

Abstract
We investigated the role of inducible NOS (iNOS) on cardiac function during the development of left ventricular hypertrophy. Hypertrophy was induced by pressure-overload via short-term (2.5 months) or long-term (6.5 months) aortic banding (AoB) in wild-type (WT) and iNOS knock out (iNOSKO) mice. Cardiac function was then assessed via echocardiography, in situ hemodynamics and papillary muscle force measurements. Quantitative RT-PCR and Western blots were used to measure expression of hypertrophic gene markers and proteins respectively. Our data demonstrate that increased afterload via AoB leads to increased expression of iNOS that is associated with cardiac dysfunction. In pressure-overload induced hypertrophy, iNOSKO delays both the expression of hypertrophic markers and contractile dysfunction without causing significant changes in the level of hypertrophy. Moreover, after long-term AoB, iNOSKO animals exhibited increased basal cardiac function and an improved response to beta-adrenergic stimulation compared to long-term AoB WT animals. In conclusion, our data demonstrate that NO production via iNOS plays an important role in modulating cardiac function after moderate AoB that mimics long-term hypertension in humans.
Keywords: Nitric Oxide, Aortic Banding, Beta-Adrenergic Stimulation, Hypertrophy
2. INTRODUCTION
The role of nitric oxide (NO) in the regulation/modulation of cardiac performance has been extensively studied since the early 1990’s (for review see Kelly et al (1). Two isoforms of NO synthase (NOS), endothelial NOS (eNOS) and neuronal NOS (nNOS), are constitutively expressed in the heart. The third isoform, inducible NOS (iNOS), is expressed under pathological conditions such as sepsis, hypertrophy or heart failure (1).
Studies performed on the molecular, cellular and whole muscle levels including in situ experiments, suggest that NO influences not only cardiac contractility, but also lusitropy, chronotropy and energetics (2-7). NO can act indirectly via the cGMP pathway (1) and directly via binding to cysteine sulfhydryls or iron groups in proteins (8). In the cell, the effects of endogenous NO are restricted to proteins localized in close vicinity to the associated NOS isoform. For example, in cardiomyocytes eNOS is found in the caveolae to associate with caveolin-3 (9,10) where it attenuates beta-adrenergic stimulation. nNOS is associated with the sarcoplasmic reticulum (SR) membranes where it modulates SR Ca2+ uptake and release (9,11). Unlike these constitutively expressed isoforms, iNOS is more ubiquitously expressed and its expression only increases following induction by events such as hypertrophy or heart failure. In such cases, iNOS appears to contribute to myocardial dysfunction and alters the myocardial response to beta-adrenergic stimulation (4,12,13). However, since NO can be produced by three different isoforms of NOS within the myocardium, it has been difficult to separate their specific effects in physiological and pathological conditions without genetic manipulations.
Recently however, different mouse models have been generated in which one or more isoforms of NOS have been knocked out or over-expressed allowing for the specific, independent effects of the NOS isoforms to be studied on cardiac function (14). In this study, we investigated the role of NO produced by iNOS in the development of left ventricular (LV) hypertrophy induced by short-and long-term pressure overload. Our results demonstrate that pressure-overload induces iNOS expression is associated with the development of hypertrophy and cardiac dysfunction. Conversely, ablation of iNOS delays hypertrophic gene expression, increases basal cardiac function and yields a greater response to beta-adrenergic stimulation than WT pressure-overloaded mice displaying similar levels of hypertrophy.
3. MATERIALS AND METHODS
3.1. Experimental animals
Inducible NOS knockout (iNOSKO) (15) and C57BL/6J (WT) male mice were purchased from Jackson Laboratories (Bar Harbor, ME). Experiments were conducted in compliance with animal care policies of the Animal Care Committee of the University of Illinois at Chicago. As a positive control for iNOS expression, WT mice were injected (I.P.) with 2.5 mg/kg of lipopolyssacharides (LPS) and sacrificed 2 days after injection. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
3.2. Induction of cardiac hypertrophy
Twelve week-old iNOSKO and WT mice were subjected to aortic constriction (AoB) to create pressure-overload as previously described (16,17) with the following modifications. The transverse thoracic aorta was banded using a 4-0 silk suture tied around a 25-gauge curved needle constructing the aorta to approximately 60% of its original lumen diameter. The iNOSKO- and WT-banded mice (iNOSKO-AoB and WT-AoB) were subjected to experimental measurements at 2.5 months (short-term AoB) or 6.5 months (long-term AoB) after surgery. Non-banded age-matched mice (WT and iNOSKO) were used as controls since we found no difference between sham-operated and age-matched control animals.
3.3. Echocardiographic measurements
At 12 weeks of age prior to AoB WT and iNOSKO mice were subjected to baseline echocardiographic measurements as previously described (18,19). Mice were randomly divided to AoB group and non-banded group. AoB groups underwent echocardiography every 4-6 weeks to measure the level of LV hypertrophy and assess LV function. Non-banded age-matched mice (WT and iNOSKO) were used as controls for comparison of echocardiographic measurements.
3.4. Hemodynamic measurements
Long-term AoB and age-matched non-banded mice were subjected to hemodynamic measurements as previously described (19) and pressure-volume loops from the left ventricle were recorded (ARIA Pressure Volume Conductance System; Millar Instruments).
3.5. Left ventricular papillary muscle experiments
LV papillary muscles were isolated and superfused using a modified protocol described by Wolska et al.(20). Muscles were superfused at 22°C with 1 mM Ca2+ and electrically stimulated at 0.2 Hz. After the initial equilibration the muscle was stretched to generate 90% of maximum developed force (DF) and stimulated at 2.0 Hz. After stable contractions were recorded, the muscles were superfused with increasing doses of isoproterenol (ISO) in nM: 2.5, 5.0, 10.0, 100.0 and 1000.0. Some muscles were pre-incubated with 10 microM of an iNOS blocker N-[[3-(aminomethyl)phenyl]methyl]-ethanimidamide, dihydrochloride (1400W) for 40 minutes. During 1400W pre-incubation, muscles were stimulated at 0.2Hz for the first 25 minutes, then at 2.0 Hz for the remaining 15 minutes. The iNOS blocker was present in perfusion solution during the entire ISO protocol. DF was normalized to baseline DF at 2.0 Hz before ISO perfusion. Time to fifty percent of the relaxation (RT50) was expressed in milliseconds (ms).
3.6. Quantitative Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
Gene expression analysis of apex samples from AoB and age-matched non-banded control mice were performed using quantitative RT-PCR with SYBR Green detection in a LightCycler thermocycler (Roche Diagnostics) as previously described (21). Expression of beta-myosin heavy chain (beta-MHC), atrial natriuretic peptide (ANP), SR CaATPase2 (SERCA2) and phospholamban (PLB) were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression.
3.7. Protein expression
Protein expression was quantified by Western blot as previously described (18,22). To assess eNOS phosphorylation levels, heart samples were separated on 8% bis/acrylamide gels and probed initially with anti-phospho-eNOS (Thr 495) antibody (2 microg/ml, Upstate). Thereafter, membranes were stripped (PIERCE, Rockford, IL) and re-probed using anti-eNOS antibody (1:10000, Transduction Laboratories). All blots were visualized with enhanced chemiluminescence. Phosphorylation of eNOS was normalized to total eNOS expression detected on the same membrane. At the end, membranes were stained with Pounceau solution to confirm loading. For quantification of changes in phospho- and total protein, expression was normalized to a WT age-matched sample. To quantify iNOS expression, samples were separated on 8% bis/acrylamide gels and probed with polyclonal anti-iNOS antibodies (1:2000, Cayman, Ann Arbor, MI; 1:500, Santa Cruz, Santa Cruz, CA). Thereafter, membranes were stripped and reprobed with anti alpha-sarcomeric actin antibody (1:10000; Sigma, St. Louis, MO) to correct for loading.
To assess cardiac alpha and beta myosin heavy chain expression mouse ventricular tissue was solubalized by Dounce homogenization in sample buffer (8 M urea, 2 M thiourea, 3% SDS, 75 mM DTT, 0.03% bromophenol blue, and 50 mM Tris-HCl, pH 6.8) and clarified by centrifugation. The resultant supernantant was heated at 100°C for 3 min, vortexed and again clarified by centrifugation. Myosin heavy chains where separated by SDS-PAGE on a 16 × 18 cm 6% (37.5:1) gel cross-linked with DATD cooled to 8°C and run until the dye front ran off the bottom (23). The resultant gel was fixed with 50% methanol and 10% acetic acid, washed with water and resultant proteins visualized by staining with Gel Code Blue Silver (Pierce, Rockford, IL). Following water washes to remove nonspecific background the gel was scanned and myosin heavy chain bands quantified using Image Quant (GE, Pittsburgh, PA). Beta-myosin heavy chain expression is expressed as percentage of total cardiac myosin heavy chain (beta-myosin heavy chain /(alpha-myosin heavy chain + beta-myosin heavy chain)).
3.8. Histology and immunofluorescence
Hearts were removed and retrograde perfused through the aorta with ice cold saline followed by Methacarn (BBC) fixation. Hearts were divided into four equal transverse blocks (5 mm) and immersion-fixed for an additional 30 minutes. Tissue was dehydrated in ethanol, cleared in xylene and embedded in paraffin. Sections (6-7 micrometer) were placed on slides, deparaffinized and rehydrated. For antigen retrieval, sections were then incubated in Retrievagen buffer (Becton Dickinson). For iNOS staining, biotin activity was blocked with an avidin/biotin blocking kit (Vector Labs). Sections were blocked in 5% goat serum and incubated overnight with 1:50 dilution of rabbit polyclonal anti-iNOS antibody (Cayman, Ann Arbor, MI) at 4°C, followed by room temperature incubation with biotinylated goat anti-rabbit IgG and then streptavidin-conjugated Alexa Fluor-594 (Molecular Probes, Carlsbad, CA) diluted 1:1000. Following staining, the slides were treated with Sudan black B to reduce autofluorescence and mounted with Vectashield containing DAPI (Vector Labs).
Images for immunofluorescence were obtained using a Zeiss LSM510 confocal microscope equipped with a 63X water immersion objective. 488 and 568 nm beams from an argon-krypton laser and a 354 nm beam from an argon UV laser were used for excitation and emission. Alexa Fluor-594 and DAPI were detected through an LP585 and BP385-470 filter respectively.
3.9. Data computation and statistical analysis
All results are presented as mean±SEM. The significance of differences between the means was evaluated by one-way ANOVA followed by Student-Newman-Keuls test. To evaluate significance in iNOS expression between short-term and long-term AoB mice, a t-test was used. A value of P<0.05 was the criterion for significance.
4. RESULTS
4.1. Expression and localization of iNOS
The expression of iNOS in WT and iNOSKO mouse hearts in response to AoB was detected by Western blotting (Figure 1) and immunofluorescence (Figure 2). Figure 1 shows that iNOS was expressed only in WT-AoB hearts after long-term AoB (panels A and B) and short-term AoB (panel B). iNOS expression in long-term WT-AoB mice was not different from short-term WT-AoB mice (0.49±0.07 (n= 6) vs. 0.34±0.08 (n= 6); P=0.18) (Figure 1C). However, iNOS expression was not detected in samples from iNOSKO-AoB hearts or in age-matched iNOSKO hearts at any time point. Expression of iNOS in long-term WT-AoB and age-matched WT hearts was confirmed using immunofluorescence (Figure 2 A-B). iNOS expression was completely absent in the iNOSKO-AoB mice (Figure 2C) and only small amounts of iNOS were detected in heart sections from WT animals (Figure 2A). Sections from long-term WT-AoB hearts showed higher expression of iNOS both in the cytoplasm and in the nuclei of cardiomyocytes (Figure 2 B). Hearts from a WT mouse injected with LPS were used as controls (Figure 2D).
Figure 1.
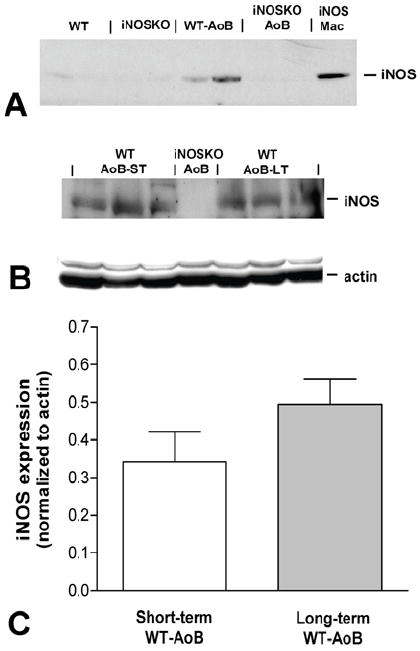
Western blots for iNOS expression in hearts from WT and iNOSKO mice after long-term AoB (panel A) and age-matched controls. Panel B shows the expression of iNOS and actin in short-term (ST) and long-term (LT) WT-AoB mice. A long-term iNOSKO-AoB was used as a negative control. Panel C represents the average iNOS expression normalized to actin expression in short-term (n=6) and long-term (n=6) WT-AoB mice. iNOS; Mac-purified iNOS from activated murine macrophage cells.
Figure 2.
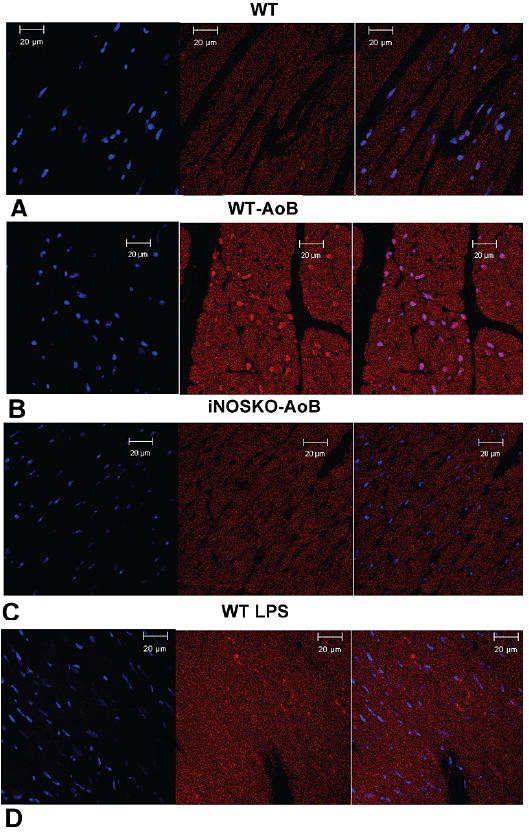
Expression of iNOS in long-term WT-AoB, iNOSKO-AoB, age-matched WT control and LPS injected WT mouse hearts detected by immunofluorescence. Representative transverse sections through A) WT heart, B) WT-AoB heart, C) iNOSKO-AoB heart and D) LPS injected WT heart. iNOS was stained using Alexa Fluor-594 (red) and nuclei were stained using DAPI (blue). The first image from the left presents the nuclei staining, the second image iNOS staining and the third is a superimposed image.
4.2. Effect of AoB on the development of hypertrophy
Table 1 summarizes the echocardiographic parameters of WT and iNOSKO mice subjected to short- and long-term AoB and age-matched non-banded WT and iNOSKO mice. After short-term AoB (11.1±1.2 weeks in WT-AoB and 9.7±1.3 weeks in iNOSKO-AoB), both groups developed similar levels of hypertrophy. LV posterior wall diastolic dimension (LVPWDd) was significantly greater in WT AoB and iNOSKO-AoB mice compared to their age-matched controls. After AoB, both groups also showed a significant increase in interventricular septum diastolic dimension (IVSDd) compared to age-matched controls. These results were confirmed by a concomitant increase in heart weight (HW) to body weight (BW) in WT-AoB and iNOSKO-AoB mice. HW/BW was 9.41±0.64 mg/g (n=11) in WT-AoB and 9.31±0.39 mg/g (n=9) in iNOSKO-AoB mice compared to 6.24±0.44 mg/g (n=5) in WT and 6.35±0.19 mg/g (n=9) in iNOSKO age-matched controls. Hypertrophy was also associated with a decrease in LV fractional shortening (FS) and circumferential shortening (VCF) in WT-AoB and iNOSKO-AoB hearts compared to age-matched control mice. Following short-term banding, there was a trend towards impairment of FS (P=0.098) and VCF (P=0.07) in WT-AoB compared to iNOSKO-AoB mice.
Table 1.
Echocardiographic measurements in WT-AoB, iNOSKO-AoB and age-matched WT and iNOSKO mice
| Group | HR (bpm) | LVIDd (mm) | LVISd (mm) | FS (%) | Vcf (circ/sec) | IVSDd (mm) | LVPWDd (mm) | E/A | IVRT(msec) |
|---|---|---|---|---|---|---|---|---|---|
| Short-term banding | |||||||||
| WT (n=5) | 464.6±18.2 | 3.80 ±0.15 | 2.21±0.09 | 41.80±1.07 | 8.68±0.85 | 0.69±0.04 | 0.65±0.02 | 1.14±0.06 | 17±2 |
| WT-AoB (n=11) | 379.8±35.7 | 3.81±0.11 | 2.64±0.15+ | 31.34±2.42*+ | 5.96±0.52*+ | 1.01±0.04*+ | 1.02±0.04*+ | 1.28±0.20 | 22±2 |
| INOSKO (n=7) | 462.3±31.3 | 3.63±0.08 | 2.00±0.09 | 45.00±1.63 | 9.73±0.61 | 0.64±0.02 | 0.65±0.02 | 1.31±0.19 | 18±1 |
| INOSKO-AoB (n=9) | 467.3±38.4 | 3.72±0.09 | 2.39±0.14 | 36.42 ±2.48+ | 7.44±0.61+ | 1.05±0.04*+ | 1.05±0.03*+ | 1.21±0.14 | 19±2 |
| Long-term banding | |||||||||
| WT (n=13) | 399.8±17.8 | 3.89±0.09 | 2.47±0.07 | 36.58±1.20 | 7.55±0.33 | 0.75±0.03 | 0.70±0.02 | 1.34±0.08 | 19±1 |
| WT-AoB (n=10) | 441.5±19.1 | 4.11±0.10 | 3.05±0.13# | 26.00±1.43*+ | 5.09±0.36*+ | 1.02±0.03*+ | 1.01±0.04*+ | 1.56±0.33 | 21±2 |
| INOSKO (n=10) | 451.6±26.3 | 3.79±0.08 | 2.41±0.08 | 37.13±1.53 | 7.67±0.44 | 0.67±0.02* | 0.66±0.01 | 1.33±0.10 | 18±1 |
| INOSKO-AoB (n=8) | 368.0±18.4 | 3.90±0.13 | 2.73±0.12 | 30.00±1.15*+ | 5.76±0.29*+ | 1.00±0.03*+ | 0.98±0.02*+ | 1.45±0.17 | 21±1 |
HR=heart rate; LVIDd=Left ventricular internal dimension in diastole; LVISd=LV internal dimension in systole; FS=LV fractional shortening; Vcf =LV circumferential fiber shortening; IVSDd=Interventricular septum dimension in diastole; LVPWDd=LV posterior wall dimension in diastole; E/A=Ratio of the maximal velocity of mitral inflow E (early LV filling) and A (atrial contraction) waves; IVRT=LV isovolumic relaxation time.
P<0.05 compared to WT;
P<0.05 compared to iNOSKO;
P<0.05 compared to all other groups
Long term AoB (27.7±2.8 weeks in WT-AoB and 26.0±3.6 weeks in iNOSKO-AoB groups) resulted in a further decreases of FS and VCF without a further increase in hypertrophy. HW/BW was 9.13±0.54 mg/g (n=10) in WT-AoB and 8.14±0.34 mg/g (n=8) in iNOSKO-AoB mice compared to 6.77±0.27 mg/g (n=13) in WT and 6.81±0.24 mg/g (n=10) in iNOSKO age-matched controls. Similar to short-term AoB, there was a trend towards reduced FS in WT-AoB compared to iNOSKO-AoB mice after long-term banding (P=0.057), but LV internal dimension in systole (LVISd) was significantly increased in WT-AoB compared to other groups.
4.3. Beta-adrenergic response in papillary muscles in short and long- term AoB
Figure 3 shows the response to increasing doses of the beta-adrenergic agonist ISO in LV papillary muscles isolated from hearts subjected to short- (panels A-D) or long-term (panels E-H) AoB and from age-matched controls. The response to ISO was similar in both WT and iNOSKO short-term AoB muscles as well as age-matched controls (Figure 3 C). However, WT-AoB muscles showed altered relaxation parameters. Time to 50% relaxation was significantly increased in basal conditions and at 1 microM ISO in WT-AoB compared to muscles from WT age-matched mice (Figure 3D). After long-term AoB we found that the response of iNOSKO-AoB muscles to ISO was larger than in the other groups, reaching 343.9±45.7% (n=6) of baseline compared to 216.8±26.5% (n=6) in WT-AoB, 168.8±14.4% (n=9) in WT and 165.3±10.1% (n=8) in iNOSKO muscles (Figure 3G).
Figure 3.
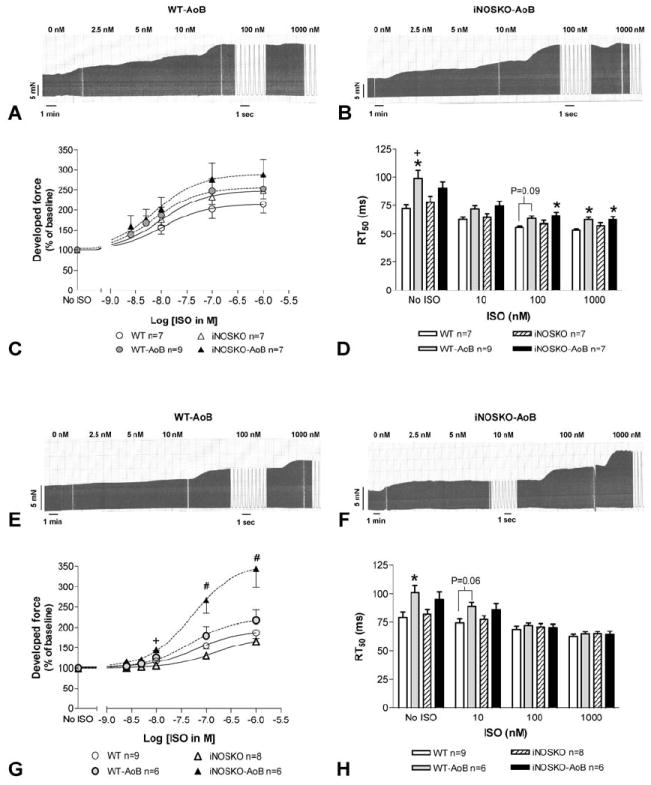
Effects of increasing doses of isoproterenol (ISO) on isometric force in LV papillary muscle isolated from short- and long-term WT-AoB and iNOSKO-AoB mice and age-matched control mice. Representative trace of the isometric force of papillary muscle isolated from WT-AoB (A) and iNOSKO-AoB (B) mice after short-term banding. Developed force (C) and time to 50% relaxation (D) in short-term AoB mice and age-matched controls. Representative trace of the isometric force of papillary muscle isolated from WT-AoB (E) and iNOSKO-AoB (F) mice after long-term banding. Developed force (G) and time to 50% relaxation (H) in long-term AoB mice and age-matched controls. *Indicates P<0.05 compared to WT; +indicates P<0.05 compared to iNOSKO; #indicates P<0.05 compared to all other groups.
To test whether the altered response to beta-adrenergic stimulation in WT-AoB muscles was due to increased NO production via iNOS, papillary muscles from WT-AoB and iNOSKO-AoB hearts subjected to long-term banding (47.8±7.9 weeks of banding in WT-AoB (n=3) and 57.5±3.9 weeks of AoB in iNOSKO-AoB (n=3) groups) were preincubated with a specific iNOS blocker (1400W) and their response to ISO determined. Following iNOS blockade, WT-AoB and iNOSKO-AoB mouse papillary muscles exhibited identical development of force in response to ISO. Moreover, muscles from both groups showed similar kinetics of contraction and relaxation (data not shown). These data suggest that NO produced by iNOS is responsible for the reduced contractile response to beta-agonist stimulation.
4.4. Hemodynamic parameters in long-term WT-AoB and iNOSKO-AoB mice
Next, we compared basal hemodynamic parameters among WT-AoB (27.9±3.6 weeks of banding), iNOSKO-AoB (26.0±3.6 weeks of banding) and their age-matched WT and iNOSKO mice (Figure 4 and Table 2). LV systolic pressure (LVSP) was significantly increased in iNOSKO-AoB mice compared to all other groups. The rate of pressure development (dP/dtmax) in iNOSKO and iNOSKO-AoB mice was also significantly increased compared to WT-AoB and iNOSKO-AoB mice exhibited an increased rate of relaxation (dP/dtmin) compared to WT-AoB group. There was a significant difference in preload-recruitable stroke work (PRSW) between iNOSKO-AoB and iNOSKO groups and a trend towards a difference between the iNOSKO-AoB and the WT-AoB groups (P=0.058).
Figure 4.
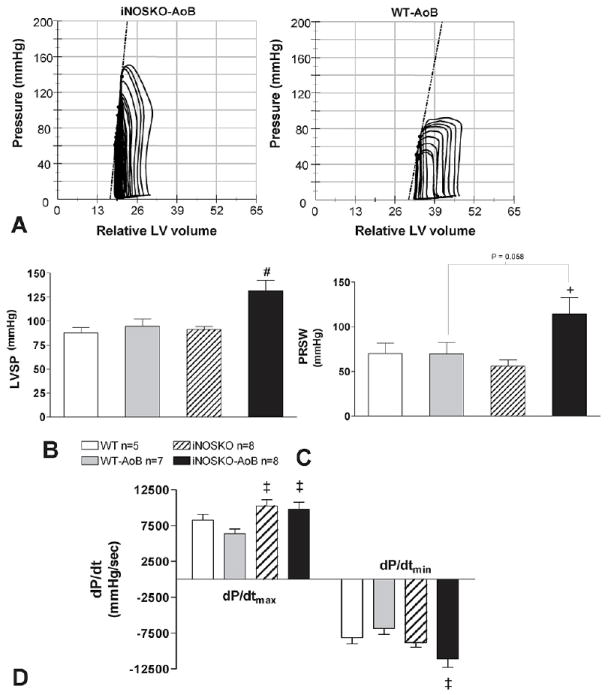
Hemodynamic parameters in long-term iNOSKO-AoB, WT-AoB mice and age-matched controls. A) Representative pressure-volume loops from iNOSKO-AoB and WT-AoB hearts. The dotted lines represent the end-systolic pressure-volume relationship (ESPVR). B) Left-ventricular systolic pressure (LVSP); C) Preload-load recruitable stroke work (PRSW); D) rates of contraction (dP/dtmax) and relaxation (dP/dtmin). +Significantly different (P<0.05) from iNOSKO; #Significantly different (P<0.05) from all other groups; ‡Significantly different (P<0.05) from WT-AoB.
Table 2.
Hemodynamic parameters in long-term WT-AoB, iNOSKO-AoB and age-matched WT and iNOSKO mice
| Group | HR (bpm) | LVSP (mmHg) | LVDP (mmHg) | dP/dtmax (mmHg/s) | dP/dtmin (mmHg/s) | ESPVR (mmHg/ml) | PRSW (mmHg) | Emax (mmHg/ml) |
|---|---|---|---|---|---|---|---|---|
| WT (n=5) | 571.4± 21.3 | 87.6±5.7 | 3.6±0.4 | 8246.4±822.1 | -8114.2±880.4 | 9.4±3.0 | 70.0±11.8 | 14.2±11.8 |
| WT-AoB (n=7) | 545.6± 24.1 | 94.7±7.3 | 3.3±0.2 | 6346.9±654.2 | -6817.4±830.8 | 12.4±3.8 | 69.4±13.1 | 22.9±11.1 |
| INOSKO (n=8) | 619.8± 13.4 | 91.0±3.6 | 3.0±0.2 | 10244.1±876.5‡ | -8794.4±670.5 | 7.2±1.5 | 56.1±6.8 | 16.2±4.7 |
| INOSKO-AoB (n=8) | 592.5± 17.6 | 131.6±11.0# | 3.3±0.2 | 9791.5±944.8‡ | -11056.6±1196.6‡ | 15.3±2.6 | 114.4±18.0+ | 29.9±4.7 |
HR=heart rate; LVSP=Left ventricular systolic pressure; LVDP=LV diastolic pressure; ESPVR=End-systolic pressure-volume relationship; PRSW=Pre-load recruitable stroke work; Emax=ESPVR slope.
P<0.05 compared to iNOSKO;
P<0.05 compared to all other groups;
P<0.05 compared to WT-AoB.
4.5. Gene expression
Quantitative real-time RT-PCR was performed to determine changes in gene expression (Figure 5). Short-term AoB resulted in a significant increase in the expression of beta-MHC and ANF in WT-AoB mice, but not in iNOSKO-AoB mice. Similarly, PLB expression was also increased in hearts from WT-AoB compared to iNOSKO-AoB mice. In short-term WT-AoB hearts the expression of SERCA was elevated compared to WT, iNOSKO and iNOSKO-AoB mice, however, this increase was not sustained in long-term AoB mice potentially representing an adaptational response.
Figure 5.
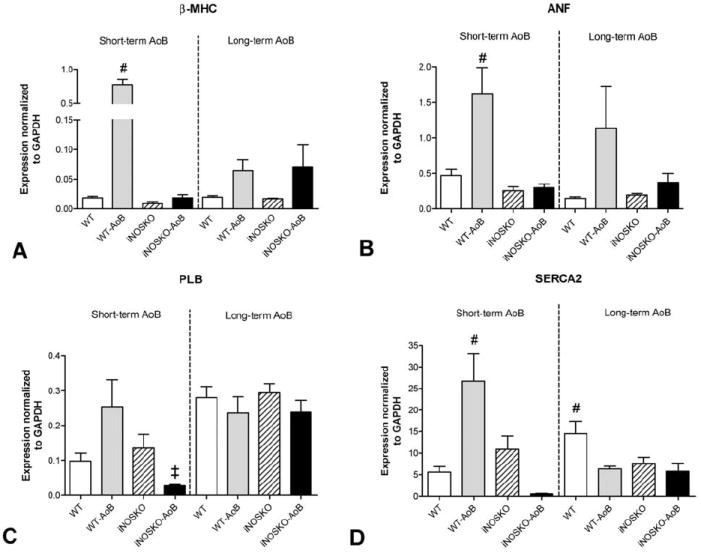
Quantitative gene expression (RT-PCR) in short- and long-term WT-AoB, iNOSKO-AoB and age-matched control mice. A) beta-myosin heavy chain (beta-MHC). B) Atrial natriuretic factor (ANF). C) Phospholamban (PLB). D) Sarcoplasmic reticulum Ca ATP-ase (SERCA2a). Expression was normalized to GAPDH expression (n=3-4). #Significantly different (P<0.05) compared to all other groups within short- or long-term AoB groups; ‡Significantly different (P<0.05) compared to WT-AoB within short- or long-term AoB groups.
4.6. Protein expression
Figure 6 shows expression of eNOS (panels A and B), PLB (panels C and D) and SERCA2a (panels E and F) in short- and long-term AoB hearts from WT, iNOSKO and age-matched controls. Total eNOS expression among the groups was not different; however, short-term iNOSKO-AoB hearts exhibited increased levels of Thr-495 phosphorylation compared to other groups. There was no difference in eNOS phosphorylation (Thr-495) in the long-term AoB group. Finally, there was no significant change in the expression of PLB or SERCA2a at the protein level. Figure 7 shows that both long term WT-AoB and iNOSKO-AoB hearts exhibit slightly increased expression of the beta-MHC isoform. This increase in beta-MHC expression was not significantly different from that observed in WT-AoB or iNOSKO-AoB age-matched control hearts.
Figure 6.
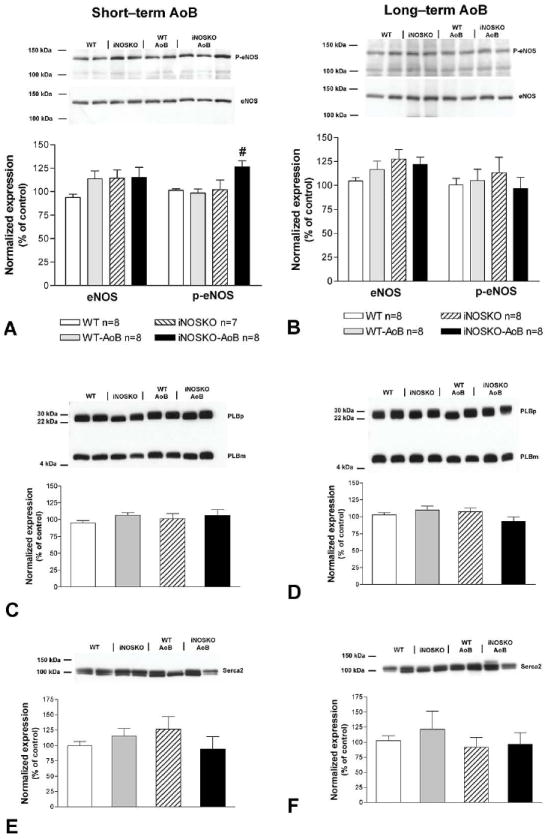
Protein level expression of eNOS, phospholamban (PLB), SERCA2 and the level of eNOS phosphorylation (p-eNOS Thr-495) in short- and long-term AoB mice and age-matched controls. Representative Western blots and summary of data for eNOS expression and p-eNOS (Thr-495) in short-term (panel A) and long-term (panel B) AoB and age-matched controls. Representative Western blot for total PLB expression (PLBm–monomeric form of PLB; PLBp–pentameric form of PLB, n=6) in short-term (panel C) and long-term (panel D) AoB and age-matched controls. Representative Western blot for total SERCA2 expression in short-term (panel E, n=5-6) and long-term (panel F, n=7-8) AoB and age-matched controls. #Indicates P<0.05 compared to all other groups.
Figure 7.
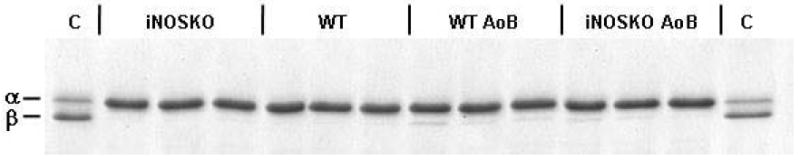
Expression of alpha- and beta-myosin heavy chain in long-term WT-AoB, iNOS-AoB and age-matched (iNOSKO and WT) control mice. Lane C represents sample prepared from 1-day old WT mouse heart containing both alpha- and beta-myosin heavy chain.
5. DISCUSSION
Our data demonstrate for the first time that the absence of iNOS during the development of hypertrophy induced by moderate aortic constriction, mimicking long-term hypertension in humans, delays both the increase in hypertrophic gene marker expression and contractile dysfunction associated with hypertrophy development. This improved function occurred in the absence of a significant effect on the development of cardiac hypertrophy itself. Moreover, after long-term AoB, iNOSKO animals exhibited increased basal cardiac function as well as an improved response to beta-adrenergic stimulation compared to WT animals. These findings support a direct role for the expression of iNOS in the development of the cardiac dysfunction, but not the hypertrophy, that results from pressure overload.
An increased expression of iNOS in failing hearts of different etiologies is well documented in humans (4, 24-26). Likewise, increased iNOS expression has been demonstrated in various animal models of HF induced by either myocardial infarction (MI) (27-30), volume overload (12) or rapid pacing (13). In samples from failing human hearts, iNOS was localized not only to cardiomyocytes, but also vascular endothelial cells, smooth muscle cells and macrophages (24,31). The level of iNOS expression and activity is varied both between hearts and within various regions of the same heart (26,32). In contrast, other studies have failed to observe iNOS expression in failing (33) or hypertrophic (34) hearts, likely due to the anatomical localization from which the biopsy specimens were taken. In our model, we found that iNOS was expressed only in WT mice, after short- or long-term AoB. In long-term WT-AoB, iNOS was most likely expressed in the cytosol and nucleus of cardiomyocytes, since Buchwalow et al previously demonstrated a similar sub-cellular localization of iNOS in both rat myocytes and the failing human heart (35).
In the current study we demonstrate that chronic pressure-overload results in LV hypertrophy in both iNOSKO and WT mice. In both groups the degree of hypertrophy, measured as IVSDd, LVPWD and HW/BW, was similar and independent of AoB duration (short- vs. long-term AoB). Interestingly, after short-term AoB mRNA expression of ANF, beta-MHC and SERCA2a were increased in WT-AoB, but not in iNOSKO-AoB hearts. However, the altered gene expression of SERCA2a, which could potentially be one of the compensatory mechanisms during the development of hypertrophy, was not manifested at the protein level since we did not detect any change in SERCA2a protein expression at any time point among the groups. Moreover, there was no change in the protein level expression of the other major Ca2+ regulatory protein, phospholamban, at any time point. These results suggest the reduced ANF and beta-MHC marker expression in iNOSKO-AoB mice was the result of an absence of iNOS and not altered calcium handling.
Although the degree of hypertrophy was the same in iNOSKO-AoB and WT-AoB hearts, the long-term iNOSKO-AoB group exhibited a preserved contractile function with increased LVSP, PRSW and better relaxation compared to WT-AoB. Other investigators have shown that iNOS blockage (12,29) or ablation (27,28,36) preserved cardiac function to increase survival following MI or volume-overload (12), and prevented hypertrophy in severe, short-term AoB (37). Sam et al. (28) reported that four months after MI, LV contractile function was improved, apoptosis decreased, and survival enhanced in iNOSKO mice compared to WT even though even though both animal groups exhibited a similar degree of hypertrophy. Interestingly, at two months post-MI they were unable to identify any significant changes in cardiac performance. In a similar study, Feng et al. (27) observed improved heart function as early as one month after MI. In contrast, Jones et al. (38) showed that iNOS deficiency did not significantly affect congestive heart failure (CHF) severity in mice one month post-MI. The discrepancy among these studies might be due to the different time points at which the mice were studied post-MI or differences in infarct size. In our hypertrophy model we report an improvement in cardiac function of iNOSKO-AoB mice compared to WT-AoB, but only after long-term AoB and not after short-term AoB. These data are not in complete agreement with results published by Zhang et al. (37), who showed that after four weeks AoB iNOS-KO mice displayed less hypertrophy, dilatation, fibrosis, and dysfunction. It is important to note, that there are major differences between Zhang’s and our studies. The level of aortic constriction and time of AoB were different in both studies. In the presented paper we performed experiments using two different time points after AoB to better mimic long-term hypertension and conducted studies up to 6.5 months. We also constricted the aorta using a 25 gauge needle, which was significantly larger than Zhang et al. (37) who used 26 and 27 gauge needles for their studies. Although, Zhang et al (37) did not measure the pressure gradient across the constriction, based on our previously published data (17) and data published by others (16,39), we can expect much higher pressure gradients across the aortic constriction in Zhang’s studies than in ours, which can explain the differences in the presented data.
Expression of iNOS in the hearts is associated with depressed sensitivity to beta-adrenergic stimulation (4,12,40). Our data demonstrate that short-term iNOSKO-AoB and WT-AoB mouse papillary muscles both exhibit a similar response to beta-adrenergic agonist. In fact, the response of AoB and age-matched control mice to ISO was also similar at this point. In contrast, papillary muscles isolated from long-term AoB WT mice demonstrate a significantly depressed response to ISO compared to iNOSKO-AoB muscles. Furthermore, this depressed response to ISO was identical in WT-AoB and iNOSKO-AoB muscles pre-treated with iNOS blocker, strongly supporting the hypothesis that iNOS expression is responsible for the depressed response to beta-stimulation.
Although altered eNOS expression has been observed in both human and animal models of HF (32,41,42), the role of eNOS in the adrenergic regulation of both normal and diseased hearts remains controversial (43-45). In our experimental model, AoB did not alter eNOS expression in either WT or iNOSKO mouse hearts. Furthermore, only a transient increase in eNOS phosphorylation at Thr-495 in short-term iNOSKO-AoB mice was observed. Phosphorylation of eNOS at this residue has been shown to decrease eNOS activity (46) and may potentiate the response to beta-adrenergic stimulation. However, we did not observe any significant differences in the response to beta-adrenergic stimulation of papillary muscles isolated from short-term iNOSKO-AoB muscles compared to other groups. This indicates the role of eNOS Thr-495 phosphorylation may only impart a small effect on cardiac function.
In summary, our data demonstrate that increased afterload via AoB induces an increased expression of iNOS that is associated with cardiac dysfunction, whereas AoB in the absence of iNOS delays the development of cardiac dysfunction. Our data provide strong rationale for further studies on new therapies involving the inhibition of iNOS activity to improve cardiac performance in hypertrophy and heart failure.
Acknowledgments
We acknowledge the Electron Microscopy Facility of the Research Resources Center at UIC for providing the use of equipment and assistance to conduct this study. This work was supported by NIH Grants HL64209, HL79032, HL91056, PO1-62426 and T32 HL07692. BMW was an Established Investigator from the American Heart Association.
References
- 1.Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circ Res. 1996;79:363–380. doi: 10.1161/01.res.79.3.363. [DOI] [PubMed] [Google Scholar]
- 2.Hare JM, Givertz MM, Creager MA, Colucci WS. Increased sensitivity to nitric oxide synthase inhibition in patients with heart failure: potentiation of beta-adrenergic inotropic responsiveness. Circulation. 1998;97:161–166. doi: 10.1161/01.cir.97.2.161. [DOI] [PubMed] [Google Scholar]
- 3.Stojanovic MO, Ziolo MT, Wahler GM, Wolska BM. Anti-adrenergic effects of nitric oxide donor SIN-1 in rat cardiac myocytes. Am J Physiol Cell Physiol. 2001;281:C342–C349. doi: 10.1152/ajpcell.2001.281.1.C342. [DOI] [PubMed] [Google Scholar]
- 4.Ziolo MT, Maier LS, Piacentino V, III, Bossuyt J, Houser SR, Bers DM. Myocyte nitric oxide synthase 2 contributes to blunted beta-adrenergic response in failing human hearts by decreasing Ca2+ transients. Circulation. 2004;109:1886–1891. doi: 10.1161/01.CIR.0000124231.98250.A8. [DOI] [PubMed] [Google Scholar]
- 5.Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89:2070–2078. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- 6.Suto N, Mikuniya A, Okubo T, Hanada H, Shinozaki N, Okumura K. Nitric oxide modulates cardiac contractility and oxygen consumption without changing contractile efficiency. Am J Physiol. 1998;275:H41–H49. doi: 10.1152/ajpheart.1998.275.1.H41. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Traverse JH, Du R, Hou M, Bache RJ. Nitric oxide modulates myocardial oxygen consumption in the failing heart. Circulation. 2002;106:273–279. doi: 10.1161/01.cir.0000021120.90970.b9. [DOI] [PubMed] [Google Scholar]
- 8.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 9.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 10.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 11.Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gealekman O, Abassi Z, Rubinstein I, Winaver J, Binah O. Role of myocardial inducible nitric oxide synthase in contractile dysfunction and beta-adrenergic hyporesponsiveness in rats with experimental volume-overload heart failure. Circulation. 2002;105:236–243. doi: 10.1161/hc0202.102015. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto S, Tsutsui H, Tagawa H, Saito K, Takahashi M, Tada H, Yamamoto M, Katoh M, Egashira K, Takeshita A. Role of myocyte nitric oxide in beta-adrenergic hyporesponsiveness in heart failure. Circulation. 1997;95:1111–1114. doi: 10.1161/01.cir.95.5.1111. [DOI] [PubMed] [Google Scholar]
- 14.Mungrue IN, Husain M, Stewart DJ. The role of NOS in heart failure: lessons from murine genetic models. Heart Fail Rev. 2002;7:407–422. doi: 10.1023/a:1020762401408. [DOI] [PubMed] [Google Scholar]
- 15.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roman BB, Geenen DL, Leitges M, Buttrick PM. PKC-beta is not necessary for cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2001;280:H2264–H2270. doi: 10.1152/ajpheart.2001.280.5.H2264. [DOI] [PubMed] [Google Scholar]
- 18.Urboniene D, Dias FA, Pena JR, Walker LA, Solaro RJ, Wolska BM. Expression of slow skeletal troponin I in adult mouse heart helps to maintain the left ventricular systolic function during respiratory hypercapnia. Circ Res. 2005;97:70–77. doi: 10.1161/01.RES.0000173849.68636.1e. [DOI] [PubMed] [Google Scholar]
- 19.Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinney RD, Geenen DL, Solaro RJ, Buttrick PM. Protein kinase C epsilon overexpression alters myofilament properties and composition during the progression of heart failure. Circ Res. 2004;95:424–432. doi: 10.1161/01.RES.0000138299.85648.92. [DOI] [PubMed] [Google Scholar]
- 20.Wolska BM, Vijayan K, Arteaga GM, Konhilas JP, Phillips RM, Kim R, Naya T, Leiden JM, Martin AF, de Tombe PP, Solaro RJ. Expression of slow skeletal troponin I in adult transgenic mouse heart muscle reduces the force decline observed during acidic conditions. J Physiol. 2001;536:863–870. doi: 10.1111/j.1469-7793.2001.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alden KJ, Goldspink PH, Ruch SW, Buttrick PM, Garcia J. Enhancement of L-type Ca2+ current from neonatal mouse ventricular myocytes by constitutively active PKC-betaII. Am J Physiol Cell Physiol. 2002;282:C768–C774. doi: 10.1152/ajpcell.00494.2001. [DOI] [PubMed] [Google Scholar]
- 22.Dias FA, Walker LA, Arteaga GM, Walker JS, Vijayan K, Pena JR, Ke Y, Fogaca RT, Sanbe A, Robbins J, Wolska BM. The effect of myosin regulatory light chain phosphorylation on the frequency-dependent regulation of cardiac function. J Mol Cell Cardiol. 2006;41:330–339. doi: 10.1016/j.yjmcc.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Warren CM, Greaser ML. Method for cardiac myosin heavy chain separation by sodium dodecyl sulfate gel electrophoresis. Anal Biochem. 2003;320:149–151. doi: 10.1016/s0003-2697(03)00350-6. [DOI] [PubMed] [Google Scholar]
- 24.Vejlstrup NG, Bouloumie A, Boesgaard S, Andersen CB, Nielsen-Kudsk JE, Mortensen SA, Kent JD, Harrison DG, Busse R, Aldershvile J. Inducible nitric oxide synthase (iNOS) in the human heart: expression and localization in congestive heart failure. J Mol Cell Cardiol. 1998;30:1215–1223. doi: 10.1006/jmcc.1998.0686. [DOI] [PubMed] [Google Scholar]
- 25.Patten RD, Denofrio D, El-Zaru M, Kakkar R, Saunders J, Celestin F, Warner K, Rastegar H, Khabbaz KR, Udelson JE, Konstam MA, Karas RH. Ventricular assist device therapy normalizes inducible nitric oxide synthase expression and reduces cardiomyocyte apoptosis in the failing human heart. J Am Coll Cardiol. 2005;45:1419–1424. doi: 10.1016/j.jacc.2004.05.090. [DOI] [PubMed] [Google Scholar]
- 26.Winlaw DS, Smythe GA, Keogh AM, Schyvens CG, Spratt PM, Macdonald PS. Increased nitric oxide production in heart failure. Lancet. 1994;344:373–374. doi: 10.1016/s0140-6736(94)91403-6. [DOI] [PubMed] [Google Scholar]
- 27.Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- 28.Sam F, Sawyer DB, Xie Z, Chang DL, Ngoy S, Brenner DA, Siwik DA, Singh K, Apstein CS, Colucci WS. Mice lacking inducible nitric oxide synthase have improved left ventricular contractile function and reduced apoptotic cell death late after myocardial infarction. Circ Res. 2001;89:351–356. doi: 10.1161/hh1601.094993. [DOI] [PubMed] [Google Scholar]
- 29.Saito T, Hu F, Tayara L, Fahas L, Shennib H, Giaid A. Inhibition of NOS II prevents cardiac dysfunction in myocardial infarction and congestive heart failure. Am J Physiol Heart Circ Physiol. 2002;283:H339–H345. doi: 10.1152/ajpheart.00596.2001. [DOI] [PubMed] [Google Scholar]
- 30.Liu YH, Carretero OA, Cingolani OH, Liao TD, Sun Y, Xu J, Li LY, Pagano PJ, Yang JJ, Yang XP. Role of inducible nitric oxide synthase in cardiac function and remodeling in mice with heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2005;289:H2616–H2623. doi: 10.1152/ajpheart.00546.2005. [DOI] [PubMed] [Google Scholar]
- 31.Haywood GA, Tsao PS, Der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH, Cooke JP, McKenna WJ, Fowler MB. Expression of inducible nitric oxide synthase in human heart failure. Circulation. 1996;93:1087–1094. doi: 10.1161/01.cir.93.6.1087. [DOI] [PubMed] [Google Scholar]
- 32.Fukuchi M, Hussain SN, Giaid A. Heterogeneous expression and activity of endothelial and inducible nitric oxide synthases in end-stage human heart failure: their relation to lesion site and beta-adrenergic receptor therapy. Circulation. 1998;98:132–139. doi: 10.1161/01.cir.98.2.132. [DOI] [PubMed] [Google Scholar]
- 33.Thoenes M, Forstermann U, Tracey WR, Bleese NM, Nussler AK, Scholz H, Stein B. Expression of inducible nitric oxide synthase in failing and non-failing human heart. J Mol Cell Cardiol. 1996;28:165–169. doi: 10.1006/jmcc.1996.0016. [DOI] [PubMed] [Google Scholar]
- 34.Satoh M, Nakamura M, Tamura G, Makita S, Segawa I, Tashiro A, Satodate R, Hiramori K. Inducible nitric oxide synthase and tumor necrosis factor-alpha in myocardium in human dilated cardiomyopathy. J Am Coll Cardiol. 1997;29:716–724. doi: 10.1016/s0735-1097(96)00567-0. [DOI] [PubMed] [Google Scholar]
- 35.Buchwalow IB, Schulze W, Karczewski P, Kostic MM, Wallukat G, Morwinski R, Krause EG, Muller J, Paul M, Slezak J, Luft FC, Haller H. Inducible nitric oxide synthase in the myocard. Mol Cell Biochem. 2001;217:73–82. doi: 10.1023/a:1007286602865. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Carretero OA, Xu J, Rhaleb NE, Wang F, Lin C, Yang JJ, Pagano PJ, Yang XP. Lack of inducible NO synthase reduces oxidative stress and enhances cardiac response to isoproterenol in mice with deoxycorticosterone acetate-salt hypertension. Hypertension. 2005;46:1355–1361. doi: 10.1161/01.HYP.0000192651.06674.3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res. 2007;100:1089–1098. doi: 10.1161/01.RES.0000264081.78659.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones SP, Greer JJ, Ware PD, Yang J, Walsh K, Lefer DJ. Deficiency of iNOS does not attenuate severe congestive heart failure in mice. Am J Physiol Heart Circ Physiol. 2005;288:H365–H370. doi: 10.1152/ajpheart.00245.2004. [DOI] [PubMed] [Google Scholar]
- 39.Rockman HA, Ono S, Ross RS, Jones LR, Karimi M, Bhargava V, Ross J, Jr, Chien KR. Molecular and physiological alterations in murine ventricular dysfunction. Proc Natl Acad Sci U S A. 1994;91:2694–2698. doi: 10.1073/pnas.91.7.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziolo MT, Katoh H, Bers DM. Expression of inducible nitric oxide synthase depresses beta-adrenergic-stimulated calcium release from the sarcoplasmic reticulum in intact ventricular myocytes. Circulation. 2001;104:2961–2966. doi: 10.1161/hc4901.100379. [DOI] [PubMed] [Google Scholar]
- 41.Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 42.Stein B, Eschenhagen T, Rudiger J, Scholz H, Forstermann U, Gath I. Increased expression of constitutive nitric oxide synthase III, but not inducible nitric oxide synthase II, in human heart failure. J Am Coll Cardiol. 1998;32:1179–1186. doi: 10.1016/s0735-1097(98)00399-4. [DOI] [PubMed] [Google Scholar]
- 43.Massion PB, Dessy C, Desjardins F, Pelat M, Havaux X, Belge C, Moulin P, Guiot Y, Feron O, Janssens S, Balligand JL. Cardiomyocyte-restricted overexpression of endothelial nitric oxide synthase (NOS3) attenuates beta-adrenergic stimulation and reinforces vagal inhibition of cardiac contraction. Circulation. 2004;110:2666–2672. doi: 10.1161/01.CIR.0000145608.80855.BC. [DOI] [PubMed] [Google Scholar]
- 44.Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation. 2001;104:3097–3102. doi: 10.1161/hc5001.101966. [DOI] [PubMed] [Google Scholar]
- 45.Danson EJ, Zhang YH, Sears CE, Edwards AR, Casadei B, Paterson DJ. Disruption of inhibitory G-proteins mediates a reduction in atrial beta-adrenergic signaling by enhancing eNOS expression. Cardiovasc Res. 2005;67:613–623. doi: 10.1016/j.cardiores.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 46.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–E75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]