

Abstract
The unique amino acid hypusine is formed exclusively in eIF5A by the successive action of deoxyhypusine synthase and deoxyhypusine hydroxylase (yeast Lia1, human DOHH). Although the first enzyme has been extensively studied, both Lia1 structure and the mechanism of action remain unclear. Hence, a multi-approach was used to evaluate Lia1 catalysis, metal/substrate binding, structural conformation and stability. Mutational analyses of Lia1 revealed fine differences in the mode of substrate binding between the human and yeast counterparts. Like human DOHH, recombinant Lia1 is an iron metalloenzyme. Iron is essential for enzyme activity since its loss renders the enzyme totally inactive. The separation of iron-free and iron-bound forms by gel filtration and native electrophoresis suggests differences in Lia1 tertiary structure related to the iron binding. The ability of Lia1 to undergo conformational changes prompted us to use a set of complementary spectroscopic approaches and SAXS to obtain detailed information on the processes underlying dissociation of iron from Lia1 at different levels of the protein organization. The additive effect of weak interactions, especially within the metal center, resulted in an active enzyme in a stabilized and compact three-dimensional fold. Loss of tertiary contacts upon iron displacement led to an elongated conformation of Lia1, in which the N- and C-terminal domains are no longer in close proximity to guarantee the proper orientation of the active groups within the active site pocket. Our results demonstrate an essential structural role for iron binding in addition to its contribution to the catalysis of hypusine formation in the eIF-5A precursor.
Keywords: eIF5A Lia1, Hypusine, Deoxyhypusine hydroxylase, HEAT-repeat containing protein, Iron metalloenzyme, Structural analysis
Introduction
Hypusine biosynthesis is a remarkable post-translational modification known to date to occur in only one single cellular protein, the putative eukaryotic translation initiation factor 5A (Cooper et al. 1983; Park et al. 1981). The unusual amino acid hypusine is generated in a two-step reaction process catalyzed consecutively by deoxyhypusine synthase (yeast Dys1/human DHS) and deoxyhypusine hydroxylase (yeast Lia1/human DOHH) (Wolff et al. 2007). The first enzyme mediates the NAD-dependent transfer of the 4-aminobutyl moiety of spermidine to the ε-amino group of one specific lysine residue (K51 in yeast protein) in the eIF5A precursor. Hydroxylation of the resulting deoxyhypusine intermediate by the second enzyme of the pathway leads to the formation of hypusine (Park 2006; Park et al. 1982). Hydroxylation not only completes hypusine biosynthesis but also eIF5A maturation, thus rendering the protein biologically active (Abbruzzese et al. 1986; Park 2006).
The hypusine residue exerts a critical role for the protein function in vivo. Disruption of deoxyhypusine synthase gene (DYS1), as well as the single mutation of the lysine at the site of hypusine formation in the eIF5A precursor, results in loss of cell viability in the yeast Saccharomyces cerevisiae (Sasaki et al. 1996; Schnier et al. 1991). Besides, an increase of cells arrested in G1 is observed upon rapid depletion of eIF5A, or shift of conditional eIF5A mutants to the restrictive temperature (Kang and Hershey 1994; Zanelli and Valentini 2005). Despite the clear correlation observed between the inhibition of hypusine formation and arrest of cell-cycle progression in yeast (Kang and Hershey 1994; Zanelli and Valentini 2005) and in mammalian cells treated with inhibitors of DOHH (Hanauske-Abel et al. 1994), the biological function of this intriguing protein still remains to be elucidated. Recently, the hypusine-dependent association of eIF5A with ribosomes actively engaged in translation has brought eIF5A back to the translation scenario (Jao and Chen 2006; Zanelli et al. 2006). Furthermore, a defect observed in polysomal profiles of temperature-sensitive mutants supports a role for eIF5A in the elongation step of translation instead of initiation (Zanelli et al. 2006; Zanelli and Valentini 2007). Finally, a defect in protein synthesis was detected in yeast cells expressing mutated forms of human or yeast eIF5A as the primary consequence of loss of protein function (Cano et al. 2008; Dias et al. 2008).
In an attempt to identify eIF5A-interacting proteins that would help us to understand its role in the cell, a two-hybrid screen was performed, and the protein encoded by the gene YJR070C, named LIA1 (Ligand of eIF5A), was identified as an eIF5A cellular partner in S. cerevisiae (Thompson et al. 2003). It was subsequently shown that the gene LIA1 encodes the enzyme responsible for the final step of hypusination, deoxyhypusine hydroxylase (Park et al. 2006).
Prediction studies of its secondary structure have revealed that Lia1 contains motifs known as HEAT-like repeats (Park et al. 2006; Thompson et al. 2003), which are found in a variety of proteins, including the four proteins Huntingtin, Elongation factor 3, the PR65/A subunit of protein phosphatase 2A and TOR1, from which “HEAT” is derived. The consensus secondary structure for an individual HEAT motif consists of a pair of anti-parallel α-helices separated by a non-helical region. Normally, HEAT repeats appear in tandem arrays to form an elongated molecule characterized by a double layer of α-helices. Interestingly, a role for mediating protein–protein interactions has been suggested for these repeats (Groves et al. 1999). Structural modeling predicts that the human homologue protein DOHH consists of eight HEAT repeats in a symmetrical dyad (four HEAT repeats in each of the N- and C- terminal arms connected by a variable loop). Two metal coordination sites have been identified at the four conserved His-Glu motifs of human DOHH (Kim et al. 2006; Park et al. 2006). Conservation of the HEAT repeat structures and the potential metal coordination sites suggests that, like human DOHH, Lia1 also possesses an iron-dependent catalytic activity.
Although the human DOHH has been characterized for its iron requirement and substrate binding through extensive mutagenesis studies (Kang et al. 2007; Kim et al. 2006), little is known about the structural aspects of Lia1 and its catalytic requirements. To better understand the secondary and tertiary structures of this distinct hydroxylase in relation to its iron binding and activity, we performed structural and functional analyses of the purified recombinant yeast Lia1, by employing in-depth spectroscopic analyses and small angle X-ray scattering (SAXS), in addition to site-directed mutagenesis to dissect the structural components of the Lia1 active site. The mutational analysis was carried out with ten proteins with ala-nine substitutions at highly conserved amino acid residues. In the spectroscopic analysis, we have exploited two structural characteristics of Lia1: the distribution of HEAT repeats along the protein, in order to follow circular dichroism signals, and the presence of a single tryptophan residue to evaluate its fluorescence properties. Finally, the conformational changes of Lia1 observed upon disruption of iron binding were analyzed by acrylamide quenching of tryptophan intrinsic fluorescence and by SAXS angle X-ray scattering (SAXS). Taken together, our data suggest fine differences in the coordination of iron and substrate at the active site between human DOHH and yeast Lia1 and support the essential contribution of iron for Lia1 hydroxylase activity as well as its tertiary structure by maintenance of its active conformation.
Experimental procedures
Purification of yeast recombinant wild type and mutant Lia1 proteins
E. coli BL21(DE3) cells harboring the plasmids expressing wild-type or mutant forms of Lia1 as GST fusion proteins were grown in 1 l of LB medium containing 100 μg/ml ampicillin. GST-Lia1 protein expression was induced when the culture reached 0.6 OD600nm by addition of 0.1 mM IPTG for 4 h at 37°C. Cells were harvested by centrifugation and suspended in 100 ml of ice-cold Tris buffer (50 mM Tris–HCl at pH 7.5 and 1 mM DTT) containing an EDTA-free protease inhibitor cocktail. Cells were lysed using an ultrasonic processor and debris was removed by centrifugation at 20,000×g at 4°C for 45 min. GST-Lia1 present in the clarified supernatant was then purified by affinity chromatography to a glutathione–Sepharose resin using a FPLC system (GE Healthcare). After thrombin cleavage of GST-Lia1, free GST protein was eliminated by a second chromatographic step using the same resin. The purified Lia1 protein was further subjected to molecular exclusion separation in a Superdex 200 gel filtration column. Fractions were grouped in pools and concentrated in centrifugal filter devices. Purified enzymes were then equilibrated in Tris buffer (50 mM Tris–HCl, pH 7.5, 1 mM DTT). A thrombin cleavage capture kit was used to release free mutant enzymes obtained from small-scale purifications.
Site-directed mutagenesis
Ten single mutants (H79A, E80A, H112A, E113A, E116A, E116D, H237A, E238A, H270A, E271A) of Lia1 from S. cerevisiae were generated at highly conserved amino acid residues using the QuickChange site-directed mutagenesis kit. pGEX-4T-1-LIA1 and pBTM116-LIA1 (Thompson et al. 2003), encoding yeast wild type Lia1 as a GST and LexA fusion protein, respectively, were used as template for the PCR with primer sets designed for the desired mutations. Each amino acid substitution was confirmed by DNA sequencing.
Circular dichroism spectroscopy
Circular dichroism measurements were carried out on a JASCO J-810 spectropolarimeter, equipped with a Peltier-type temperature controller and a thermostated cell holder, interfaced with a thermostatic bath. Far-UV spectra were recorded in a 0.1 cm path length quartz cell at a protein concentration of 0.15 mg/ml in 20 mM Tris–HCl pH 7.5. Five consecutive scans were accumulated and the average spectra stored. The data were corrected for the baseline contribution of the buffer and the observed ellipticities were converted into the mean residue ellipticities [θ] based on a mean molecular mass per residue of 112 Da. Thermal denaturation experiments were performed by increasing the temperature from 20 to 95°C at 1°/min, allowing temperature equilibration for 5 min before recording each spectrum. Tm represents the temperature at the midpoint of the unfolding transition. (GdnHCl)-induced denaturation curves at constant temperature were obtained by recording the CD signal at 222 nm for each sample.
Fluorescence spectroscopy
Steady-state fluorescence measurements were carried out in an Aminco BOWMAN series 2 spectrofluorometer. Excitation and emission bandwidths were 8 and 16 nm, respectively. Tryptophan fluorescence was measured with an excitation wavelength of 295 nm and the emission spectra were recorded between 310 and 400 nm. All measurements were performed in 20 mM Tris–HCl, pH 7.5 at 25°C and at a protein concentration of 0.15 mg/ml. Stern–Volmer quenching was performed using 295 nm excitation and 342 nm emission. Fluorescence intensities were corrected for dilution and contribution of the quenchers and plotted as the ratio of initial fluorescence intensity (Fo) to intensity (F) at each quencher concentration (Fo/F vs. [Q].) The slope determined by linear regression was the Stern–Volmer quenching constant, Ksv. Iodide stock solutions (KI 1 M) were made fresh before use, held on ice, and protected from light.
Calculation of the apparent fractional extent of unfolding
The observed CD or fluorescence signal at any given temperature or GdnHCl concentration was converted to apparent fractional extent of unfolding (Fapp) using the equation: Fapp = (Yobs − YU)/(YU − YN), where Yobs is the observed CD or fluorescence signal, and YN and YU refer to the native and unfolded states, respectively. The values of YN and YU were obtained by linear extrapolation of the temperature or perturbing agent dependence from the regions before and after the transition.
Small angle X-ray scattering
Small angle X-ray scattering experiments were performed at the D011A-SAXS1 beamline at the National Laboratory of Synchrotron Light (Campinas, Brazil). Data were collected using a MAR CCD 165 (MAR Research) area detector, and 10 min exposure time for each measurement at room temperature. The wavelength used in the experiments was 1.488 Å. The distance between the sample and detector was 1,074.35 mm. Scattered intensities were recorded in the angular range 0.0501 < q < 0.3085 Å−1, where q is the scattering vector (4πsinθ/λ), and 2θ and λ are the scattering angle and wavelength of the X-rays, respectively. Data were analyzed using the software package FIT2D. Intensity data were reduced to I(q) versus q using standard procedures to correct for incident beam intensity, sample absorption and blank subtraction. The radius of gyration, Rg, was calculated using the entire scattering curve with the program GNOM (Semenyuk and Svergun 1991), which also provides the distance distribution function P(r) and the maximum dimension of the scattering particles. The low-resolution model was calculated using the program DAMMIN in slow mode (Svergun 1999). In order to increase the reliability of the results, the final models for the dummy atom modeling were obtained by a spatial average of six independent models, calculated with the program DAMAVER (Volkov and Svergun 2003).
Results
Lia1 is an iron metalloenzyme
The yeast recombinant protein Lia1 purified by affinity chromatography on glutathione–Sepharose resin was subjected to size-exclusion chromatography in order to evaluate its purity. The elution profile of Lia1 during gel filtration on Superdex 200 column (Fig. 1a) revealed a small shoulder corresponding to the first four eluted fractions, in addition to a major peak corresponding to the last five fractions. All fractions showed a single band of approximately 36 kDa when analyzed by SDS-PAGE (Fig. 1b). However, when the same samples were evaluated by non-denaturing electrophoresis (Fig. 1c), two different bands were clearly distinguished, a diffuse upper band (dashed arrow), present in all fractions, and a better resolved band (solid arrow), which migrates faster, present only in the last five fractions (fractions 32–36). The fractions were pooled into three different groups: group 1, fractions 28–31, displaying mainly the upper diffuse band; group 2, fractions 32 and 33 with a mixture of the two observed bands; and group 3, fractions 34–36, composed mainly of the lower band.
Fig. 1.

Biochemical characterization of recombinant wild type Lia1 protein. a Elution profile of Lia1 recombinant protein during size exclusion chromatography on Superdex 200 column. b All fractions corresponding to the peak generated in the chromatographic step were evaluated by SDS-PAGE. c Electrophoresis under non-denaturing conditions. Wild-type enzyme resolved into two major species: a compact, fast moving band (solid arrow) and a second more diffuse, slow moving band (dashed arrow). d In vitro hydroxylase activity assay. The purified protein fractions were pooled into three different groups: Group 1 (28–31), Group 2 (32–33) and group 3 (34–36). Each pool was assayed for deoxyhypusine hydroxylase activity and enzyme activity is presented as the amount of hypusine formed relative to the total radioactivity (hypusine + deoxyhypusine). e Analysis of iron content of each pool by inductively coupled high-plasma resolution mass spectrometry. mAU milliabsorbance units, Hpu hypusine, Fr fractions
When Lia1 activity was measured in these three groups (Fig. 1d), the enzyme activity was markedly reduced (nearly no enzymatic activity) in group 1, and was moderate in group 2 (35% of substrate deoxyhypusine converted to hypusine). On the other hand, purified Lia1 from group 3 showed the highest activity with nearly 70% conversion of the substrate protein, eIF5A([3H]Dhp), to the hypusine form, eIF5A([3H]Hpu) (Fig. 1d). The enzyme activity was evaluated as herein (Park et al. 1984). eIF5A([3H]Dhp) was prepared as described previously (Park et al. 2003).
Analysis of metal content was then performed by inductively coupled plasma-high resolution mass spectrometry. Iron, out of nine metals analyzed (cadmium, cobalt, chromium, copper, iron, magnesium, manganese, nickel and zinc) was found to be the only metal associated with Lia1 in the samples. Metal contents were 0.1, 0.3 and 1.2 mol of iron per mol of enzyme, for pool 1, 2 and 3, respectively (Fig. 1e). The essential nature of iron for Lia1 activity is evident as loss of metal (in pool 1) results in a totally inactive enzyme. Taken together, these data suggest that the lower band of the enzyme upon native gel represents the metal-bound holoenzyme, whereas the diffuse upper band corresponds to the inactive iron-free apoenzyme. The presence of both the apoenzyme and the holoenzyme in groups 2 and 3 may be due to a gradual metal dissociation from Lia1 or due to its inability to obtain a full content of iron. In contrast to the human enzyme, no reconstitution of the holoenzyme from apoenzyme was observed with addition of 0.2 mM ferrous ammonium sulfate or ferric chloride (data not shown). Separation of the Lia1 apoenzyme and the holoenzyme by gel filtration and by native gel electrophoresis demonstrates that the apoenzyme has a larger hydrodynamic size than the holoenzyme.
Mapping amino acid residues critical for iron and/or substrate binding and catalysis
In order to identify amino acid residues of Lia1 active site that contribute to metal binding and catalysis, a mutational analysis was undertaken by site-directed mutagenesis. Target residues were selected at the four conserved His-Glu motifs (the predicted iron binding site) and an additional highly conserved residue E116. Thus, ten mutant proteins were generated: H79A, E80A, H112A, E113A, E116A, E116D, H237A, E238A, H270A, E271A. All the eight mutant proteins with alanine substitutions at the conserved His-Glu motifs were completely inactive in deoxyhypusine hydroxylation (Fig. 2a). Interestingly, while the mutant E116A was inactive, substitution at the same residue with aspartate rendered low levels of enzyme activity (3% of wild type). The metal content for six mutants (H79A, H112A, E113A, H237A, H270A and E271A) was very low (nearly zero), indicating that loss of iron binding contributes to enzyme inactivation. On the other hand, mutants E80A and E238A, despite the lack of enzymatic activity, displayed a relatively high metal content (0.6 and 1.2 mol/mol, respectively) and E116A and E116D, a modestly reduced level of iron (0.4 mol/mol).
Fig. 2.

Mutational and functional analyses of Lia1 protein. a In vitro hydroxylase activity assay of wild-type and mutant recombinant Lia1 proteins. b Evaluation of the migration pattern by electrophoresis under non-denaturing and denaturing conditions. c Mapping the amino acid residues critical for Lia1 binding to eIF5A. Tenfold serial dilutions of the two-hybrid reporter strain L40 containing both pBTM-LIA1, encoding wild-type or mutant proteins, and pACT-eIF5A were spotted onto −Leu/Trp (−LT) and −Leu/Trp/His (−LTH) plates and incubated at 30°C for 2 days. For β-galactosidase assay, 4 μL of cell suspension were dropped onto a nitrocellulose membrane laying on top of a −Leu/Trp plate and incubated 24 h at 30°C prior to the assay. The L40 strain transformed with the plasmids pBTM.LIA1/pACT (−), pBTM.TIF51A/pACT.LIA1 (+) or pBTM.TIF51A/pACT.DYS1 (++) was used as control
The activity and iron contents of the His-Glu site mutants of Lia1 are fairly similar to the pattern observed for the corresponding mutants of the human protein reported (Kang et al. 2007; Kim et al. 2006). Yet the involvement of the same six residues of the conserved His-Glu motifs (H79, H112, E113, H237, H270 and E271 for the yeast Lia1) in iron binding reveals the critical importance of these set of amino acid residues for Lia1 catalysis. However, analysis by native gel electrophoresis not only revealed some differences in mobility between the human mutant enzymes and the yeast counterparts, but also showed some disparities among the yeast mutants (Fig. 2b). The slow and diffused upper band of Lia1 wild type suggest heterogeneous population of apoenzymes with different tertiary structures and varying degrees of openness of the dyad arms. The fast moving tight band, in turn, suggests that the holoenzymes are homogeneous in compact structures. Although all the iron deficient mutants of human DOHH behaved like the human wild type apoenzyme upon native gel electrophoresis (Kim et al. 2006), some iron deficient Lia1 mutants did not. Lia1 mutants H79A, H112A and E113A resolved as a sharp band around the same position of the wild type apoenzyme (Fig. 2b), suggesting that these mutants have opened dyad arms, but are homogeneous in the degree of openness and tertiary structure. In contrast, E271A resolved as a much broader band than the apo form of the wild type Lia1, suggesting that two dyad arms are more flexible and free to move in this mutant. Curiously, the two other iron binding deficient mutants, H237A and H270A also exhibited a smear of apoenzyme, but an additional fast moving band of higher resolution could be detected (Fig. 2b), suggesting that a portion of this mutant still retains a compact holoenzyme-like structure. E116A/D displayed both apo and holo forms of the enzyme with reduced holoenzyme band consistent with its reduced iron content. These results demonstrate that the yeast Lia1 mutant proteins, in the absence of iron binding, display structural variations depending on the specific mutations.
Finally, to determine whether enzyme inactivity was a result of impaired substrate binding, the same Lia1 mutants were generated as LexA-fusion proteins. Expression of two-hybrid reporter genes, HIS3 and LacZ, was used to monitor the interaction between eIF5A fused to Gal4 activating domain and the mutant enzymes (Fig. 2c) (Thompson et al. 2003). E113A, E116A, E238A, H270A, E271A and E274A mutants did not grow in medium lacking histidine and also were negative for β-galactosidase assay, suggesting loss of interaction with eIF5A. On the other hand, the mutant proteins H79A, E80A, H112A, E116D, H237A and E274D were able to interact with eIF5A as they induced expression of the reporter genes and supported growth fully or partially in the absence of His. The finding that the E116D and E274D with Asp substitution are partially active in eIF5A binding whereas those with Ala substitutions are inactive, suggests the importance of these acidic residues of Lia1 in anchoring the substrate protein. Thus, the data reveal that Glu residues E113, E116, E238, E271 and E274 are critical for anchoring eIF5A in the enzyme structure.
Structural characterization of Lia1 by spectroscopic techniques
Prediction studies of Lia1 secondary structure revealed the presence of motifs known as HEAT repeats (Thompson et al. 2003). These motifs are individually composed of a pair of α-helices and seem to assemble in a repetitive fashion throughout the Lia1 protein. In this context, circular dichroism spectroscopy in the far-UV region (190–250 nm) was initially used to evaluate the backbone conformation of Lia1. The CD spectrum of Lia1 at 25°C (Fig. 3a) showed two relative minima at 208 and 222 nm, and a maximum around 192 nm. This spectrum is typical of proteins with a high content of α-helices, and thus, confirms the structural prediction. In addition, an estimation of the secondary structure content using the program CDNN showed the presence of 59.3% α-helix, 6.6% β-sheet, 15.7% turn and 22.5% remainder.
Fig. 3.

Circular dichroism and tryptophan fluorescence of yeast recombinant Lia1. a Circular dichroism spectroscopy in the far-UV region (190–250 nm). b Lia1 intrinsic fluorescence characteristics. Emission spectra were obtained after excitation at 280 nm (full line) and 295 nm (dashed line)
Lia1 contains a single tryptophan residue located at position 155. According to the structural model of the human protein (Park et al. 2006), this residue is positioned in the fourth HEAT repeat in the vicinity of a variable loop, which connects the N- (HEAT repeats 1–4) and C-terminal (HEAT repeats 5–8) halves of this protein. The hinge loop is believed to be a region of great flexibility, and thus, the single tryptophan W155 was used as a reporter group to monitor structural properties of the yeast Lia1 enzyme. The tryptophan maximum emission wavelengths (λmax) observed after excitation of Lia1 protein at 280 and 295 nm were, respectively, 339 and 342 nm (Fig. 3b). These values suggest a low exposure of the indole ring to an aqueous environment. As a means to determine the degree of exposure of this tryptophan residue, quenching studies of Lia1 intrinsic fluorescence were carried out using collisional quenchers. The Stern–Volmer constants (KSV) obtained for acrylamide and potassium iodide (KI) were 2.45 ± 0.04 and 2.99 ± 0.15 M−1, respectively. In contrast, the reported Stern–Volmer constants for acrylamide and KI quenching of N-acetyl-tryptophan-amide (Ac-Trp-NH2), a model compound for a completely solvent-exposed fluorophore, were 20.8 and 10.5 M−1, respectively. The low values of KSV observed for Lia1 quenching confirmed that its tryptophan residue (W155) is buried in the native protein molecule, thus not readily accessible to acrylamide or iodide.
Effects of pH on Lia1 catalytic activity and tertiary structure
As pH can alter the ionization state of amino acid side chains, thereby affecting either the reactivity of catalytic groups or the conformation of the enzyme active site, we sought to investigate the effects of pH on Lia1 enzyme activity and tertiary structure. The purified recombinant yeast enzyme was able to catalyze in vitro the conversion of eIF5A([3H]Dhp) to eIF5A([3H]Hpu) in a pH range from 6.0 to 10.0 (Fig. 4a). Although the protein was active in this wide pH range, the maximum activity was observed at pH 7.5. Low pH values (<6.0) resulted in complete loss of activity, whereas at higher pH values (>8.0) the enzyme partially retained its activity. To evaluate whether loss of enzyme activity was related to dissociation of iron, native gel electrophoresis was used to analyze the conformational state adopted by Lia1 at different pH. As shown in Fig. 4b, at pH 2.0 and 3.0 the enzyme displayed only the upper band, characteristic of the apoenzyme. This is in accordance with the lack of activity at these pH values. Conversely, the lower band was mainly detected at pH > 7.0, likely due to the presence of iron in the active site of the holoenzyme. Although the two bands representing the apo- and the holo-forms are present at pH 4.0 and 5.0, no enzymatic activity was detected. The lack of activity at these pH values might be a consequence of the low iron content combined with the protonation of the histidine and glutamate residues of the active site and conformational changes of Lia1 due to global changes in protonation status. Finally, at pH 6.0 both forms coexisted and partial activity was observed.
Fig. 4.

pH dependence of Lia1 active conformation. a Analysis of Lia1 enzymatic activity under the pH range 2–10 by the in vitro hydroxylase assay. b Analysis of the conformational state adopted by Lia1 under different pHs by native and denaturing gel Electrophoresis. c Quenching of Lia1 native tryptophan fluorescence. Quenching experiments were carried out by titration of Lia1 protein solutions at pH 3.0 (circles), 5.5 (triangles) and 7.5 (squares) with increasing concentrations of acrylamide. After excitation at 295 nm, fluorescence intensities values at kmax were used to obtain Stern–Volmer plots
As shown earlier by collisional quenching studies with acrylamide and KI, the only tryptophan residue present in Lia1 protein seems to be buried from the solvent. In order to determine if the structural changes are associated with iron disruption from the Lia1 active site, quenching experiments with acrylamide were carried out at pH 3.0, 5.5 and 7.5 (Fig. 4c). Fluorescence intensities values at λmax were used to obtain Stern–Volmer plots and to calculate KSV (Table 1). At low pH values, 5.5 and 3.0, quenching of Lia1 fluorescence proceeds as a biphasic process. For both pH values, the first phase showed an anomalous increase, in contrast to the KSV obtained for Lia1 at pH 7.5, since the KSV was close to that of free tryptophan. The KSV calculated for the second stages of both pH values 3.0 and 5.5 were approximately 2 and 2.5 times higher than the one obtained at pH 7.5 (Table 1). Thus, these results suggest that tryptophan accessibility, which was low at pH 7.5, increased upon disruption of compact Lia1 structure as the pH was lowered.
Table 1.
Stern–Volmer constants for the fluorescence quenching of Lia1 by acrylamide
| KSV (M−1) | |
|---|---|
| pH 7.5 | 2.45 ± 0.04 |
| pH 5.5 - Slope 1 | 17.08 ± 2.45 |
| pH 5.5 - Slope 2 | 6.27 ± 0.18 |
| pH 3.0 - Slope 1 | 12.61 ± 1.13 |
| pH 3.0 - Slope 2 | 5.51 ± 0.07 |
Stability of Lia1 as judged by heat- and GdnHCl-induced unfolding
The spectral features of Lia1 shown above suggest that changes in CD signals and fluorescence can be used to characterize the global structural changes occurring during the unfolding of this protein. As the temperature was raised to 95°C, Lia1 lost the characteristics of an α-helical protein, giving rise to a different CD spectrum at each of the pH values analyzed (data not shown). At pH 7.5, both minima signals at 208 and 222 nm shifted to 204 and 220 nm, respectively; while the relative maximum at 192 nm decreased its intensity to nearly zero (Fig. 5a). However, temperature-induced changes of the ellipticity at pH 7.5 began at early stages (~35°C) and unfolding of the protein was completed once the temperature reached 65°C (Fig. 5b). The detection of an inflexion point around 45°C might indicate the existence of a stable unfolding intermediate. The presence of an excess of iron (100 μM) in the solution seems to change this point slightly (Fig. 5b). A significant decrease in protein stability under acidic conditions was observed (Fig. 5c), as reflected by the decrease of the midpoint of denaturation. Although at pH 5.5 Lia1 began to unfold at similar temperatures observed for higher pH values, the midpoint of denaturation was achieved 10°C lower (Fig. 5c), revealing the cooperativity of the unfolding profile under this pH. At the lowest pH studied (3.0) the effect was even more drastic, since unfolding of the protein started at 25°C and was saturated at 55°C with the unfolding midpoint at approximately 30°C. At pH 9.6 the inflexion point detected at pH 7.5 became more pronounced (Fig. 5c). These data reinforce the existence of a stable unfolding intermediate.
Fig. 5.

Conformational stability of Lia1 during thermal and GdnHCl-induced equilibrium unfolding. a Thermal unfolding profile of Lia1 by far-UV circular dichroism spectroscopy at pH 7.5. b Thermal unfolding profile of Lia1 at pH 7.5 and in the presence of excess of iron (100 mM). c Thermal unfolding profile of Lia1 at pH 3.0 (squares), pH 5.5 (circles), pH 7.5 (triangles) and pH 9.6 (octagon). In both b and c, the ellipticity at 222 nm was studied as a function of temperature increase to measure unfolding ratio. d Chemical unfolding transitions at pH 4.5 and 7.5 were monitored by following the changes of tryptophan maximum emission wavelength, center of mass and ellipticity at 222 nm as a function of GdnHCl concentration increase
GdnHCl-induced unfolding of Lia1 was monitored at pH 7.5 and 4.5 by following changes in intrinsic fluorescence of the tryptophan residue, reflected by fluorescence maximum intensity and center of mass, as well as CD signals (Fig. 5d). The changes occurring in Lia1 secondary structure monitored at 222 nm showed a GdnHCl-concentration-dependent loss of ellipticity in both pH 7.5 and 4.5. The transition curves shown in Fig. 5d demonstrated that Lia1 at pH 7.5 began to unfold with 1.0 M GdnHCl and complete unfolding of the protein was achieved in 4.5 M GdnHCl. In contrast, at pH 4.5 the midpoint of denaturation was achieved at lower GdnHCl concentration. Similar to the thermal unfolding, in all the three profiles an inflexion point between 2.5 and 3 M GdnHCl was observed. Thus, the GdnHCl-induced transition curves of the enzyme displayed a more complex unfolding pathway, suggesting the accumulation of intermediate specie(s) in equilibrium with native and unfolded states. Taken together, the data demonstrate higher protein instability at lower pH, which can be correlated with loss of iron from its coordination sites.
Topology of Lia1 in its iron-free and iron-bound states by SAXS
Small angle scattering profiles, obtained for Lia1 wild-type at two different pH values (7.5 and citrate 4.5) and for the mutant apoenzyme E113A are shown in Fig. 6 together with their corresponding pair distance distribution functions. The asymmetric shape of the P(r) functions revealed that the protein is slightly elongated. The radius of gyration of Lia1 wild-type at pH 7.5, calculated from the second moment of the P(r) function, was 24.17 ± 0.01 Å. In contrast, in the absence of iron the radii of gyration were 27.07 ± 0.04 Å (WT, pH 4.5) and 25.80 ± 0.02 Å (E113A). These numbers are in good agreement with the hydrodynamic radius obtained by dynamic light scattering and indicate a more compact conformation of the iron-bound state.
Fig. 6.

Determination of Lia1 topology by small angle X-ray scattering, SAXS. The small angle scattering parameters for Lia1, in the absence (WT pH4.5 and E113A) and in the presence of iron (WT pH7.5). a Scattering intensity as function of scattering vector q for the apo- and holo-forms of Lia1 enzyme. b Pair distances distribution functions obtained by indirect Fourier formation of the scattering data displayed in a
A low-resolution model was calculated using a dummy atom modeling procedure (Svergun 1999). The models obtained for Lia1 are shown in Fig. 7, together with the crystal structure of the protein YIBA from E. coli (PDB Id. 1OYZ), used for comparison. This protein has a HEAT-repeat folding and a molecular mass of 31.9 kDa, close to that of Lia1. The models are the average of six independent calculations in order to give the most probable structure (Volkov and Svergun 2003). The overall shape of iron-free of Lia1 (WT-pH 4.5 and E113A) is less compact and more elongated than that observed for the holoenzyme (WT-pH 7.5), suggesting that both arms are further separated in the apoenzyme. These data are in accordance with the tryptophan fluorescence experiments and support the notion that Lia1 conformation is dependent on iron binding or release.
Fig. 7.
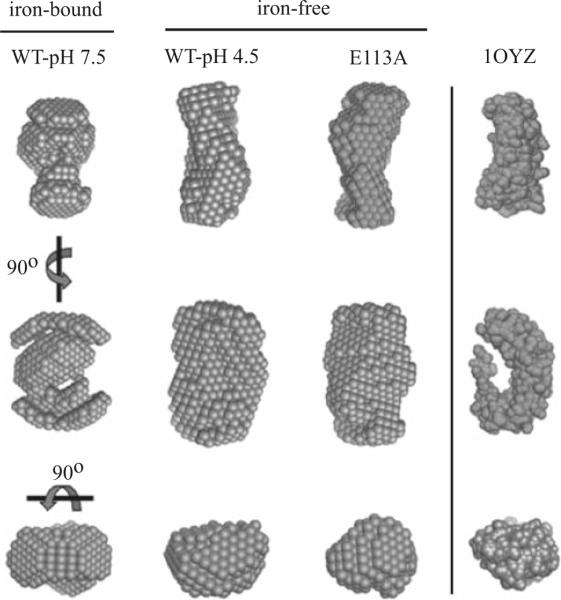
Conformational change of Lia1 in its iron-bound and iron-free states by SAXS. Three-dimensional reconstruction of Lia1 in the presence (WT-pH 7.5) and the absence of iron (WT-pH 4.5 and E113A) from SAXS measurements. The crystal structure of the protein YIBA from E. coli (PDB Id. 1OYZ) is shown for comparison
Discussion
Lia1 catalyzes the final step of hypusine synthesis, the hydroxylation of deoxyhypusine in eIF5A. Our results demonstrate that Lia1 is a novel HEAT repeat containing metalloenzyme with a structure distinct from that of other protein hydroxylases. The sequence of Lia1 is highly conserved in all eukaryotes from fungi to human and its structure is predicted to consist of eight HEAT repeats, with four repeats in symmetrical N- and C-terminal dyad arms, as was modeled for the human protein (Park et al. 2006). Mutational analyses of Lia1 reveal apparent differences in the binding of substrate protein at the active sites of yeast Lia1 and human DOHH. Furthermore, we have carried out extensive structural analyses of Lia1 employing complementary spectroscopic techniques (CD and quenching of tryptophan fluorescence) and SAXS to obtain detailed information on the process of structural changes associated with thermal and chemical denaturation, low pH and loss of iron. The results indicate a dual role of iron binding in Lia1, for catalysis as well as for structural integrity.
The yeast recombinant Lia1, like human DOHH, was shown to be an iron metalloenzyme consisting of a mixture of apoenzyme (iron-free form) and holoenzyme (iron-bound form). The essential nature of this metal for enzyme activity was demonstrated by the fact that Lia1 apoenzyme was totally devoid of activity. The identification and separation of apo- and holo-forms of the human enzyme (Kim et al. 2006) and Lia1 by molecular exclusion chromatography and native electrophoresis suggest structural differences in the tertiary structure depending on the presence or absence of metal. The super helical structure of DOHH is entirely different from the beta-jellyroll structure, termed double stranded beta helix (DSBH), found in the majority of other protein hydroxylases in the family of the Fe(II)-and 2-oxoacid-dependent dioxygenases, e.g., prolyl and lysyl hydroxylases. While both DOHH and DSBH enzymes are dependent on molecular oxygen as a source of hydroxyl group, there are clear differences in reaction mechanisms. There is no requirement for 2-oxoacid for DOHH catalysis and no site identified for 2-oxoacid binding (Kim et al. 2006; Park et al. 2006). Moreover, DOHH has a di-iron active center, whereas DSBH enzymes have a mono-iron active center. The mode of iron binding at the active site of human DOHH analyzed by Mössbauer spectroscopy (in collaboration with Dr. Lawrence Que, University of Minnesota, unpublished results) suggests a reaction mechanism similar to that of methane monoamine oxidase hydroxylase (MMOH), which has a di-iron active center, coordinated by His and Glu/Asp residues contributed from four alpha helical bundles (Nordlund and Eklund 1995). Although a number of iron chelators and their conjugates have been shown to be effective inhibitors of human DOHH in cells and to cause cell cycle arrest at the G1/S boundary (Hanauske-Abel et al. 1994), no specific inhibitors are currently available. Thus, elucidation of the structure and reaction mechanism of DOHH will facilitate the design and development of specific inhibitors of the human enzyme.
Substitution of any residue at the four conserved His-Glu motifs with Ala resulted in total inactivation of Lia1 activity. Iron binding was diminished in six mutant enzymes, H79A, H112A, E113A, H237A, H270A and E271A, but not in E80A and E238A, suggesting involvement of H79, H112, E113, H237, H270 and E271 in coordination of iron. We tested the ability of these eight mutant proteins and Ala and Asp substitution mutants at two additional highly conserved Glu sites, E116 and E274, close to the second and fourth His-Glu motifs, to bind its substrate in vivo by a two hybrid assay. In case of human DOHH mutants, substrate binding carried out in vitro by pull-down of human eIF5A(Dhp) by GST-human DOHH suggested the importance of all Glu residues of the His-Glu motifs and the conserved Gly residues neighboring the first and third His-Glu motifs (Kang et al. 2007). Although the two methods used are different, the data are consistent in indicating the importance of the acidic Glu residues of Lia1 and DOHH active sites in the binding of eIF5A(Dhp) substrate. Obviously, the carboxyl side chains of these Glu residues (E113, E116, E238, E271 and E274) of Lia1 must be important in anchoring not only the deoxyhypusine side chain, but also other basic amino acid residues of the hypusine site loop (S47KTGK51HGHAK56) containing the deoxyhypusine residue. It is curious that E80 of Lia1 is not required for eIF5A binding in vivo, since E57 of human DOHH (corresponding to E80 of Lia1) was critical for eIF5A(Dhp)/hDOHH binding in vitro (Kang et al. 2007). Furthermore, H270 of Lia1 appears to be important for substrate binding, whereas the matching residue in human DOHH (H240) is not. Thus, there seem to be fine differences in the mode of substrate binding between the human and the yeast enzymes, while both interactions remain to be extremely selective. Although His and Glu residues of Lia1 active site that are critical for binding of iron and/or the protein substrate have been identified, we cannot yet assign the specific coordination sites for the two iron atoms, oxygen and the protein substrate.
The knowledge of Lia1 structural features is necessary not only to understand how this enzyme interacts with its substrate, but also to elucidate the mechanism by which the deoxyhypusine-containing eIF5A intermediate is promptly converted to the mature hypusinated form. As catalytic metals have been shown to play structural roles in metalloenzymes, mainly conferring stability to the protein (Arnold and Zhang 1994), a set of experiments were designed in order to investigate the possible role of the metal in the structure of this protein. Recombinant Lia1 was sensitive to thermal or chemical unfolding. It began to unfold at relatively low temperatures (~35°C) and low chemical agent concentration (1 M GdnHCl), indicating a poor protein stability. Thermal unfolding profiles showed the presence of more than one inflexion point indicating the possible presence of more than two different states (native and unfolded) in the denaturation process. The analysis of the circular dichroism and fluorescence data showed different unfolding profiles for different parameters of the protein, indicating the existence of intermediate states in the unfolding process. Removal of the iron from the recombinant protein by either reducing the pH of the solution, below the pKa of the histidines that hold the metal, or addition of sodium citrate caused a drastic reduction in Lia1 stability against thermal treatment or chemical agents. These data point to the important role of the metal atoms in maintaining the structure of Lia1 by bridging the two dyad arms of the protein to result in a closed, compact conformation. This role of iron is further supported by the increased accessibility of the tryptophan residue to collisional quenchers upon removal of bound metal and also by the models obtained from SAXS data.
The model obtained from SAXS data is compatible with the horseshoe model of the human DOHH (Park et al. 2006) and shows that Lia1 exists as a monomer in solution. However, due to the low resolution of SAXS measurements, our model does not rule out the possibility that Lia1 might assume another conformation. SAXS analyses of members of the karyopherin β family, nucleocytoplasmic transporters known to be constituted of 20 HEAT repeats, revealed that they undergo large conformational changes upon ligand binding (Fukuhara et al. 2004). This is in agreement with the conformational differences between iron-bound and iron-free Lia1. Determination of crystal structures of Lia1 apo and holoenzymes and its complex with the protein substrate will provide further information on the structural rearrangements that this enzyme undergoes upon iron displacement from its active site, unveil the precise topology of the complex interactions and provide insights into the reaction mechanism of this unique protein hydroxylase.
Acknowledgments
We thank Edith C. Wolff (NIDCR/NIH) and Cleslei F. Zanelli (UNESP) for helpful discussion and critical reading of our manuscript. This work was supported by grants to S. R. V. from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). We also thank FAPESP, CNPq and CAPES for fellowships awarded to V. S. P. C.
Abbreviations
- eIF5A
Eukaryotic initiation factor 5A
- eIF5A(Dhp)
Deoxyhypusine-containing intermediate
- eIF5A(Hpu)
Mature form of eIF5A containing hypusine
- Dys1
Deoxyhypusine synthase
- Lia1
Yeast deoxyhypusine hydroxylase
- DOHH
Human deoxyhypusine hydroxylase
- YPD
Rich medium containing glucose
- DTT
Dithiothreitol
- WT
Wild type
- NAD
Nicotinamide adenine nucleotide
- CD
Circular dichroism
- SAXS
Small-angle X-ray scattering
- IPTG
Isopropyl-beta-D-thiogalactopyranoside
- GST
Glutathione S-transferase
- KSV
Stern–Volmer constant
- GdnHCl
Guanidinium hydrochloride
References
- Abbruzzese A, Park MH, Folk JE. Deoxyhypusine hydroxylase from rat testis. Partial purification and characterization. J Biol Chem. 1986;261(7):3085–3089. [PubMed] [Google Scholar]
- Arnold FH, Zhang JH. Metal-mediated protein stabilization. Trends Biotechnol. 1994;12(5):189–192. doi: 10.1016/0167-7799(94)90081-7. [DOI] [PubMed] [Google Scholar]
- Cano VS, Jeon GA, Johansson HE, Henderson CA, Park JH, Valentini SR, Hershey JW, Park MH. Mutational analyses of human eIF5A-1—identification of amino acid residues critical for eIF5A activity and hypusine modification. FEBS J. 2008;275(1):44–58. doi: 10.1111/j.1742-4658.2007.06172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HL, Park MH, Folk JE, Safer B, Braverman R. Identification of the hypusine-containing protein hy + as translation initiation factor eIF-4D. Proc Natl Acad Sci USA. 1983;80(7):1854–1857. doi: 10.1073/pnas.80.7.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias CA, Cano VS, Rangel SM, Apponi LH, Frigieri MC, Muniz JR, Garcia W, Park MH, Garratt RC, Zanelli CF, Valentini SR. Structural modeling and mutational analysis of yeast eukaryotic translation initiation factor 5A reveal new critical residues and reinforce its involvement in protein synthesis. FEBS J. 2008;275(8):1874–1888. doi: 10.1111/j.1742-4658.2008.06345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N, Fernandez E, Ebert J, Conti E, Svergun D. Conformational variability of nucleo-cytoplasmic transport factors. J Biol Chem. 2004;279(3):2176–2181. doi: 10.1074/jbc.M309112200. [DOI] [PubMed] [Google Scholar]
- Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96(1):99–110. doi: 10.1016/s0092-8674(00)80963-0. [DOI] [PubMed] [Google Scholar]
- Hanauske-Abel HM, Park MH, Hanauske AR, Popowicz AM, Lalande M, Folk JE. Inhibition of the G1-S transition of the cell cycle by inhibitors of deoxyhypusine hydroxylation. Biochim Biophys Acta. 1994;1221(2):115–124. doi: 10.1016/0167-4889(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Jao DL, Chen KY. Tandem affinity purification revealed the hypusine-dependent binding of eukaryotic initiation factor 5A to the translating 80S ribosomal complex. J Cell Biochem. 2006;97(3):583–598. doi: 10.1002/jcb.20658. [DOI] [PubMed] [Google Scholar]
- Kang HA, Hershey JW. Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. J Biol Chem. 1994;269(6):3934–3940. [PubMed] [Google Scholar]
- Kang KR, Kim YS, Wolff EC, Park MH. Specificity of the deoxyhypusine hydroxylase-eukaryotic translation initiation factor (eIF5A) interaction: identification of amino acid residues of the enzyme required for binding of its substrate, deoxyhypusine-containing eIF5A. J Biol Chem. 2007;282(11):8300–8308. doi: 10.1074/jbc.M607495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Kang KR, Wolff EC, Bell JK, McPhie P, Park MH. Deoxyhypusine hydroxylase is a Fe(II)-dependent, HEAT-repeat enzyme. Identification of amino acid residues critical for Fe(II) binding and catalysis [corrected] J Biol Chem. 2006;281(19):13217–13225. doi: 10.1074/jbc.M601081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund P, Eklund H. Di-iron-carboxylate proteins. Curr Opin Struct Biol. 1995;5(6):758–766. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- Park MH. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A) J Biochem (Tokyo) 2006;139(2):161–169. doi: 10.1093/jb/mvj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, Cooper HL, Folk JE. Identification of hypusine, an unusual amino acid, in a protein from human lymphocytes and of spermidine as its biosynthetic precursor. Proc Natl Acad Sci USA. 1981;78(5):2869–2873. doi: 10.1073/pnas.78.5.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, Cooper HL, Folk JE. The biosynthesis of protein-bound hypusine (N epsilon -(4-amino-2-hydroxybutyl)lysine). Lysine as the amino acid precursor and the intermediate role of deoxyhypusine (N epsilon -(4-aminobutyl)lysine) J Biol Chem. 1982;257(12):7217–7222. [PubMed] [Google Scholar]
- Park MH, Liberato DJ, Yergey AL, Folk JE. The biosynthesis of hypusine (N epsilon-(4-amino-2-hydroxybutyl)lysine). Alignment of the butylamine segment and source of the secondary amino nitrogen. J Biol Chem. 1984;259(19):12123–12127. [PubMed] [Google Scholar]
- Park JH, Wolff EC, Folk JE, Park MH. Reversal of the deoxyhypusine synthesis reaction. Generation of spermidine or homospermidine from deoxyhypusine by deoxyhypusine synthase. J Biol Chem. 2003;278(35):32683–32691. doi: 10.1074/jbc.M304247200. [DOI] [PubMed] [Google Scholar]
- Park JH, Aravind L, Wolff EC, Kaevel J, Kim YS, Park MH. Molecular cloning, expression, and structural prediction of deoxyhypusine hydroxylase: a HEAT-repeat-containing metalloenzyme. Proc Natl Acad Sci USA. 2006;103(1):51–56. doi: 10.1073/pnas.0509348102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Abid MR, Miyazaki M. Deoxyhypusine synthase gene is essential for cell viability in the yeast Saccharomyces cerevisiae. FEBS Lett. 1996;384(2):151–154. doi: 10.1016/0014-5793(96)00310-9. [DOI] [PubMed] [Google Scholar]
- Schnier J, Schwelberger HG, Smit-McBride Z, Kang HA, Hershey JW. Translation initiation factor 5A and its hypusine modification are essential for cell viability in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1991;11(6):3105–3114. doi: 10.1128/mcb.11.6.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenyuk AV, Svergun DI. GNOM-a program package for small-angle scattering data processing. J Appl Crystallogr. 1991;24:537–540. [Google Scholar]
- Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson GM, Cano VS, Valentini SR. Mapping eIF5A binding sites for Dys1 and Lia1: in vivo evidence for regulation of eIF5A hypusination. FEBS Lett. 2003;555(3):464–468. doi: 10.1016/s0014-5793(03)01305-x. [DOI] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff EC, Kang KR, Kim YS, Park MH. Post-translational synthesis of hypusine: evolutionary progression and specificity of the hypusine modification. Amino Acids. 2007;33(2):341–350. doi: 10.1007/s00726-007-0525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanelli CF, Valentini SR. Pkc1 acts through Zds1 and Gic1 to suppress growth and cell polarity defects of a yeast eIF5A mutant. Genetics. 2005;171(4):1571–1581. doi: 10.1534/genetics.105.048082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanelli CF, Valentini SR. Is there a role for eIF5A in translation? Amino Acids. 2007;33(2):351–358. doi: 10.1007/s00726-007-0533-0. [DOI] [PubMed] [Google Scholar]
- Zanelli CF, Maragno AL, Gregio AP, Komili S, Pandolfi JR, Mestriner CA, Lustri WR, Valentini SR. eIF5A binds to translational machinery components and affects translation in yeast. Biochem Biophys Res Commun. 2006;348(4):1358–1366. doi: 10.1016/j.bbrc.2006.07.195. [DOI] [PubMed] [Google Scholar]