

Abstract
Arginine-rich peptides and small-molecule intercalating agents utilize distinct molecular mechanisms for RNA recognition. Here, we combined these distinct binding modules in an effort to create conjugate ligands with enhanced affinity and specificity using the bacteriophage λ N peptide/boxB interaction as a model system. We first designed and synthesized a series of peptide-acridine conjugates using portions of the RNA-binding domain of N protein (11- and 22- residue peptide segments), then compared the binding affinity, specificity, salt dependence, and structural properties of the RNA-peptide and RNA-peptide-acridine conjugate complexes using steady-state fluorescence, CD spectroscopy, NMR, and native gel mobility shift assays (GMSA). These analyses revealed that the full-length peptide-acridine conjugate displayed substantially improved RNA-binding affinity (~80-fold; Kd ~ 15 pM) relative to the peptide alone (Kd ~ 1.2 nM). In accordance, we also observed specificity enhancement (~25-fold) as determined by comparing binding of the best conjugate to a cognate λ boxB RNA with that to a noncognate P22 RNA hairpin (80-fold vs. 3.2-fold enhancement). Furthermore, the observed binding enhancement was unique to the full length conjugate with a flexible linker, implying that the structural context of acridine presentation was critical. Taken together, our observations support the idea that peptide- and intercalation-based binding can be combined to create a new class of high-affinity, high-specificity RNA-binding ligands.
Keywords: acridine-peptide conjugate, N peptide, boxB RNA recognition, 2-aminopurine, multiple binding modules
The design of proteins and other ligands with the capacity to recognize nucleotide sequences with high affinity and specificity has been the subject of extensive research in recent years; for example, designing novel zinc finger proteins (1–3) and polyamides (4–7) were found to target the major and the minor groove of DNA, respectively. In comparison, RNA molecules often fold into complex and unique three dimensional shapes and perform versatile functions in diverse biological processes, thus prompting great interest in designing sequence or structure-specific RNA-binding molecules. Enhanced understanding of sequence-specific RNA binding molecules and the expanding structural databases for RNA and RNA/protein complexes, together with the development of new synthetic tools, have provided new opportunities for the design of peptide or small molecule RNA binders.
One approach to achieve enhanced RNA binding is to combine different binding modules for interaction sites that coexist in a given RNA molecule. In principle, if two binding sites are in close proximity, a dimeric derivative ligand can bind simultaneously to the two sites, resulting in binding affinity greater than either module alone (8–9). Furthermore, such modular design of RNA-binding ligands has been successfully developed as multivalent ligands to target triplet repeat RNAs that are implicated in myotonic muscular dystrophy, and shown to inhibit RNA-protein complex formation (10).
As proof of principle, recent studies have shown acridine derivatives as a useful intercalating module (11–14). In a new class of HIV-1 Tat antagonist, a Tat-TAR binding inhibitor consisting of a substituted acridine and a polyamine moiety was demonstrated to inhibit Tat function (15). Specifically, the modular design principle was based on the stacking ability of the aromatic moiety of acridine and the contacts with the TAR RNA phosphate backbone by a polycationic anchor. The two modules are linked by an aliphatic linker. Aside from the compound stacking between two bases, direct hydrogen-bond contacts with a GC base pair were also involved.
Two other studies further illustrate the utility of acridine modules in RNA binding. A series of cationic small molecules were synthesized and their binding abilities to defined RNA duplexes with and without bulged bases were investigated (16–17). Complex stabilization and selectivity for an RNA duplex containing a single bulged base over a normal RNA duplex have been obtained with a ligand consisting of a chloroacridine moiety covalently attached to 2,6-diaminopurine through an aminoalkyl linker. It is believed that the chloroacridine moiety intercalates into the RNA duplex and the 2,6-diaminopurine interacts with the bulged base. Another example of this type of binding enhancement is illustrated by a neomycin-acridine conjugate, synthesized by covalently linking neomycin B to 9-aminoacridine via a short spacer, as a potent inhibitor of Rev-RRE binding (18). Its affinity to the RRE is about 50-fold higher than that of the parent neomycin B and approaches that of the Rev peptide. These results demonstrate that the combination of different binding modes (e.g., ionic and intercalation) within one ligand is a powerful approach to enhancing RNA binding.
Acridine conjugation has also been used in designing functional molecules. For example, attachment of an acridine moiety to a catalytic tripeptide produced a RNase mimic (19), and to a base-linker construct generated abasic site recognition and cleavage functions (20). Acridine has also been used in conjunction with an oligonucleotide to facilitate site-selective RNA hydrolysis (21), where acridine was hypothesized to push the bulged base out of the helix and present a scission site.
In light of the versatile and modular functions of acridine, we reasoned that the same principle may also apply to the N peptide/boxB RNA complex system. Bacteriophage N proteins play an essential role in transcriptional antitermination, which is critical for phage development). The inhibition of transcription termination at both intrinsic (22 and Rho-dependent terminators by N proteins depends on recognition of a cis-acting RNA-element called Nut (N utilization) on the nascent transcript. Together with four phage Escherichia coli host factors (NusA, NusB, NusG, and ribosomal protein S10), N protein-Nut initiate the formation of a ribonucleoprotein a termination-resistant form ( complex that converts the RNA polymerase into 22–23).
The Nut site consists of a 5′-single-stranded RNA element (boxA) and a 3′ hairpin (boxB)). The boxB from lambda phage (24–25 is a 15-mer RNA stem-loop hairpin containing a purine-rich pentaloop. The RNA-binding domain of the N protein consists of an arginine-rich motif located at the N-terminus (26). The 22-residue short peptide recognizes the cognate boxB RNA with similar specificity and affinity as the intact N protein (27). Upon complex formation, four of the pentaloop nucleotides adopt a canonical GNRA tetraloop fold (28) with the fourth adenine extruded (29–30); the peptide forms a bent α helix and binds tightly to the major groove of the RNA (29–31).
The RNA–protein interface of the λN22/boxB complex is dominated by electrostatic interactions and hydrophobic contacts (29). The five arginines and two lysines of λN22 create a positively charged surface on one face of the α-helix that interacts with the negatively charged phosphodiester backbone of the boxB RNA. Hydrophobic interactions are also important for boxB recognition. Ala-3 and Trp-18 are involved in crucial hydrophobic interactions. In addition, the roles of arginine and lysine residues are not restricted to ionic interaction, some of the aliphatic portions of these side chains also contact the RNA bases or sugars.
Given the above detailed characterization, the λN22/boxB complex is an ideal model system for testing new binding modes, e.g., intercalation. In this paper, we report the use of acridine-peptide conjugates to enhance binding of boxB RNA targets by the N peptide.
Materials and Methods
Synthesis of peptides and acridine-peptide conjugates
Crude peptides with or without linkers were constructed by automated solid phase peptide synthesis using Fmoc protected monomers (ABI) on an Applied Biosystems 432A peptide synthesizer. A series of full length λN22 and truncated λN11 acridine-peptide conjugates were manually synthesized on the resin using the chemistry outlined in Scheme 1. Two types of flexible linkers, a Gly-Gly linker and an ethylene glycol linker, were used. Crude peptides and crude acridine-peptide conjugates were deprotected and cleaved from the resin and purified by reverse-phase HPLC on a C18 column. The purity of the products was checked by analytical HPLC and their identity confirmed by MALDI–TOF mass spectrometry. Concentrations of peptide stocks were determined by UV absorption at 280 nm for free peptides containing either tryptophan or tyrosine, or at 412/434 nm for acridine-peptide conjugates using extinction coefficients (ε=1.32×104cm−1M−1 at 412 nm, and 1.12×104cm−1M−1 at 434 nm) determined for a water soluble acridine derivative (3-acridin-9-ylamino-propanol) synthesized from 9-chloroacridine and 3-amino-1-propanol as described (18).
Scheme 1.

Synthesis and quantification of acridine-peptide conjugates.
Synthesis of 2AP-labeled RNA oligomers
Crude RNA oligomers with fluorescent 2- aminopurine (2AP) label substituted for adenine at the 2nd, 3rd, and 4th base positions of the pentaloop (denoted 2AP-2, 2AP-3, and 2AP-4, respectively) were constructed by automated synthesis using 2-aminopurine-TOM-CE phosphoramidite (Glen Research, Sterling, VA). Oligomers were deprotected and purified by 20% urea-PAGE. Purified oligomers were desalted on a NAP-25 column and quantified by UV absorption at 260 nm.
Steady-State Fluorescence Measurements
Steady-state fluorescence measurements were made on a Shimadzu RF-5301PC Spectrofluorophotometer as described (27, 32). Aliquots of concentrated stocks of free peptide or acridine-peptide conjugates were added stepwise to a stirred solution of 2AP-containing RNA maintained at various temperatures in a series of buffer conditions. Fluorescence signal of 2AP was monitored at 370 nm with excitation at 310 nm. Dissociation constants (Kd) were calculated for a one step binding mechanism by nonlinear least squares regression using the computer program DynaFit (33), as previously reported (34).
Band Shift Analysis
Free boxB RNA or complexes of peptide-RNA were preformed in NMR buffer (50 mM NaCl, 10 mM Phosphate, 0.5 mM EDTA, pH 6), and diluted by TBE buffer before loading to 20% non-denaturing PAGE gel maintained at 10–15 °C. Free RNA and complex bands were visualized by UV.
One Dimensional NMR Spectroscopy
Unlabeled 15mer boxB RNA 5′GCCCUGAAAAAGGGC3′ (bases in the loop are underlined) was synthesized by in vitro transcription using T7 RNA polymerase (35). The RNA was purified by 20% urea-PAGE and desalted on a NAP-25 column. Purified RNA oligomer was resuspended in NMR buffer (50 mM NaCl, 10 mM Phosphate, 0.5 mM EDTA, pH 6, 90:10 H2O/D2O). Spectra were collected on a Varian INOVA 600 MHz NMR spectrometer at 25 °C. Spectrum of free boxB RNA was collected first; titration of concentrated peptide or acridine peptide conjugate into the boxB RNA was monitored by inspecting the imino proton region of RNA.
CD Spectroscopy
Spectra were taken on an Aviv 62 DS CD spectrometer at 20°C. The samples contained 5 μM RNA and 6 μM peptide in 10 mM potassium phosphate buffer (pH 7.9). The spectra of the bound peptides were determined by subtracting the spectra for free RNA and excess free peptide from that of the complex.
Results
In the acridine peptide conjugate–RNA complex, the high affinity λ N peptide is expected to bind to the major groove of the boxB RNA with the same molecular interactions as in the wild-type complex (29–30). Consequently, the acridine moiety may be positioned proximal to the lower part of the RNA stem where it can either intercalate into the stem (Figure 1) or simply associate with the RNA extrahelically.
Figure 1.

Design of acridine-peptide conjugate binding to boxB RNA. Shown in the middle is the NMR structure (PDB: 1QFQ) of boxB RNA/N peptide complex drawn by Pymol. The coordinates of acridine moiety were modeled around the stem region for schematic purpose. A. Sequences and secondary stem-loop structures of boxB RNA from λ bacteriophages used in this study. B. Sequences of λN22 full length and truncated λN11 peptides. The 11-mer was tagged with gy for quantification purposes. C. Structure of acridine. D. Structure of linkers.
Kd values
Dissociation constants (Kd) were determined by monitoring the change in the fluorescence of 2-aminopurine (2AP) incorporated at variable positions within the loops of the RNA hairpins. This base was found to be an extremely useful probe for nucleic acid structure and dynamics (36), as well as RNA-peptide interactions (32). The fluorescence intensity of 2AP is highly sensitive to local environment, exhibiting decrease or increase when stacked or solvent exposed, respectively (37). In most cases, peptide binding of the RNA can be detected by either a fluorescence increase or decrease of more than 20% from the starting value for the free boxB RNA (27, 38).
Table 1 shows the Kd values directly determined at 50 mM K+, 20 mM Tris for acridine-λ N11, and acridine-λ N22 peptide conjugates, compared to those of WT peptides alone, for boxB RNA targets from both the λ and P22 phages. Binding affinities for truncated λ N11 peptide-acridine conjugates constructed with either a Gly-Gly linker 1 or an ethylene glycol linker 2 were similar and slightly weaker than the WT λ N11 peptide. However, the binding affinities were enhanced when the longer and more flexible linker 2 was used on the full length peptide. The enhancement was dependent on the actual construct and RNA targets and the conjugates displayed increased binding specificity. For example, an acridine-full length peptide conjugate showed 0.015 nM affinity to the 17mer boxB RNA with 2AP-4 labeling when extrapolated from high salt measurement to 50 mM K+ (Table 2, Figure 2), corresponding to a 80-fold enhancement over the wild type full length peptide (1.2 nM). The longer and more flexible linker seemed to confer affinity enhancement. Full length acridine peptide conjugates constructed with the shorter and more rigid linker 1 exhibited less binding enhancement; therefore, further investigations were focused on the linker 2. In line with this view, we also observed specificity enhancement (~25-fold) determined by comparing enhanced binding affinity of the acridine-linker 2-λ N22 peptide conjugate to a cognate λ boxB RNA (80-fold) with a modest enhancement (3.2-fold by comparing 257 nM with 80 nM) to a noncognate P22 RNA hairpin.
Table 1.
Dissociation constants (Kd) for peptides and acridine-peptide conjugates against boxBR RNAa
| λ boxBR RNA targets (17mers) | P22 BoxB RNA | |||
|---|---|---|---|---|
| λ-2AP-2 | λ-2AP-3 | λ-2AP-4 | 2AP-2 | |
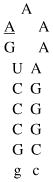 |
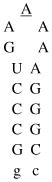 |
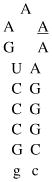 |
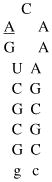 |
|
| Peptide | Kd (nM) | Kd (nM) | Kd (nM) | Kd (nM) |
| λN11 | 1126 | 1480 | 1000 | |
| Acr-Link1-λN11 | 3945 | 2304 | 3504 | |
| Acr-Link2-λN11 | 3082 | 2200 | 3245 | |
| λN22 | 1.9 | 1.0 | 1.2 | 257 |
| Acr-Link1-λN22 | 2.7 | 1.3 | 2.0 | |
| Acr-Link2-λN22 | 0.025b | 0.019b | 0.015b | 80 |
Binding constants were determined for standard condition: 20°C; 50 mM KOAc, 20 mM Tris.OAc, pH 7.5. Individual isotherms were fit to a one-step reaction with less than 10% error. Hairpin base positions substituted with 2AP are underlined. Acr- refers to acridine moiety.
Table 2.
Salt dependence*
| λ boxBR RNA target λ-2AP-4(17mer) | ||||||||
|---|---|---|---|---|---|---|---|---|
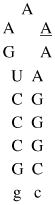 |
||||||||
| Total monovalent cation concentration (mM) | ||||||||
| Peptide | 70 | 95 | 120 | 175 | 220 | 320 | 420 | 620 |
| Kd (nM) | ||||||||
| λN22 | 1.2 | 1.9 | 4.2 | 8.5 | 19.8 | 43.5 | ||
| Acr-Link2-λN22 | 0.5 | 1.1 | 2.1 | 10 | ||||
Total monovalent cation concentration is sum of K+ and Tris. Binding constants were determined for standard condition: 20°C; 50–600 mM KOAc, 20 mM Tris.OAc, pH 7.5. Individual isotherms were fit to a one-step reaction with less than 10% error. Hairpin base positions substituted with 2AP are underlined. Acr- refers to acridine moiety.
Figure 2.

Salt dependence of λ full length peptide (square) and acridine-full length peptide conjugate (triangle) binding to 17mer λ boxB RNA (2AP-4). Measurements were made at 20 °C.
Salt dependence of binding
In general, protein-nucleic acid interactions become weaker with increasing salt concentration due to the electrostatic nature of binding. This principle likely also applies to our system. Since the N peptide is arginine-rich, the electrostatic interactions between positively charged peptide side chains and negatively charged RNA phosphate backbone are critical for binding (29). Consequently, these electrostatic interactions are mitigated by increasing concentration of salt. Within a range of cation concentrations, plots of log (Kd) vs. log [M+] give a linear relationship. The slope of these plots corresponds to the number of counterions that are released upon peptide binding. In general, the slope is related to the net charge of the peptide and ranges from 2.5 to 5 for the N peptide sequences that have been tested (34). Representative salt-dependent curves of dissociation constants for the acridine full length N peptide conjugate–boxB RNA complex were shown using the 17mer RNA with 2AP-4 labeling (Table 2, Figure 2). Most of the isothermal curves showed biphasic transitions with the second transition starting after peptide conjugate reaches 1:1 stoichiometry with the RNA target. The Kd values were fit to the first transition. Indeed, higher salt decreased the binding affinity of acridine-peptide conjugate to boxB RNA as observed for the WT peptide. The salt dependence graph showed linear relationship at higher salt concentrations (Figure 2) as observed previously (39–40). The derived slope value of 2.9 was slightly higher than the value of 2.4 for the wild type N peptide, indicating roughly three cations were released upon binding. The Kd value extrapolated by salt dependence curve to 50 mM K+, 20mM Tris was 0.015 nM. A roughly 80–fold binding enhancement was observed across a broad range of ionic concentrations. Since the conjugate showed slightly higher slope of salt dependence than the free peptide, the enhancement was even higher when extrapolated to lower salt concentrations.
CD Spectra
The CD spectra collected for boxB RNA binding by the acridine-linker 2-λ N22 peptide conjugate and free full length λ N22 peptide are shown in Figure 3. Neither the peptide nor the acridine-peptide conjugate showed any appreciable structure in the absence of RNA. The difference spectra of the two complexes indicated that both peptides folded into α helices when bound to the RNA. Although globally similar, the two complexes displayed specific differences in regions indicative of peptide folding (200–225nm) and RNA folding (260–300nm). The acridine conjugate/RNA complex appeared less α-helical.
Figure 3.

CD spectra comparison of free peptides and peptide/boxB RNA complexes.
NMR Spectroscopy
NMR experiments can provide useful structural information on the boxB RNA and the N peptide, especially the structural change upon complex formation. In particular, imino protons from RNA base pairs in the stem and Trp18 indole NH proton produce unique and easily detectable signals that are easy to monitor. NMR spectra were collected in order to distinguish the boxB RNA binding between the acridine-peptide conjugate and the free peptide (Figure 4).
Figure 4.

One dimensional NMR spectra for peptide, boxB RNA, and complexes. A. Comparison of λN11 and λN22 peptide/boxB complexes. a: free 15-mer boxB RNA; b: λN11 peptide/boxB complex; c: acridine-λN11 peptide conjugate/boxB complex; d: λN22 peptide or acridine-λN22 peptide conjugate alone; e: λN22 peptide/boxB complex; f: acridine-λN22 peptide conjugate/boxB complex. B. Comparison of 1:1 peptide/boxB complex and 2:1 peptide/boxB complex. a: 1:1 λN22 peptide/boxB complex; b: 2:1 λN22 peptide/boxB complex; c: 1:1 acridine-λN22 peptide conjugate/boxB complex; d: 2:1 acridine-λN22 peptide conjugate/boxB complex. The asterisk indicates the additional peak.
The free 15-mer boxB RNA (Figure 4A, a) has 5 base pairs in the stem. The 1D imino proton NMR spectrum showed 3 peaks (13.25 ppm, 12.65 ppm, and 12.55 ppm) corresponding to the imino protons of G12, G13, and G14 from the three middle CG base pairs. The terminal GC pair was not observable due to fraying, and the U imino proton from loop closing UA pair was also missing, presumably due to the flexibility of the unstructured pentaloop in the free RNA. Binding by either full length N peptide or truncated amino-terminal 11-mer induced similar RNA structural changes, evidenced by shifting of the three CG imino protons to higher field (Figure 4A, b and e). Beside these changes, there were additional imino protons at 13.5 ppm corresponding to U5 of the UA pair, and a broader peak at 10.7 ppm attributable to the G6 imino proton from the sheared GA base pair characteristic of GNRA tetraloop type of folding. This indicates that the loop has been stabilized by peptide binding. In addition, the Trp18 indole NH proton in the full length peptide has a large shift from 10 ppm (Figure 4A, d) in the free peptide to 9.2 ppm in the complex (Figure 4A, e), which is due to the stacking interaction between Trp18 and the RNA loop (30).
In comparison, the spectrum for the acridine-linker 2-λN11 conjugate bound to the boxB RNA (Figure 4A, c) did not show these characteristic signal changes as observed in λN11 peptide/RNA complex, except for the broadening of the original three peaks. This indicated that the acridine-λN11 conjugate did not bind the RNA in the same mode as the λN11 peptide. Since acridine compound 3-acridin-9-ylamino-propanol was found to bind to the boxB RNA with about 15 μM affinity, similar to the affinity between the λN11 peptide and the boxB RNA, it is possible that the two modules compete for the RNA binding site, and the acridine moiety may associate with the RNA non-specifically, thus disrupting the specific complex structure between the λN11 peptide and the boxB RNA. The imino protons of U5 and G6 are therefore broadened due to the heterogeneity of the complex, as observed in other cases (41).
The spectrum for 1:1 acridine-linker 2-full length peptide conjugate/boxB RNA complex (Figure 4A, f) showed very similar imino proton signals for RNA component and characteristic Trp18 indole proton shift as seen in the spectrum for λN22 peptide complex. Within the acridine-full length peptide conjugate, the peptide portion appeared to bind the RNA in the same mode as in the WT λN22 peptide-RNA complex, thus delivering the acridine moiety to the lower part of boxB RNA stem.
Since the acridine-full length peptide conjugate showed biphasic binding to the boxB RNA, we further investigated the potential second binding site on the boxB RNA for the conjugate. Over titration of the conjugate produced a 2:1 ratio of conjugate/RNA complex. Comparison of the spectra for 1:1 and 2:1 complexes of both WT peptide and the acridine-peptide conjugate revealed interesting discrepancies (Figure 4B). The imino protons for U5 moved further down field, and G6 and G14 moved up field, respectively; furthermore, there was an additional peak at 12.4 ppm. These features suggest a specific second binding site for the acridine-peptide conjugate.
Band Shift Analysis
Observations from NMR experiments were consistent with the band shift analysis shown in Figure 5. A band shift was observed indicative of specific binding of the boxB RNA by the λN11 peptide (lane 2 of Figure 5A). On the other hand, the smear for the acridine-linker 2-λN11 peptide conjugate with the boxB RNA (lane 3 of Figure 5A) indicated non-specific interaction, possibly random intercalation of acridine either in the stem or in the loop region which compete with the binding of peptide portion.
Figure 5.

Gel shift analysis of peptide/boxB complexes for WT peptides and acridine-peptide conjugates. A. λN11/RNA and acridine-λN11/RNA complexes. B. λN22/RNA and acridine-λ N22/RNA complexes.
The acridine-linker 2-λN22 full length conjugate, however, not only showed a 1:1 complex to the boxB RNA 15-mer (lane 3 of Figure 5B) as in the peptide-RNA complex (lane 2 of Figure 5B), but a higher stoichiometric complex as well. These results, likely suggestive of a second binding site for the acridine-λN22 peptide conjugate, may explain the profile of the fluorescence titration curves, which sometimes show biphasic transitions with the second transition apparently following equimolar titration.
Discussions
To achieve higher RNA binding affinities, the design of a ligand consisting of two moieties connected by a linker is a promising approach (8–9). Higher binding affinities could result from a favorable entropic factor compared with the binding of the two monomeric counterparts. Under certain conditions, the length and nature of the linker is critical. The molecule can be a dimeric form of a known binder (42) or consist of two distinct moieties that bind to RNA in different modes, e.g., groove binding and intercalation (15–18). To apply this idea to the N peptide system, a series of λ N peptide-based peptide-acridine conjugates were designed and synthesized. Their binding affinities to boxB RNAs with stem-loop hairpin structures were determined by steady-state fluorescence measurements.
Binding affinity and specificity enhancement
Acridine compound 3-acridin-9-ylamino-propanol binds the boxB RNA with 15 μM affinity, potentially conferring significant enhancement when linked to another binding module. From comparing salt dependence of binding, our acridine-linker 2-λN22 peptide conjugation showed 80-fold binding enhancement over a broad range of salt concentrations. This result is similar to binding enhancement observed for a neo-acridine construct over neomycin B alone binding to a RRE RNA (18). This observed binding enhancement was unique to the full length conjugate with a more flexible ethylene glycol linker tested, implying that the structural context of acridine presentation was critical. The same construct showed only 3-fold tighter binding to a related P22 boxB RNA target. Compared to λ boxB, P22 RNA displays flipped orientation for one base pair in the stem, and has a cytidine in the loop instead of adenine (Table 1). Homologous hydrophobic interactions occur between the boxB hairpin stem and conserved alanine residues within the N peptide amino-terminal module. Distinct hydrophobic interactions are involved between the boxB hairpin loops and carboxy-terminal modules of the N peptides. In λ, a tryptophan residue stacks on the boxB loop; in P22, non-polar alanine and isoleucine residues interact with an extruded pyrimidine. In both phage λ and P22, the bound pentaloops adopt stable GNRA tetraloop folds by extruding either loop base 4 (4-out) in the λ complex, or loop base 3 (3-out) in the P22 complex. Both λN22 and P22N21 full-length peptides discriminate strongly between their cognate RNA hairpins and other boxB targets (34, 38). Our acridine-linker 2-λN22 peptide conjugate showed 25-fold binding specificity enhancement between these two similar RNA structures.
In contrast, the acridine to the λN11 peptide conjugate displayed 3-fold weaker binding than the λN11 peptide alone. The NMR and gel shift data showed that the conjugate did not form specific complex with the boxB RNA (Figure 4A and 5A), presumably due to the fact that the non-specific binding of the acridine moiety to the boxB RNA showed similar affinity compared to the the λN11 peptide. Therefore, the acridine moiety competed with peptide moiety for RNA binding sites. Alternatively, it is also possible that in the acridine-λN11 conjugate, non-specific electrostatic attraction by Arg residues in the peptide to the RNA become important as the origin of low binding affinity of acridine-λN11 conjugate. This result suggests that when designing a ligand with multiple binding modules, it is important to avoid a non-specific binding module that can compete for binding to multiple sites intended for other modules. Furthermore, a module with higher affinity for one site will help anchor the entire ligand. Taken together, our observations indicate that the specific binding peptide module of the full length acridine-peptide conjugate exhibits much higher affinity for the λboxB RNA, thereby maintaining the native contacts with RNA (Figure 4A) and facilitating delivery of the acridine moiety to the intended site.
Acridine intercalating site
Previous structural studies on DNA intercalated with acridine related compounds have consistently shown intercalation sites at alternating base steps, particularly at the terminal base steps (43). This is likely due to the fact that terminal base steps have much greater conformational freedom than internal base steps, thus more receptive to intercalation. Recent studies on DACA, an antitumor acridine derivatized with variable functional substitutions at position 4, showed that they intercalate into DNA with side chains positioned in the major groove (44–46). However, the crystal structure of an acridine-tetraarginine conjugate with the peptide attached to position 9 showed intercalation of acridine into the minor groove of central AA/TT base step of a long DNA fragment, leaving the peptide in the minor groove (47).
In these cases, it appeared that specific hydrogen bonding interactions between the lateral chain of acridine and DNA may affect how the acridine moiety intercalates into nucleic acid. Interestingly, a recent study also showed that in peptide-acridine conjugates, the point of attachment on acridine affected the conjugates’ affinity for the nucleic acid target (11).
Our acridine-N peptide conjugates feature attachment of the peptide to acridine at position 9, similar to the acridine-tetraarginine case. The binding mode of acridine in these conjugates is currently unknown. If the acridine moiety derivatized at position 9 with a peptide prefers intercalation into the minor groove as in the case of acridine-tetraarginine (47), our construct may preclude intercalation because the full length N peptide binds to the boxB RNA in the major groove with high affinity. With respect to the acridine-λ N11 conjugate, the acridine may be able to intercalate into the minor groove; however, such intercalation is probably not site-specific. Alternatively, binding of the N peptide, especially the full length version, to the boxB RNA peptide may induce RNA folding. There are extensive interactions between the peptide and the RNA stem, some of which may be disrupted by an intercalated acridine moiety in the stem. Although the acridine may still be able to intercalate at the terminal base step, the inability to directly observe the imino proton from the terminal base pair precludes direct confirmation of this possibility.
A second specific binding site for the acridine-N peptide conjugate on the boxB RNA
Similar to the neo-acridine case (18) which involved a second binding site, our fluorescence, NMR, and gel shift data also suggested a second specific binding site for the acridine-N peptide conjugate on the boxB RNA. The nature of the second site is not known, but may involve either the major or the minor groove. Changes of the imino proton chemical shifts were observed throughout the RNA structure (Figure 4B and 5), suggesting extensive interactions.
Alternative approaches of enhancing RNA binding affinity are also available. N peptides bind to the boxB RNA in an -helical conformation but exhibit little helical structure when free in solution (31). Stabilizing the helical form of such peptides is expected to favor RNA binding by virtue of pre-organization. For example, Cα-substituted amino acids has long been recognized as a means of introducing local conformational restriction in peptides (48). Another approach to stabilize the α-helical form of peptides is through the incorporation of covalent linkages between constituent amino acid side chains. It was found that substantial helix stabilization was achieved when the linkage was placed between the i and i+4 residue in the peptide backbone by ring closing metathesis (49–52). Therefore, cyclic helical N peptides wherein ring closing metathesis is used to incorporate a carbon-carbon tether between appropriate residue side chains is expected to enhance binding. In line with this view, our analyses revealed that the full-length peptide-acridine conjugate had substantially improved RNA-binding affinity (~80-fold) and specificity (~25-fold) relative to the peptide alone. Our work supports the idea that peptide- and intercalation-based binding can be combined to create a new class of high-affinity, high-specificity RNA-binding ligands. This approach provides straightforward optimization to enhance the affinity of known modules and facilitate the discovery of powerful new hybrid ligands with novel functionalities.
Acknowledgments
The authors thank Drs. T. T. Takahashi and Ryan J. Austin for helpful discussions.
This research is financially supported by NIH grant GM60416 (R.W.R.).
Abbreviations
- TAR
trans-activating response region
- Tat
trans-activators of transcription
- Nut
N-utilization
- 2-AP
2-aminopurine
References
- 1.Klug A. Zinc finger peptides for the regulation of gene expression. J Mol Biol. 1999;293:215–218. doi: 10.1006/jmbi.1999.3007. [DOI] [PubMed] [Google Scholar]
- 2.Segal DJ, Barbas CF., 3rd Design of novel sequence-specific DNA-binding proteins. Curr Opin Chem Biol. 2000;4:34–39. doi: 10.1016/s1367-5931(99)00048-4. [DOI] [PubMed] [Google Scholar]
- 3.Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 4.White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB. Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands. Nature. 1998;391:468–471. doi: 10.1038/35106. [DOI] [PubMed] [Google Scholar]
- 5.Dervan PB. Molecular recognition of DNA by small molecules. Bioorg Med Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 6.Fechter EJ, Dervan PB. Allosteric inhibition of protein--DNA complexes by polyamide--intercalator conjugates. J Am Chem Soc. 2003;125:8476–8485. doi: 10.1021/ja030125e. [DOI] [PubMed] [Google Scholar]
- 7.Fechter EJ, Olenyuk B, Dervan PB. Design of a sequence-specific DNA bisintercalator. Angew Chem Int Ed Engl. 2004;43:3591–3594. doi: 10.1002/anie.200454231. [DOI] [PubMed] [Google Scholar]
- 8.Michael K, Tor Y. Designing novel RNA binders. Chemistry-a European Journal. 1998;4:2091–2098. [Google Scholar]
- 9.Cheng AC, Calabro V, Frankel AD. Design of RNA-binding proteins and ligands. Curr Opin Struct Biol. 2001;11:478–484. doi: 10.1016/s0959-440x(00)00236-0. [DOI] [PubMed] [Google Scholar]
- 10.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. Rational Design of Ligands Targeting Triplet Repeating Transcripts That Cause RNA Dominant Disease: Application to Myotonic Muscular Dystrophy Type 1 and Spinocerebellar Ataxia Type 3. J Am Chem Soc. 2009;131:9767–9779. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson CB, Beal PA. Point of attachment and sequence of immobilized Peptide-acridine conjugates control affinity for nucleic acids. J Am Chem Soc. 2002;124:8510–8511. doi: 10.1021/ja026029f. [DOI] [PubMed] [Google Scholar]
- 12.Carlson CB, Spanggord RJ, Beal PA. Selection of small-molecule mediators of the RNA regulation of PKR, the RNA-dependent protein kinase. Chembiochem. 2002;3:859–865. doi: 10.1002/1439-7633(20020902)3:9<859::AID-CBIC859>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 13.Carlson CB, Vuyisich M, Gooch BD, Beal PA. Preferred RNA binding sites for a threading intercalator revealed by in vitro evolution. Chem Biol. 2003;10:663–672. doi: 10.1016/s1074-5521(03)00147-9. [DOI] [PubMed] [Google Scholar]
- 14.Gooch BD, Beal PA. Recognition of duplex RNA by helix-threading peptides. J Am Chem Soc. 2004;126:10603–10610. doi: 10.1021/ja047818v. [DOI] [PubMed] [Google Scholar]
- 15.Hamy F, Brondani V, Florsheimer A, Stark W, Blommers MJ, Klimkait T. A new class of HIV-1 Tat antagonist acting through Tat-TAR inhibition. Biochemistry. 1998;37:5086–5095. doi: 10.1021/bi972947s. [DOI] [PubMed] [Google Scholar]
- 16.Wilson WD, Ratmeyer L, Cegla MT, Spychala J, Boykin D, Demeunynck M, Lhomme J, Krishnan G, Kennedy D, Vinayak R, Zon G. Bulged-Base nucleic acids as potential targets for antiviral drug action. New J Chem. 1994;18:419–423. [Google Scholar]
- 17.Wilson WD, Mizan S, Tanious FA, Yao S, Zon G. The interaction of intercalators and groove-binding agents with DNA triple-helical structures: the influence of ligand structure, DNA backbone modifications and sequence. J Mol Recognit. 1994;7:89–98. doi: 10.1002/jmr.300070206. [DOI] [PubMed] [Google Scholar]
- 18.Kirk SR, Luedtke NW, Tor Y. Neomycin-acridine conjugate: A potent inhibitor of Rev-RRE binding. J Am Chem Soc. 2000;122:980–981. [Google Scholar]
- 19.Tung CH, Wei Z, Leibowitz MJ, Stein S. Design of peptide-acridine mimics of ribonuclease activity. Proc Natl Acad Sci U S A. 1992;89:7114–7118. doi: 10.1073/pnas.89.15.7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fkyerat A, Demeunynck M, Constant J-F, Michon P, Lhomme J. A New Class of Artificial Nucleases That Recognize and Cleave Apurinic Sites in DNA with Great Selectivity and Efficiency. J Am Chem Soc. 1993;115:9952–9959. [Google Scholar]
- 21.Kuzuya A, Mizoguchi R, Morisawa F, Machida K, Komiyama M. Metal ion-induced site-selective RNA hydrolysis by use of acridine-bearing oligonucleotide as cofactor. J Am Chem Soc. 2002;124:6887–6894. doi: 10.1021/ja025653p. [DOI] [PubMed] [Google Scholar]
- 22.Greenblatt J, Nodwell JR, Mason SW. Transcriptional antitermination. Nature. 1993;364:401–406. doi: 10.1038/364401a0. [DOI] [PubMed] [Google Scholar]
- 23.Das A. Control of transcription termination by RNA-binding proteins. Annu Rev Biochem. 1993;62:893–930. doi: 10.1146/annurev.bi.62.070193.004333. [DOI] [PubMed] [Google Scholar]
- 24.Salstrom JS, Szybalski W. Coliphage lambdanutL-: a unique class of mutants defective in the site of gene N product utilization for antitermination of leftward transcription. J Mol Biol. 1978;124:195–221. doi: 10.1016/0022-2836(78)90156-0. [DOI] [PubMed] [Google Scholar]
- 25.Olson ER, Tomich CS, Friedman DI. The nusA recognition site. Alteration in its sequence or position relative to upstream translation interferes with the action of the N antitermination function of phage lambda. J Mol Biol. 1984;180:1053–1063. doi: 10.1016/0022-2836(84)90270-5. [DOI] [PubMed] [Google Scholar]
- 26.Lazinski D, Grzadzielska E, Das A. Sequence-specific recognition of RNA hairpins by bacteriophage antiterminators requires a conserved arginine-rich motif. Cell. 1989;59:207–218. doi: 10.1016/0092-8674(89)90882-9. [DOI] [PubMed] [Google Scholar]
- 27.Barrick JE, Takahashi TT, Ren J, Xia T, Roberts RW. Large libraries reveal diverse solutions to an RNA recognition problem. Proc Natl Acad Sci U S A. 2001;98:12374–12378. doi: 10.1073/pnas.221467798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heus HA, Pardi A. Structural features that give rise to the unusual stability of RNA hairpins containing GNRA loops. Science. 1991;253:191–194. doi: 10.1126/science.1712983. [DOI] [PubMed] [Google Scholar]
- 29.Legault P, Li J, Mogridge J, Kay LE, Greenblatt J. NMR structure of the bacteriophage lambda N peptide/boxB RNA complex: recognition of a GNRA fold by an arginine-rich motif. Cell. 1998;93:289–299. doi: 10.1016/s0092-8674(00)81579-2. [DOI] [PubMed] [Google Scholar]
- 30.Scharpf M, Sticht H, Schweimer K, Boehm M, Hoffmann S, Rosch P. Antitermination in bacteriophage lambda. The structure of the N36 peptide-boxB RNA complex. Eur J Biochem. 2000;267:2397–2408. doi: 10.1046/j.1432-1327.2000.01251.x. [DOI] [PubMed] [Google Scholar]
- 31.Su L, Radek JT, Hallenga K, Hermanto P, Chan G, Labeots LA, Weiss MA. RNA recognition by a bent alpha-helix regulates transcriptional antitermination in phage lambda. Biochemistry. 1997;36:12722–12732. doi: 10.1021/bi971408k. [DOI] [PubMed] [Google Scholar]
- 32.Lacourciere KA, Stivers JT, Marino JP. Mechanism of neomycin and Rev peptide binding to the Rev responsive element of HIV-1 as determined by fluorescence and NMR spectroscopy. Biochemistry. 2000;39:5630–5641. doi: 10.1021/bi992932p. [DOI] [PubMed] [Google Scholar]
- 33.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 34.Austin RJ, Xia T, Ren J, Takahashi TT, Roberts RW. Differential modes of recognition in N peptide-boxB complexes. Biochemistry. 2003;42:14957–14967. doi: 10.1021/bi0351312. [DOI] [PubMed] [Google Scholar]
- 35.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menger M, Eckstein F, Porschke D. Dynamics of the RNA hairpin GNRA tetraloop. Biochemistry. 2000;39:4500–4507. doi: 10.1021/bi992297n. [DOI] [PubMed] [Google Scholar]
- 37.Rachofsky EL, Osman R, Ross JB. Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment on fluorescence. Biochemistry. 2001;40:946–956. doi: 10.1021/bi001664o. [DOI] [PubMed] [Google Scholar]
- 38.Austin RJ, Xia T, Ren J, Takahashi TT, Roberts RW. Designed arginine-rich RNA-binding peptides with picomolar affinity. J Am Chem Soc. 2002;124:10966–10967. doi: 10.1021/ja026610b. [DOI] [PubMed] [Google Scholar]
- 39.Record MT, Jr, Lohman ML, De Haseth P. Ion effects on ligand-nucleic acid interactions. J Mol Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 40.Record MT, Jr, Ha JH, Fisher MA. Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol. 1991;208:291–343. doi: 10.1016/0076-6879(91)08018-d. [DOI] [PubMed] [Google Scholar]
- 41.Robertson SA, Harada K, Frankel AD, Wemmer DE. Structure determination and binding kinetics of a DNA aptamer - Argininamide complex. Biochemistry. 2000;39:946–954. doi: 10.1021/bi9915061. [DOI] [PubMed] [Google Scholar]
- 42.Campisi DM, Calabro V, Frankel AD. Structure-based design of a dimeric RNA-peptide complex. Embo J. 2001;20:178–186. doi: 10.1093/emboj/20.1.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams LD, Egli M, Gao Q, Rich A, editors. Nucleic Acids. I. Adenine Press; Schenectady, NY: 1992. [Google Scholar]
- 44.Todd AK, Adams A, Thorpe JH, Denny WA, Wakelin LP, Cardin CJ. Major groove binding and ‘DNA-induced’ fit in the intercalation of a derivative of the mixed topoisomerase I/II poison N-(2-(dimethylamino)ethyl)acridine-4-carboxamide (DACA) into DNA: X-ray structure complexed to d(CG(5-BrU)ACG)2 at 1.3-A resolution. J Med Chem. 1999;42:536–540. doi: 10.1021/jm980479u. [DOI] [PubMed] [Google Scholar]
- 45.Adams A, Guss JM, Denny WA, Wakelin LP. Crystal structure of 9-amino-N-[2-(4-morpholinyl)ethyl]-4-acridinecarboxamide bound to d(CGTACG)2: implications for structure-activity relationships of acridinecarboxamide topoisomerase poisons. Nucleic Acids Res. 2002;30:719–725. doi: 10.1093/nar/30.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams A, Guss JM, Collyer CA, Denny WA, Wakelin LP. Crystal structure of the topoisomerase II poison 9-amino-[N-(2-dimethylamino)ethyl]acridine-4-carboxamide bound to the DNA hexanucleotide d(CGTACG)2. Biochemistry. 1999;38:9221–9233. doi: 10.1021/bi990352m. [DOI] [PubMed] [Google Scholar]
- 47.Malinina L, Soler-Lopez M, Aymami J, Subirana JA. Intercalation of an Acridine-Peptide Drug in an AA/TT Base Step in the Crystal Structure of [d(CGCGAATTCGCG)](2) with Six Duplexes and Seven Mg(2+) Ions in the Asymmetric Unit. Biochemistry. 2002;41:9341–9348. doi: 10.1021/bi020135c. [DOI] [PubMed] [Google Scholar]
- 48.Liff MI, Kopple KD, Tian Z, Roeske RW. Effects of C alpha-methyl substitution on the conformation of linear GnRH antagonist analogs. Int J Pept Protein Res. 1994;43:471–476. doi: 10.1111/j.1399-3011.1994.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 49.Miller SJ, Grubbs RH. Synthesis of Conformationally Restricted AminoAcids and Peptides Employing Olefin Metathesis. J Am Chem Soc. 1995;117:5855–5856. [Google Scholar]
- 50.Lynn DM, Mohr B, Grubbs RH. Living ring-opening metathesis polymerization in water. J Am Chem Soc. 1998;120:1627–1628. [Google Scholar]
- 51.Blackwell HE, Grubbs RH. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Ange Chem-Int Ed. 1998;37:3281–3284. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 52.Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]