of the Human Protein Atlas is that it contains proteins that have been imaged in various different cell lines and tissues. Recently, the Atlas has been significantly augmented by the addition of confocal microscope images for many antibodies [4]. In this work, we seek to extend subcellular pattern recognition across different cell types via development and evaluation of new features.
2. Methods
2.1. Image Collection
The HPA confocal images were analyzed in the form of uncompressed, 8-bit TIFFs, with each file a single data channel, and four such files comprising a single image field. The channels are: protein, nucleus, microtubules, and endoplasmic reticulum (ER). The protein channel was obtained by immunofluorescence labeling with monospecific antibodies, while the other channels were acquired using standard stains [5]. Up to two image fields were taken for each protein in three different cell lines, A-431, U-251MG, and U-2 OS, using a confocal microscope. 1902 proteins were imaged.
After images were acquired, they were visually inspected for the amount of staining as well as the patterns depicted in the images. Based on this, we ignored images with low levels of staining. For classification analysis, we selected proteins that localized specifically major subcellular locations.
2.2. Cell Segmentation
We sought to analyze protein patterns in single cell regions, requiring the segmentation of images into single cell regions. Because we had nuclear and whole cell references- in the form of the DAPI and ER stains- we used seeded watershed for segmentation, based on earlier work [6]. Our seeding procedure specifically involved thresholding the nuclear image; the threshold was determined as the intensity of the most common pixel in the image. Next, the binary nuclear image was eroded and very small objects were removed. Then, objects whose areas were outside an acceptable range were labeled as erroneous seeds. Finally, the ER channel was used to determine background seeds; large areas in the ER image that were filled with pixels with zero intensity were set as background seeds. The seeds, along with an inverted ER image, were then used in the seeded watershed algorithm, and resulting regions corresponding to background or erroneous seeds were removed.
2.3. Feature Extraction
We extracted various types of features from both multicell, whole images and segmented cell regions. Haralick texture [7], edge [8], threshold adjacency statistics [9], non-object fluorescence [10], and object features [8] were chosen as they have been used in subcellular pattern recognition in the past. Additionally, we calculated overlap, mutual information, and correlation features between the protein and three reference channels, as such features were thought to be useful in distinguishing between the nuclear, microtubule, and ER locations. Additionally, skeleton features [10] and object features in relation to the three references were calculated on the single cell regions.
2.4. Support Vector Machine Classification
Support vector machine (SVM) classification with a radial basis function kernel was evaluated using N-fold cross-validation. For each fold, stepwise discriminant analysis (SDA) was performed in order to select informative features. SVM parameters g, the kernel parameter, and C, the slack penalty, were set to 64 and 0.25. A five-fold cross validation routine was used to determine the top K SDA ranked features which gave the best classification on the training fold. To account for the unbalanced class membership across multiple classes, class weighting was used. Classification was implemented using the LIBSVM toolbox (http://www.csie.ntu.edu.tw/cjlin/libsvm/).
Since the classifiers output the probabilities that each sample belongs to a class, we boosted classification accuracy for the segmented data by summing class probabilities for all samples that originated from the same image field, and then assigning all of these samples the label corresponding to the resulting maximum value.
2.5. Random Forest Classification
As a alternative approach to classification we used a random forest (RF) classifier, which makes its decision over an aggregate of several classification trees [11]. We used 500 trees and on each tree used 11 features at each decision node. RF is a bootstrapping method, which allowed us to evaluate the classifier on each of the image samples. Moreover, since the variables selected for optimal partitioning over class-labels can be examined from a variable importance plot which indicates which variables are most discriminatory between various classes [11], we used RF as a feature selection approach as well. The output of this classification system is thus a classifier, series of discriminative features, and– like SVM– probabilities that each sample belongs to each class. Classification was implemented using the version 4.5-30 of the randomForest package (http://cran.r-project.org/web/packages/randomForest/index.html).
2.6. Software
All image processing and analysis was performed in Matlab 7.4 with the exception of random forest, which was run in R.
3. Results
3.1. Evaluation of Support Vector Machine Classification
We chose images that depict one of nine specific patterns (an example of a pattern is shown in Figure 1.A). These pattern classes are: centrosome, cytoskeleton, ER, Golgi apparatus, punctate patterns- which includes lysosomes, peroxisomes, endosomes-, mitochondria, nucleoli, nucleus, and plasma membrane (PM), and 834 proteins out of 1902 showed just one of these patterns in this dataset. Each class has at least 10 proteins, and the total number of image fields in this resulting dataset is 3557 samples. Images from the three different cell lines are considered.
Fig. 1.
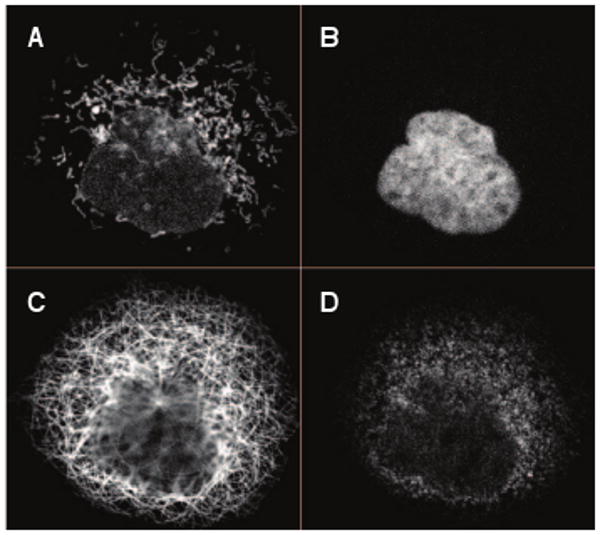
Example of a segmented single cell region. A protein (Atlas ID 1915) exhibiting a mitochondrial pattern (A), the parallel nuclear (B), microtubule (C), and endoplasmic reticulum channels (D). Unprocessed image fields consist of multiple cells.
We first classified images using field features with 10-fold cross validation. Classification accuracies range from 30.0–96.3% accuracy between the classes, with an overall accuracy of 84.8% (Table 1, column 4). Classes with fewer samples have lower accuracies. 19.4% of the nucleolar samples and 20.0% of the centrosome samples were confused with the nuclear class (data not shown), and the centrosome class was also highly confused with the Golgi class. The cytoskeletal, ER, Golgi, and PM classes each had more than 10% of their samples confused with the mitochondria class (data not shown).
Table 1.
Comparison of classification approaches.
| Classes | #field img. | #cell img. | SVM field | SVM cell | SVM voting | RF field | RF cell | RF voting |
|---|---|---|---|---|---|---|---|---|
| Centrosomes | 40 | 289 | 30.0 | 11.4 | 7.5 | 45.0 | 29.4 | 40.0 |
| Cytoskeleton | 260 | 2035 | 61.2 | 58.2 | 67.8 | 67.3 | 66.5 | 70.3 |
| ER | 226 | 1818 | 77.9 | 71.6 | 81.3 | 81.9 | 78.9 | 85.5 |
| Golgi apparatus | 218 | 1800 | 66.1 | 57.8 | 74.5 | 68.3 | 66.4 | 75.5 |
| Punctate patterns | 180 | 1497 | 76.1 | 60.3 | 73.1 | 70.0 | 68.6 | 72.6 |
| Mitochondria | 612 | 4905 | 86.4 | 80.0 | 91.6 | 91.2 | 87.4 | 96.2 |
| Nucleoli | 247 | 2114 | 70.9 | 65.4 | 72.1 | 76.9 | 74.1 | 81.2 |
| Nucleus | 1720 | 14183 | 96.3 | 95.7 | 98.3 | 98.0 | 98.0 | 99.2 |
| Plasma membrane | 54 | 458 | 53.7 | 27.3 | 28.8 | 46.3 | 46.5 | 40.0 |
| Overall accuracy | — | — | 84.8 | 80.6 | 87.4 | 87.5 | 85.8 | 89.8 |
We next applied classification at the single cell level. Since each image contains multiple cells, we first segmented the images into single cell regions. This increased the number of samples to 29099 regions. Classification of these samples using 5-fold cross validation yielded an overall accuracy of 80.6%, and class accuracies ranged from 11.4–95.7% (Table 1, column 5). Using the classifier probability outputs to choose a single, maximum probability label for all cells belonging to the same image, we found that we could boost classification accuracy to 87.4%. In doing this, all class accuracies save that of the centrosome class improved over the single cell classifier (Table 1, column 6). Moreover, classification accuracy improved over the simple field level analysis for seven of the nine location classes.
We performed precision-recall analysis on these latter results. We sorted the labeled samples by the magnitude of the maximum probability value for each sample. Generally, as only more confident assignments are considered, classification accuracy increases (Figure 2). At a recall of 60%, the classification accuracy is 98.5%.
Fig. 2.
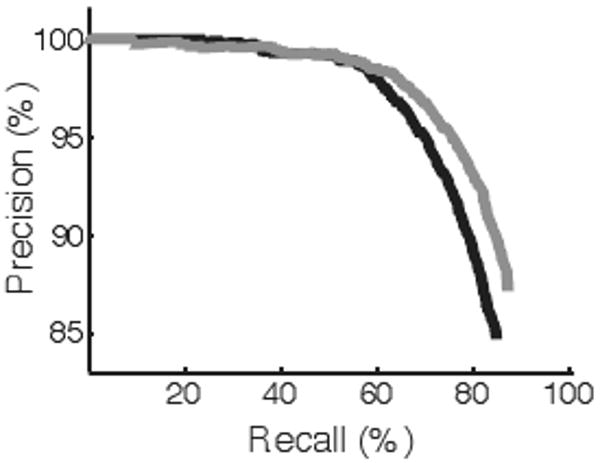
Precision-recall curve for cell labels with voting across image field. The black profile denotes SVM, while gray shows RF performance. At a recall of 60%, the precision for both approaches is 98.5%.
3.2. Evaluation of Random Forest Classification
We next applied the RF classification framework to analyze the field and cell level images. The overall accuracy using field features was 87.5%, with class accuracies ranging from 45.0-98.0% (Table 1, column 7). Compared to SVM using field features, RF performs better on seven of the nine classes. At the cell level, RF performs better than SVM on all classes and achieved an 85.8% accuracy (Table 1, column 8). Finally, RF with voting performs better than SVM with voting in overall accuracy and in eight of the nine classes (Table 1, column 9). As more confident assignments are considered using field features, classification accuracy increases (Figure 2). At a recall of 60%, accuracy is 98.5%.
3.3. Comparison of Feature Selection Methods
SDA has proved to be an effective method for feature selection in subcellular pattern recognition [12]. One drawback to SDA, however, is that it is highly sensitive to training samples; the addition or subtraction of a few samples can affect the ranking and selection of features. RF is less sensitive to this issue. We compared the features selected by RF to features selected by SDA. Both RF and SDA were applied to all of the field level data. RF identifies 16 features as especially discriminative, while SDA returns 107 features. All of the 16 RF features appear in the top 64 ranked SDA features, and the top ranked features in both selection methods match (Table 2). Of these 16 features, nine are related to the nucleus, ER, or tubulin reference channels.
Table 2.
Comparison of feature selection methods on field level features. The first two columns show rankings by different selection methods. RF returned 16 features while SDA with SVM classifier tuning returned 107 features.
| RF | SDA | Feature name |
|---|---|---|
| 1 | 1 | Corr. between prot. and nuc. channels |
| 2 | 2 | Corr. between prot. and ER channels |
| 3 | 64 | Prot. and ER obj. overlap int. ratio |
| 4 | 30 | Threshold adjacency statistic #13 |
| 5 | 29 | Prot. and nuc. obj. int. overlap ratio |
| 6 | 11 | Threshold adjacency statistic #12 |
| 7 | 7 | Prot. and nuc. obj. area overlap ratio |
| 8 | 10 | Prot. and tub. obj. int. overlap ratio |
| 9 | 4 | Non-object fluorescence |
| 10 | 3 | Texture #16: corr. (4× downsampling) |
| 11 | 37 | Variance of # of pixels per object |
| 12 | 35 | Texture #3: corr. (4× downsampling) |
| 13 | 5 | Corr. between prot. and tub. channels |
| 14 | 16 | Info. between prot. and nuc. channels |
| 15 | 12 | Info, between prot. and ER channels |
| 16 | 28 | Ratio of largest to smallest obj. size |
4. Discussion
We have proposed two different approaches for the location classification of Human Protein Atlas immunofluorescence images. Both work with accuracies greater than 80% for over 800 proteins across three different cell lines. Moreover, the images exhibited differing levels of staining (visually assessed at the time of image collection). Taken together, these indicate that our results are a promising approach to analyzing subcellular patterns at a large scale.
Our results indicate that the RF approach generally performs better than the SVM classification with SDA for feature selection. However, in our current implementation with SVM we have shown that the SVM can achieve high accuracies when only confident images are considered.
Moreover, a simple comparison between the feature selection methods shows that they are both finding similar features. Both methods show that the features related to reference channels are very important in classification. This highlights the benefit of acquiring multiple data channels during proteomic studies.
We have analyzed over 3000 images in the Atlas. However, there are over 4000 more images that exhibit mixed patterns. As we have shown our features to be informative in identifying nine major subcellular patterns, we can now turn to unsupervised learning approaches to analyze the rest of the Atlas using these selected features.
Acknowledgments
The research described here was supported in part by NSF grant EF-0331657 and NIH grant GM075205. A.R. was supported by a postdoctoral fellowship from the Lane Fellows program.
References
- 1.Human Proteome Resource Consortium. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4(no. 12):1920–1932. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Glory E, Murphy RF. Automated subcellular location determination and high-throughput microscopy. Dev Cell. 2007;12(no. 1):7–16. doi: 10.1016/j.devcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Chen SC, Zhao T, Gordon GJ, Murphy RF. Automated image analysis of protein localization in budding yeast. Bioinformatics. 2007;23(no. 13):i66–71. doi: 10.1093/bioinformatics/btm206. [DOI] [PubMed] [Google Scholar]
- 4.Barbe L, Lundberg E, Oksvold P, Stenius A, Lewin E, Bjorling E, Asplund A, Ponten F, Brismar H, Uhlen M, Andersson-Svahn H. Toward a confocal subcellular atlas of the human proteome. Mol Cell Proteomics. 2008;7(no. 3):499–508. doi: 10.1074/mcp.M700325-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Lundberg E, Sundberg M, Graslund T, Uhlen M, Svahn H Andersson. A novel method for reproducible fluorescent labeling of small amounts of antibodies on solid phase. J Immunol Methods. 2007;322(no. 1-2):40–49. doi: 10.1016/j.jim.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 6.Velliste M, Murphy RF. Automated determination of protein subcellular locations from 3D fluorescence microscope images. Proc IEEE Int Symp Biomed Imaging; Washington, DC. 2002. pp. 867–870. [Google Scholar]
- 7.Boland MV, Markey MK, Murphy RF. Automated recognition of patterns characteristic of subcellular structures in fluorescence microscopy images. Cytometry. 1998;33(no. 3):366–375. [PubMed] [Google Scholar]
- 8.Boland MV, Murphy RF. A neural network classifier capable of recognizing the patterns of all major subcellular structures in fluorescence microscope images of hela cells. Bioinformatics. 2001;17(no. 12):1213–1223. doi: 10.1093/bioinformatics/17.12.1213. [DOI] [PubMed] [Google Scholar]
- 9.Hamilton NA, Pantelic RS, Hanson K, Teasdale RD. Fast automated cell phenotype image classification. BMC Bioinformatics. 2007;8:110. doi: 10.1186/1471-2105-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy RF, Velliste M, Porreca G. Robust numerical features for description and classification of subcellular location patterns in fluorescence microscope images. J VLSI Signal Process Syst. 2003;35(no. 3):311–321. [Google Scholar]
- 11.Breiman L. Random forests. Machine Learning. 2001;45(no. 1):5–32. 10. [Google Scholar]
- 12.Huang K, Velliste M, Murphy RF. Feature reduction for improved recognition of subcellular location patterns in fluorescence microscope images. Proc SPIE. 2003;4962:307–318. [Google Scholar]