

Abstract
Background
Mutation in the progranulin gene (GRN) can cause frontotemporal dementia (FTD). However, it is unclear whether some rare FTD-related GRN variants are pathogenic and whether neurodegenerative disorders other than FTD can also be caused by GRN mutations.
Objectives
To delineate the range of clinical presentations associated with GRN mutations and to define pathogenic candidacy of rare GRN variants.
Design
Case-control study.
Setting
Clinical and neuropathology dementia research studies at 8 academic centers.
Participants
Four hundred thirty-four patients with FTD, including primary progressive aphasia, semantic dementia, FTD/amyotrophic lateral sclerosis (ALS), FTD/motor neuron disease, corticobasal syndrome/corticobasal degeneration, progressive supranuclear palsy, Pick disease, dementia lacking distinctive histopathology, and pathologically confirmed cases of frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U); and 111 non-FTD cases (controls) in which TDP-43 deposits were a prominent neuropathological feature, including subjects with ALS, Guam ALS and/or parkinsonism dementia complex, Guam dementia, Alzheimer disease, multiple system atrophy, and argyrophilic grain disease.
Main Outcome Measures
Variants detected on sequencing of all 13 GRN exons and at least 80 base pairs of flanking introns, and their pathogenic candidacy determined by in silico and ex vivo splicing assays.
Results
We identified 58 genetic variants that included 26 previously unknown changes. Twenty-four variants appeared to be pathogenic, including 8 novel mutations. The frequency of GRN mutations was 6.9% (30 of 434) of all FTD-spectrum cases, 21.4% (9 of 42) of cases with a pathological diagnosis of FTLD-U, 16.0% (28 of 175) of FTD-spectrum cases with a family history of a similar neurodegenerative disease, and 56.2% (9 of 16) of cases of FTLD-U with a family history.
Conclusions
Pathogenic mutations were found only in FTD-spectrum cases and not in other related neurodegenerative diseases. Haploinsufficiency of GRN is the predominant mechanism leading to FTD.
Frontotemporal dementia (FTD) is a heterogeneous group of diseases that present with language and/or behavioral problems frequently in association with a movement disorder. It consists of a spectrum of clinical and pathological diagnoses that includes corticobasal degeneration, FTD with amyotrophic lateral sclerosis (FTD/ALS) or motor neuron disease, progressive supranuclear palsy, Pick disease, and dementia lacking distinctive histopathology.
Frontotemporal dementia is familial in 25% to 50% of cases and can occur as an autosomal dominantly inherited disorder with high penetrance.1-5 Genes causing this type of FTD include MAPT, the gene encoding tau, CHMP2B, and GRN, the gene that encodes progranulin (PGRN). In other families, autosomal dominant FTD is caused by as yet unidentified genes located at 9p12-p216,7 and 9q21-q11.8 In MAPT mutation cases, the predominant autopsy feature is aggregated tau as neurofibrillary and, in some cases, glial tangles. In GRN mutation cases, the predominant deposited protein is ubiquitinated TAR DNA binding protein (TARDBP or TDP-43), and tau tangles are rarely seen. Most of the known GRN mutations are either nonsense mutations that result in a premature stop codon or splicing mutations that alter the reading frame of the messenger RNA (mRNA), resulting in a premature stop codon downstream from the mutation.9-12 The result is that the mutant mRNA is degraded by nonsense-mediated decay (NMD), no protein is produced from the mutant gene, and FTD is caused by haploinsufficiency of PGRN. A single missense mutation (p.A9D) that is clearly pathogenic is known.11,13 This mutation is located within the signal peptide sequence and leads to cytoplasmic missorting, low PGRN levels, and haploinsufficiency. Mutations in the TARDBP gene can cause either ALS or FTD.
In this study, we screened a large number of subjects with FTD-related neurodegenerative disorders for mutations in GRN. The goal was 2-fold. First, we wanted to further delineate the range of clinical presentations associated with GRN mutations. The initial studies of GRN were on subjects with FTD, all of whom had TDP-43 deposits. Subsequent work showed that TDP-43 inclusions are also found in idiopathic ALS,14 frontotemporal dementia with inclusion body myopathy,15 Guam ALS/parkinsonism dementia complex,16 and some cases of Alzheimer disease (AD).17 Therefore, additional cases were included in this study to encompass a broad range of clinical phenotypes including a subset of non-FTD cases, some of which have been associated with TDP-43 pathological features. Second, we sought to evaluate the pathogenicity and potential molecular mechanism of rare GRN variants associated with the disease. We observed 8 new GRN mutations that are clearly pathogenic, including nonsense mutations, deletions, or splice-site mutations that generate premature stop codons. One large (193 base pair [bp]) deletion observed previously by others18 had unusual neuropathological findings including abnormal tau deposits. In addition, we observed other variants that may be pathogenic, including a silent mutation that potentially alters splicing and a missense variant that alters a critical amino acid that is part of the conserved consensus sequence for granulin peptides. Our findings show that pathogenic GRN mutations are only found in FTD-spectrum cases and that haploinsufficiency is their predominant mechanism leading to FTD.
METHODS
STUDY SAMPLES FOR GRN MUTATION SCREENING AND CONTROLS
We screened 545 cases from the clinical and neuropathology dementia research studies at the University of Washington, University of Pennsylvania, University of California at San Diego, University of California at San Francisco, University of Southern California, Oregon Health & Science University, University of British Columbia in Vancouver, and Center for Neuropathology and Prion Research in Munich, Germany (Table 1). We selected cases on the basis of having a clinical or autopsy-confirmed diagnosis of FTD and FTD-related disorders. Cases of FTD with known MAPT mutations and cases of ALS with known SOD1 mutations were excluded. Control samples were from neurologically healthy control participants, who were of a similar age and ethnic origin, from the University of California at San Diego and Veterans Affairs Medical Center, San Diego; the Coriell Institute, Camden, NJ (neurologically normal white control panels); clinical controls from the Alzheimer Disease Center at the University of Pennsylvania; and brain autopsy samples without evidence of neurodegenerative diseases from the University of Pennsylvania. All subjects were studied with institutional review board approval for human subjects from each institution.
Table 1. Summary of Proband Cases Screened.
| Diagnosis, No.a |
Family History, No. |
Age at Onset, y |
No. of Subjects |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Clinical | Neuropathological | Positiveb | Sporadicc | Unknownd | Mean (SD) | Range | Ne | Screened | Pathogenic Mutation |
|
| FTD series | 434 | |||||||||
| FTD | 279 | NA | 113 | 76 | 90 | 59.2 (10.3) | 25-86 | 215 | 279 | 19 |
| FTLD-U | NA | 42 | 16 | 19 | 7 | 56.1 (12.0) | 28-79 | 37 | 42 | 9 |
| CBS/CBD | 45 | 6 | 17 | 29 | 5 | 59.8 (9.6) | 47-82 | 38 | 51 | 2 |
| FTD/ALS or FTD/MND | 9 | 21 | 13 | 8 | 9 | 54.8 (11.1) | 30-69 | 11 | 30 | 0 |
| PSP | 4 | 1 | 3 | 1 | 1 | 54.8 (25.8) | 17-75 | 4 | 5 | 0 |
| Pick or tauopathy | 2 | 19 | 8 | 8 | 5 | 62.0 (10.9) | 42-83 | 15 | 21 | 0 |
| DLDH | NA | 6 | 5 | 1 | 0 | 46.8 (5.3) | 40-56 | 6 | 6 | 0 |
| Other related disorders | 111 | |||||||||
| ALS | 26 | 44 | 24 | 10 | 36 | 57.0 (12.1) | 30-78 | 24 | 70 | 0 |
| Guam ALS | 1 | 5 | 6 | 0 | 0 | 50.5 (4.1) | 47-58 | 6 | 6 | 0 |
| Guam PDC/PDC/ET | 2 | 5 | 7 | 0 | 0 | 61.8 (9.3) | 45-73 | 6 | 7 | 0 |
| AD/DLB/dementia NOS | 10 | 6 | 7 | 5 | 4 | 60.1 (14.3) | 29-79 | 10 | 16 | 0 |
| MSA | 4 | 4 | 2 | 5 | 1 | 53.8 (12.5) | 35-72 | 8 | 8 | 0 |
| AGD | 0 | 4 | 0 | 0 | 4 | No information | 4 | 0 | ||
| Total | 382 | 163 | 221 | 162 | 162 | 545 | 30 | |||
Abbreviations: AD, Alzheimer disease; AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; CBD, corticobasal degeneration; CBS, corticobasal syndrome; DLB, dementia with Lewy bodies; DLDH, dementia lacking distinctive histopathology; ET, essential tremor; FTD, frontotemporal dementia; FTLD-U, frontotemporal lobar degeneration with ubiquitin-positive inclusions; MND, motor neuron disease; MSA, multiple system atrophy; NA, not applicable for clinical or neuropathological diagnosis; NOS, not otherwise specified; PDC, parkinsonism dementia complex; PSP, progressive supranuclear palsy.
In cases with an autopsy, neuropathological diagnosis is given, whereas in cases with only clinical information, clinical diagnosis is given.
Similar disease in first-degree relative.
Family history known and negative.
None or insufficient family history information available.
Number of subjects with available age-at-onset information.
DNA SEQUENCING AND GENOTYPING
The 13 exons of GRN (GenBank NM_002087.2) and at least 80 bp of their flanking introns were fully sequenced in both directions. These fragments were polymerase chain reaction (PCR) amplified from genomic DNA by means of primers that were selected by Primer3 software19; primer sequences were reported in previous publications from our group.20,21 The PCR was carried out by mixing 20 pmol of primers and 20 ng of genomic DNA with 10 μL of DNA PCR master mix (HotStarTaq; Qiagen, Valencia, California) in a final volume of 20 μL. After 32 cycles, 2 μL of a PCR cleanup reagent (ExoSAP-IT; USB, Cleveland, Ohio) was added to the PCR products and digested for 1 hour at 37°C to remove the residual primers and deoxyribonucleotide triphosphates. Four microliters of the reagent-treated PCR fragments was sequenced with a terminator cycle sequencing kit (BigDye; Applied Biosystems Inc, Foster City, California) in a final volume of 10 μL and 25 cycles. The sequence information was collected and analyzed (3100 Genetic Analyzer; Applied Biosystems), and nucleotide variants were identified by sequence alignment using SEQUENCHER software (Gene Codes Corp, Ann Arbor, Michigan) or Mutation Surveyor (Soft Genetics LLC, State College, Pennsylvania). Genetic variants were designated in both DNA and protein levels according to the nomenclature guidelines of the Human Genome Variation Society (http://www.hgvs.org/mutnomen). For genotyping, allele discrimination assays were custom made (TaqMan; Applied Biosystems) and genotyped in 384 control individuals. Genotyping was performed on 384-well plates with 5 ng of genomic DNA, 0.075 μL of 20× single-nucleotide polymorphism genotyping assay (TaqMan), 1.5 μL of PCR assay mix (TaqMan Universal PCR Master Mix), and 1.425 μL of distilled water in each well. The PCR was carried out with an amplification system (GeneAmp PCR System 9700, Applied Biosystems) with a profile of 95°C for 10 minutes and then 50 cycles at 92°C for 15 seconds and 60°C for 90 seconds. Plates were then subjected to end-point read in a real-time quantitative PCR system (7900 Real-Time PCR System, Applied Biosystems). The results were first evaluated by cluster variations; the allele calls were then assigned automatically before being transferred and integrated into the genotype database.
We defined polymorphism as “a change found at a frequency of 1% or higher in the controls.”22 Minor allele frequencies of the variants were generated either by genotyping in our 760 control chromosomes or by combining the genotyping results of previously published references (Table 2). A selected number of mutations were evaluated in an additional series of 732 control chromosomes as indicated in Table 2.
Table 2. GRN Genetic Variants Observed in This Study.
| Coding DNA NM_002087.2 | Predicted Protein NP_002078.1 |
MAF (Count) |
Case Information |
Sourced | |||||
|---|---|---|---|---|---|---|---|---|---|
| Casesa (n=868) |
Controls 1b (n=760) |
Controls 2c | ID | Dx | AAO, y | FH | |||
| Premature Termination Mutations (Pathogenic) | |||||||||
| Nonsense mutations | |||||||||
| c.328C>T | p.R110X | 0.001 (1) | NA | NA | UP74 | FTD | 52 | Possible | 21 |
| c.347C>A | p.S116X | 0.001 (1) | 0.000 (0) | NA | UP455 | FTD | 57 | Yes | Novel |
| c.911G>A | p.W304X | 0.001 (1) | NA | 0.000 (0/400) | UPA118 | FTLD-U | 73 | Yes | 11, 21 |
| c.1009C>T | p.Q337X | 0.001 (1) | NA | NA | UPA148 | FTLD-U | 62 | Possible | 21 |
| c.1157G>A | p.W386X | 0.001 (1) | NA | 0.000 (0/990) | UBA1 | FTD | 59 | Yes | 9, 11 |
| c.1252C>T | p.R418X | 0.001 (1) | NA | 0.000 (0/400) | UPA43e | FTLD-U | 58 | Yes | 9, 11, 21 |
| c.1402C>T | p.Q468X | 0.001 (1) | NA | 0.000 (0/590) | FPS47 | FTD | 59 | Yes | 9 |
| c.1477C>T | p.R493X | 0.007 (6) | 0.000 (0) | 0.000 (0/400) | DHH1 | CBD | 60 | Yes | 11, 18, 21, 23-26 |
| ORBF2 | FTD | Unk | Yes | ||||||
| VAJ1 | FTD | 62 | Yes | ||||||
| VXM1 | FTLD-U | 59 | Yes | ||||||
| UPA251 | FTLD-U | 56 | Possible | ||||||
| UP403e | FTD | 45 | Yes | ||||||
| Frameshift by indel mutations | |||||||||
| c.90_91insCTGC | p.C31LfsX35 | 0.001 (1) | NA | 0.000 (0/400) | BQL01 | FTD | Unk | Unk | 9, 11 |
| c.299delC | p.P100HfsX156 | 0.001 (1) | 0.000 (0) | NA | LRR1 | FTD | 60 | Yes | Novel |
| c.388_391del CAGT | p.Q130SfsX125 | 0.001 (1) | NA | 0.000 (0/400) | BKM1 | FTD | 65 | Yes | 9, 11 |
| c.592_593delAG | p.R198GfsX19 | 0.001 (1) | 0.000 (0) | NA | MAQ1 | FTD | 58 | Yes | Novel |
| c.675_676delCA | p.S226WfsX28 | 0.001 (1) | NA | 0.000 (0/400) | UP525 | FTD | 56 | Possible | 11, 21 |
| c.813_816delCACT | p.T272SfsX10 | 0.001 (1) | 0.000 (0) | NA | UP791 | FTD | 68 | Possible | 25, 27 |
| c.846_852dupGAAATGT | p.D285EfsX3 | 0.001 (1) | 0.000 (0) | NA | LXR1 | FTLD-U | 53 | Yes | Novel |
| c.1317_1318delCA | p.D441HfsX4 | 0.001 (1) | 0.000 (0) | NA | UP946 | CBS | 55 | Yes | Novel |
| Frameshift by large genomic deletion mutation | |||||||||
| c.1414-16_1590del | p.A472VfsX10 | 0.001 (1) | 0.000 (0) | NA | ARC62 | CBD | 50 | No | 18 |
| Frameshift by splice site mutations | |||||||||
| c.264+2T>C | p.V90SfsX67 | 0.001 (1) | 0.000 (0) | NA | RPN17 | FTD | 55 | Yes | 28 |
| c.348A>C | p.A89VfsX139 | 0.001 (1) | 0.000 (0) | NA | UPA573 | FTLD-U | 67 | Yes | Novel |
| c.709-2A>G | p.A237TfsX6 | 0.002 (2) | 0.000 (0) | NA | EFe | FTLD-U | 57 | Yes | 20, 25, 26, 29 |
| VHAe | FTD | 65 | Yes | ||||||
| c.1179+2T>C | p.V395YfsX29 | 0.001 (1) | 0.000 (0) | NA | UPA203 | FTLD-U | 59 | Possible | Novel |
| c.1414-2A>G | p.A472VfsX10 | 0.001 (1) | 0.000 (0) | NA | UP52e | FTD | 52 | Yes | Novel |
| Missense Mutations (Pathogenic) | |||||||||
| Altered protein translocation | |||||||||
| c.26C>A | p.A9D | 0.001 (1) | NA | 0.000 (0/500) | HDDD | FTD | 62 | Yes | 11, 30 |
| Altered granulin motif (probable pathogenic) | |||||||||
| c.313T>C | p.C105R | 0.001 (1) | 0.000 (0) | NA | SMF1 | FTD | 39 | Yes | 11 |
| Other Coding/Exon Variations (Pathogenic Potential Unknown) | |||||||||
| Missense variants (MAF >0.01) | |||||||||
| c.208G>A | p.G70S | 0.001 (1) | 0.001f (1) | NA | UP192 | FTD | 55 | Possible | Novel |
| c.229G>A | p.V77I | 0.001 (1) | 0.000 (0) | NA | EF46 | At risk | Unk | Yes | Novel |
| c.545C>T | p.T182M | 0.003 (3) | 0.000 (0) | 0.000 (0/380) | ARC43 | MSA | 57 | Yes | 30, 31 |
| NCM1 | At risk | Unk | Yes | ||||||
| FTD12 | Pick | 58 | No | ||||||
| c.634C>T | p.R212W | 0.001 (1) | 0.001 (1) | NA | FTD134 | FTD | Unk | Yes | Novel |
| c.752C>G | p.T251S | 0.001 (1) | 0.000 (0) | NA | CCW1 | FTD | 44 | Yes | Novel |
| c.827C>T | p.A276V | 0.001 (1) | 0.000 (0) | NA | UPA573 | Depressiong | 50 | Unk | Novel |
| c.893G>A | p.R298H | 0.001 (1) | 0.000 (0) | NA | UPA147 | FTLD-U | 70 | Unk | Novel |
| c.970G>A | p.A324T | 0.001 (1) | NA | 0.008 (3/367) | FPS47 | FTD, AD | 59 | Yes | 31 |
| c.1058G>A | p.S353N | 0.001 (1) | 0.001 (1) | NA | UPA158 | FTLD-U | 65 | None | Novel |
| c.1070C>G | p.P357R | 0.001 (1) | 0.000 (0) | NA | UPA148 | FTLD-U | 62 | Possible | Novel |
| c.1253G>A | p.R418Q | 0.002 (2) | NA | 0.002 (2/1046) | FRD17 | FTD | 83 | Unk | 11, 30, 32 |
| UPC1793 | FTLD/MND | Unk | No | ||||||
| c.1297C>T | p.R433T | 0.006 (5) | 0.001 (1) | 0.009 (8/930) | RPN17 | FTD | 55 | Yes | 11, 25, 31, 32 |
| UPA147 | FTLD-U | 70 | Unk | ||||||
| UPA203 | FTD | 59 | Possible | ||||||
| UP528 | FTD | 61 | None | ||||||
| UPC1590 | ALS | 74 | Unk | ||||||
| c.1544G>C (rs25647) | p.G515A | 0.001 (1) | 0.005f (4) | 0.004 (4/1046) | GRR1 | FTD | 57 | Yes | 11, 31 |
| Silent variants (MAF <0.01) | |||||||||
| c.99C>T | p.D33 | 0.001 (1) | NA | 0.008 (11/1314) | UPC1658 | ALS | 57 | Unk | 11, 31, 32 |
| c.159G>A | p.L53 | 0.001 (1) | 0.000 (0) | NA | UP192 | FTD | 55 | Possible | Novel |
| c.903G>A | p.S301 | 0.003 (3) | 0.000 (0) | 0.006 (4/646) | HEG1 | FALS | 60 | Yes | 11, 32 |
| UPA148 | FTLD-U | 62 | Possible | ||||||
| UP684 | FTD | 60 | Unk | ||||||
| 3′UTR variants | |||||||||
| c.*30G>Ah | NC | 0.001 (1) | NA | NA | UP59 | FTD | 65 | No | Novel |
| c.*301delTh | NC | 0.001 (1) | NA | NA | PCK1 | Pick | Unk | No | Novel |
Abbreviations: AAO, age at onset; AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; CBD, corticobasal degeneration; CBS, corticobasal syndrome; Dx, diagnosis; FALS, familial ALS; FH, family history; FTD, frontotemporal dementia; FTLD-U, frontotemporal lobar degeneration with ubiquitin-positive inclusions; ID, identifier; indel, insertion-deletions; MAF, minor allele frequency; MND, motor neuron disease; MSA, multiple system atrophy; NA, not available; NC, no change; Unk, unknown; UTR, untranslated region.
Frequency in 868 chromosomes of FTD series cases; numbers in parentheses indicate counts.
Frequency in our 760 (or 1510) control chromosomes; numbers in parentheses indicate counts.
Combined control frequency from the references; numbers in parentheses indicate counts over chromosomes screened.
Numbers represent reference numbers.
Cases with same mutation also found in additional affected family member.
There were 1510 control chromosomes.
Diagnosis was originally FTD and later changed to depression.
*Stop codon.
EX VIVO SPLICING ASSAYS
Heterologous constructs contained a single GRN exon and its flanking 100-bp intronic sequences, inserted between heterologous exons. Both alleles of the target locus were PCR amplified from a heterozygous subject’s genomic DNA by means of primers that were integrated with restriction enzyme sites on their 5′ end. The PCR-amplified fragments were double-digested with restriction enzymes XhoI and BamHI and inserted into the same double-digested expression vector pSPES.33 Multiple clones were isolated and fully sequenced to select for clones representing both alleles of the variation without additional PCR artifacts. Minigene constructs contained 3 exons and their 2 internal introns with the target exon located in the middle, representing the actual exon-intron setting of the original gene structure. The same cloning procedure for heterologous construct was followed except that the vector pRc/RSV (Invitrogen, Carlsbad, California) and restriction enzyme sites of HindIII and XbaI were used in the minigene constructs. These constructs were then transiently transfected into a rat pheochromocytoma cell line (PC12). PC12 cells were cultured in Dulbecco modified Eagle medium supplemented with 5% fetal bovine serum, 10% horse serum, and penicillin (100 units/mL)/streptomycin (100 μg/mL) and seeded in triplicate 25-cm2 flasks to achieve 50% to 70% confluency 1 day before transfection. PC12 cells were transfected with 5 μg of plasmid DNA and 60 μL of a transfection reagent (Lipofectamine; Invitrogen) in 1.5 mL of medium (Opti-MEM; Invitrogen) for 4 hours at 37°C (5% carbon dioxide), after which 1.5 mL of Dulbecco modified Eagle medium supplemented with 20% horse serum and 10% fetal bovine serum was added. Transfected PC12 cells were harvested 24 hours after transfection. Total RNA was isolated (RNAeasy; Qiagen). Transcripts from transiently transfected splicing vectors were analyzed by reverse-transcription PCR assays by means of either SD6/SA2 primer pairs (for heterologous constructs) or GRN exon primers (for minigene constructs). Amplified reverse-transcription PCR products were resolved on agarose gel electrophoresis and visualized with ethidium bromide staining. These products were excised and extracted from the gel (GFX Gel Band Purification Kit; GE Healthcare, Piscataway, New Jersey) and then subjected to DNA sequencing to determine their nucleotide content. Primer sequences of constructs and PCR conditions will be provided on request.
RESULTS
STUDY SUBJECT
We screened GRN for mutations in a spectrum of patients with FTD (n=434) that included some subjects with primary progressive aphasia, semantic dementia, FTD/ALS, FTD with motor neuron disease, corticobasal syndrome (CBS)/corticobasal degeneration, progressive supranuclear palsy, Pick disease, dementia lacking distinctive histopathology, and pathologically confirmed cases of frontotemporal lobar degeneration with ubiquitin-positive inclusion (FTLD-U) (Table 1). Cases known to have mutations in MAPT, APP, PSEN1,or PSEN2 were excluded; however, not all cases were screened for mutations in these genes. In our cohort, 55.2% (175 of 317) of the patients with FTD for whom information was available had a definite or possible family history of a similar neurodegenerative disease. To date, all patients with GRN mutations evaluated pathologically had TDP-43 deposits. Therefore, we also screened non-FTD cases in which TDP-43 deposits were a prominent neuropathological feature. These cases included subjects with ALS, Guam ALS and/or parkinsonism dementia complex, Guam dementia, AD, multiple system atrophy, and argyrophilic grain disease (Table 1). All GRN exons, along with at least 80 bp of the flanking introns, were sequenced.
GRN VARIANTS
We observed 58 different genetic variants, of which 46 (79.3%) were single-nucleotide substitutions and 12 were insertion-deletions (indel) (Table 2 and eTable 1 [http:///www.archneurol.com]). Of these, 26 are novel variants not previously described.* We considered a variant as potentially pathogenic if it was not observed in controls and if the variant was in the coding region of the gene. Thus, the 8 variants found in controls and 3 in the 3′ untranslated region were not considered in the subsequent analysis. Of the 434 FTD cases screened, 41 (9.4%) had a rare variant not observed in controls.
FRAMESHIFT PATHOGENIC MUTATIONS
We observed 22 mutations that resulted in a premature stop codon and were thus considered pathogenic. Eight were nonsense mutations, including 1 not previously observed (p.S116X). We also found 8 small indels in coding sequences that caused frameshifts resulting in premature stop codons. Four of these are novel and are predicted to produce truncated proteins that are missing the 148, 297, 306, and 337 C-terminal amino acids of PGRN (Table 2). We also found a large deletion (c.1414- 16_1590del), which is described in more detail in the next paragraph. We identified 5 variants (3 of which are novel) in either the 5′ splice site or the 3′ splice site of GRN exons (Table 2 and eTable 2). All are within 2 bp of the exon-intron junctions and alter the consensus sequence for splice sites. All are predicted by in silico methods39 to reduce use of the affected splice site, cause exon skipping, and generate a downstream premature stop codon. We used ex vivo splicing assays to test these predictions, and all 5 mutations showed altered splicing. Four of the mutations completely inactivated the splice site, and the exon was completely excluded from the resulting transcript. The remaining mutation (c.1414-2A>G) resulted in only a partial reduction in use of the affected splice site. The proband for the c.1414-2A>G mutation, subject UP52a, had 2 affected siblings (neither of whom were available for testing). However, 1 of these siblings was an obligate carrier because this person had a child who carried the same variant and who was affected with semantic dementia with onset at 52 years of age. Thus, the splicing data and cosegregation of this mutation support the conclusion that this splice site change is pathogenic. Gel image of splicing assays will be provided on request.
We observed a large deletion (c.1414-16_1590del) that removed 193 bp, starting in intron 10 (I10) and extending 177 bp into exon 11 (E11). The effect of this mutation on mRNA splicing was tested in ex vivo splicing experiments with the use of a minigene that contained the E10-I10-E11-I11-E12 segment of GRN. Constructs were generated that had sequences either from the normal or from the deletion allele. Splicing of the normal allele removed I10 and I11, leaving E10-E11-E12 correctly spliced. The deletion allele produced 2 products, 1 of which was unspliced with both I10 and I11 retained, and a second product with I10 retained and I11 removed. In vivo, both products would cause a frameshift and a predicted protein truncated at amino acid 481, resulting in haploin-sufficiency. Clinically, this patient (ARC62) had a “left alien hand” and a diagnosis of CBS. The neuropathological findings in this case are somewhat atypical for a GRN mutation. Cortical atrophy was most severe in frontal and temporal lobes but extended to include the parietal lobes. Basal cerebral structures appeared normal, although the substantia nigra was pale. Hematoxylin-eosin–stained sections of all cerebral lobes showed dramatic cortical neuron loss, vacuolization, and intense gliosis with marked degeneration of underlying white matter and gliosis that was worst in the frontal lobes (Figure 1). Similarly prominent neuron loss and gliosis also were present in the globus pallidus and substantia nigra. The hippocampus was spared. Tau immunoreactivity (IR) in cerebral cortical sections showed scattered small rounded or curved cytoplasmic neuronal inclusions but not neurofibrillary tangles, as well as abundant glial tau IR that in several instances appeared as “tufted” astrocytes. More numerous tau IR deposits were present in the underlying cortical white matter. Ubiquitin- and TDP-43–IR neuronal cytoplasmic inclusions were identified in affected regions of cerebral cortex and in the granule neurons of the hippocampus. Ubiquitin, but not TDP-43, IR inclusions and deposits were numerous in affected cerebral white matter. This same mutation was reported in an FTD case with ubiquitin-positive inclusions.18
Figure 1.
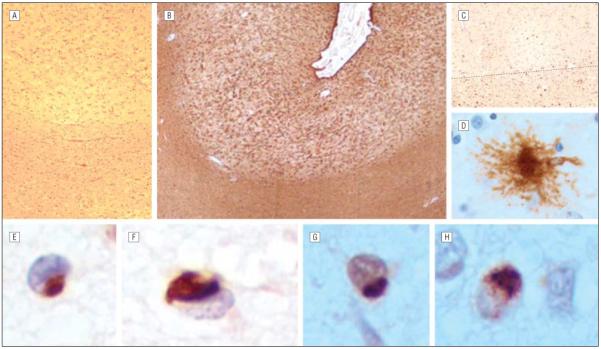
Neuropathological results of case ARC62 with GRN c.1414-16_1590del mutation. Photomicrographs of middle frontal gyrus showed extensive astrogliosis of cortex and less so of underlying white matter (A, hematoxylin-eosin, original magnification ×100; B, glial fibrillary acidic protein, original magnification ×40), extensive tau immunoreactivity in gray and white matter (C, tau2, original magnification ×100; gray matter above dotted line), a cortical “tufted” astrocyte (D, tau2 immunohistochemistry, original magnification ×400), and cytoplasmic intraneuronal inclusions that were immunoreactive for TDP-43 (E and F, original magnification ×600) and ubiquitin (G, original magnification ×600). Similar cytoplasmic inclusions immunoreactive for ubiquitinated TAR DNA binding protein and ubiquitin (H, original magnification ×600) were present in the granule neurons of the dentate gyrus of hippocampus.
MISSENSE VARIANTS
We identified 16 missense changes, of which 8 are novel (Table 2, eTable 1, and eTable 3). Of these, 8 were considered potentially pathogenic because they did not appear in controls (p.A9D, p.V77I, p.C105R, p.T182M, p.T251S, p.A276V, p.R298H, and p.P357R). The p.A9D variant is clearly pathogenic because it cosegregates with disease in a large FTD kindred.34 We performed in silico analysis using the PMut,40 PolyPhen,41 and SIFT42 programs to predict whether the variants potentially altered protein function. Among the 16 missense variants, one, p.R212W, was predicted by all 3 algorithms to alter protein function (eTable 3). Another 6 variants (p.A9D, p.R19W, p.G70S, p.C105R, p.T182M, and p.R433W) were predicted to be deleterious by 2 algorithms. The C105R change alters a conserved cysteine that is part of the consensus sequence that defines granulin peptides. Thus, p.C105R potentially alters granulin peptide function after secretion, a mechanism not previously implicated in GRN mutation pathogenesis. Eight other missense mutations also fall within the granulin peptide sequences (eTable 3).
Missense variants could also be pathogenic by altering exon-splicing enhancers (ESEs) or exon-splicing silencers, thereby causing exon skipping and downstream premature stop codons. We tested 7 candidate missense variants in ex vivo splicing assays and found that A276V altered splicing. Variant A276V (c.827C>T) showed an altered splicing pattern in the heterologous construct experiment, but this splicing alteration was not observed in the minigene construct. Because it was not observed in the controls, A276V is a potential pathogenic mutation.
SILENT VARIATIONS
We identified 4 silent changes in coding exons of GRN and one of these, c.159G>A (p.L53), was not present in controls. These changes, like the missense mutations described in the previous section, have the potential to alter splicing. We analyzed the variant sequences in silico for potential effects on ESEs by means of the RESCUE-ESE algorithm43 (eTable 4). We also tested these variants in ex vivo splicing assays. The in silico analysis predicted that the rare alleles of c.99C>T (E1) and c.159G>A (E2) would add ESE sites that could enhance exon inclusion. However, these variants did not affect splicing in the ex vivo assays. In contrast, c.903G>A (E8) did show dramatic alteration of splicing in the heterologous, but not the minigene, construct experiments. Thus, the c.903G>A change is also potentially a pathogenic mutation. Because it has also been observed in controls,11,32 it may be either a reduced-penetrance mutation or not pathogenic.
GENOTYPE-PHENOTYPE CORRELATIONS OF GRN PATHOGENIC MUTATIONS
Detailed clinical information was available for 31 GRN mutation–positive patients from 28 different families. The mean age at onset was 57.7 years, with a wide range (39-73 years). Clinical features of the 28 positive probands and their affected family members (3 additional family members tested from families UP52, UPA43, and UP403) at their first examination by a neurologist included impairments in language (21 of 26), praxis (5 of 13), and behavior (17 of 23). Nine of 22 patients exhibited parkinsonism. The most common clinical diagnosis was FTD (24 of 31) or subtypes of FTD including primary progressive aphasia (3 patients) and CBS (1 patient). Three patients were initially diagnosed as having AD, whereas 1 patient carried a dual clinical diagnosis of AD and Parkinson disease. One patient had a clinical diagnosis of dementia with Lewy bodies. In the 5 cases in which the clinical diagnosis was not FTD or a subtype of FTD, however, the neuropathological diagnosis was FTLD-U.
COMMENT
We screened GRN for genetic variants in 545 patients with a collection of different clinically and/or pathologically defined neurodegenerative disorders (Table 1). Because all GRN mutation cases reported to date, with pathological evaluation, have TDP-43 deposits, we included disorders in which TDP-43 neuropathological findings exist for screening. We also screened some other neurodegenerative disorders because PGRN plays a role in neuronal stabilization,44,45 neuronal survival,46 and neurogenesis.47 The majority of cases evaluated (434 of 545 [79.6%]) belonged to the FTD spectrum. GRN mutations that were clearly pathogenic were identified only in FTD-spectrum cases. Pathogenic variants were scattered throughout the entire GRN gene, consistent with the previously published spectrum of GRN mutations.† Almost all of the pathogenic variants (≥98% in estimation) were located between exons 1 and 11 of the GRN genomic sequence, which is basically the entire gene.
GENETIC VARIANTS AND PATHOGENICITY
The goal of the foregoing work was to identify the range of clinical and neuropathological presentations associated with GRN mutations. Because a large number of candidate GRN mutations were identified, to make clear the clinical-neuropathological correlations, it is necessary to determine which genetic variants are pathogenic and which are benign with respect to causation of neurodegenerative disease. We identified a number of variants that are clearly pathogenic. These are either nonsense changes or variants that shift the reading frame by insertion or deletion to produce a downstream premature stop codon. Changes within the splice site acceptor and donor sequences were mostly predicted to alter splicing. Because some of these mutations altered splice site nucleotides that are not completely invariant (eg, c.348A>C), it was important to test each by ex vivo splicing experiments. These experiments showed empirically that each splice site variant altered splicing and each resulted in a frameshift and a downstream premature stop codon (eTable 2). Thus, these mutations, listed in eTable 2, are all pathogenic. Previous work showed that premature stop codons in GRN mRNA cause NMD. All the nonsense and frameshift mutations described in the first section of Table 2 meet the “position of an exon-exon junction” rule for NMD.48 That is, for NMD to occur, a premature termination codon has to be located at more than 50 to 55 nucleotides upstream of the last exon-exon junction. The 3′-most premature termination codon is generated from variant c.1414-16_1590del, and it is 100 nucleotides upstream of the exon 11-12 junction. Therefore, we predict that all the premature termination variants identified in this study (first section of Table 2) cause NMD and that these mutations act by a common haploinsufficiency mechanism whereby reduced production of the GRN gene product, PGRN, causes FTD.
For the missense variants observed, it is difficult to predict whether these changes are pathogenic. The exception is the c.26C>A (p.A9D) change, which cosegregates with FTD in the large hereditary dysphasic disinhibition dementia kindred.34 Shankaran et al13 further demonstrated that this mutation prevents PGRN from being secreted. The result is haploinsufficiency caused by failure of the mutant protein to reach its site of biological activity. Among the other missense variants observed, one (p.R212W) was predicted by in silico methods to have a high probability of being deleterious. However, this variant was also observed in the controls and with a frequency similar to that in the FTD cases (Table 2); thus, it is unlikely to be a mutation. Another 6 missense variants (p.A9D, p.R19W, p.G70S, p.C105R, p.T182M, and p.R433W) had a moderate probability of being deleterious (eTable 3). Of these, the p.C105R variant is interesting because it alters one of the most conserved cysteine residues in G granulin. Absence of this variant in the controls also supported the hypothesis that this is a deleterious change. However, others reported that this variant does not cosegregate with disease in a family with FTLD-U,11 suggesting that if it is pathogenic it is a low-penetrance allele. Also, this change affects only 1 of the 7 granulin peptides and thus may have only a minor effect on the total biological activity of PGRN. Interestingly, the p.A9D (c.26C>A) variant was not predicted to be a strong candidate by the in silico analyses, yet it is the only established pathogenic locus among the 16 missense variants. Thus, the in silico predictions must be interpreted with caution. Additional work is needed to examine the effects of these variants on protein localization and secretion, proteolytic cleavage of PGRN, and granulin peptide activity.
Missense and silent variants can also affect splicing by impairing ESEs, altering exon-splicing inhibitor sequences, or generating cryptic splice sites. In fact, many recent studies show that silent variations can affect splicing and cause human diseases.49-51 Also, these types of changes can alter mRNA folding, translational control, and gene regulation.52-54 We used in silico predictions and ex vivo assays to evaluate the effects of missense and silent changes on RNA splicing. The missense variant (p.A276V, c.827C>T) and 1 of the silent variants (c.903G>A) appear to affect splicing in at least 1 of the assays used in this study. Thus, these changes potentially affect splicing and have the potential to produce downstream stop codons and induce NMD.
From the results of splice site analysis and ex vivo splicing assays, we also identified 2 candidate exons (E3 and E8) that could be involved in naturally occurring alternative splicing of GRN. According to the in silico analysis (eTable 2), the 3′ end of GRN exon 3 carries a weak splice donor site with a score of 0.26 (of a total of 1.0) in the NNSPLICE prediction. A potential biological consequence of such a weak splice site is the presence and usage of alternative splice sites in the vicinity of this exon 3 donor site. In addition, our ex vivo splicing assays with the c.1179 + 2T>C minigene construct suggested the presence of an alternatively spliced site within exon 8 of GRN. Searching through the expressed sequence tag database, we identified multiple GRN expressed sequence tags that were alternatively spliced around exons 3 and 8 (Figure 2). Overall, these results suggest that alternatively spliced isoforms of GRN could have distinct functions. Thus, some of the missense and silent coding variants identified in this study could influence the regulation of alternative splicing of GRN transcripts.
Figure 2.

GRN expressed sequence tags (ESTs) with alternatively spliced exons. The EST data were extracted from University of California Santa Cruz genome browser (Genome Bioinformatics Group). RefSeq indicates reference sequence.
FREQUENCY OF PATHOGENIC GRN MUTATIONS
In this study, pathogenic mutations in GRN, all heterozygous, were detected in 30 of 434 unrelated patients (6.9%) with clinically or pathologically defined FTD (including CBS with corticobasal degeneration, ALS/FTD, FTD with motor neuron disease, progressive supranuclear palsy, Pick disease, and dementia lacking distinctive histopathology) and FTLD-U. Pathogenic mutations were not detected in patients with other related diseases (Table 1). Twenty-eight of the 30 cases identified with GRN mutations occurred in families with a history of a similar neurodegenerative disorder, whereas 1 had insufficient history to be definitively determined (p.C31LfsX35) and another appeared to be sporadic (p.A472VfsX10). Overall, the frequency of GRN mutations among individuals with clinical FTD and a family history of an FTD-like illness was 16.0% (28 of 175), and it was 6.9% (30 of 434) in all cases of clinical FTD. If only cases with autopsy-confirmed FTLD-U are considered, the GRN mutation frequency is 56.2% (9 of 16) in individuals with a family history and 21.4% (9 of 42) among all FTLD-U cases.
DISEASE MECHANISM CONSIDERATIONS
From this study, it is apparent that the majority of GRN mutations introduce a premature termination codon, suggesting that their corresponding mRNAs will be degraded by the NMD mechanism. These results support the notion that most FTD-associated GRN mutations are expected to create functional null alleles. It also implies that normal PGRN levels are required for neuronal survival. Although the intracellular accumulation of pathological forms of TDP-43 is a consistent feature of FTD cases with GRN mutations, reduction of PGRN level does not appear to induce mislocalization or generation of pathologic TDP-43 species such as C-terminal fragments.13 Therefore, the mechanisms by which GRN haploinsufficiency leads to neurodegeneration in FTD remains unclear. It is also possible that other, currently unknown, molecular mechanisms account for the intrafamilial and interfamilial clinical heterogeneity associated with GRN mutations.
Supplementary Material
Acknowledgments
Funding/Support: This work was funded by Veterans Affairs Biomedical Laboratory Research Development Merit Review, the National Institutes of Health (grants AG10124, AG17586, AG005136-22, AG14382, and AG008017), and the German Federal Ministry of Education and Research (grant 01GI0704).
Footnotes
REFERENCES
- 1.Stevens M, Van Duijn CM, Kamphorst W, et al. Familial aggregation in frontotemporal dementia. Neurology. 1998;50(6):1541–1545. doi: 10.1212/wnl.50.6.1541. [DOI] [PubMed] [Google Scholar]
- 2.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. 1999;56(7):817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Work Group on Frontotemporal Dementia and Pick’s Disease. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58(11):1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 4.Bird T, Knopman D, VanSwieten J, et al. Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol. 2003;54(suppl 5):S29–S31. doi: 10.1002/ana.10572. [DOI] [PubMed] [Google Scholar]
- 5.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(pt 9):2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 6.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129(pt 4):868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 7.Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66(6):839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 8.Hosler BA, Siddique T, Sapp PC, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA. 2000;284(13):1664–1669. doi: 10.1001/jama.284.13.1664. [DOI] [PubMed] [Google Scholar]
- 9.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 10.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 11.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15(20):2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 12.Le Ber I, van der Zee J, Hannequin D, et al. French Research Network on FTD/FTD-MND. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat. 2007;28(9):846–855. doi: 10.1002/humu.20520. [DOI] [PubMed] [Google Scholar]
- 13.Shankaran SS, Capell A, Hruscha AT, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283(3):1744–1753. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- 14.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 15.Seelaar H, Schelhaas HJ, Azmani A, et al. TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without progranulin mutations. Brain. 2007;130(pt 5):1375–1385. doi: 10.1093/brain/awm024. [DOI] [PubMed] [Google Scholar]
- 16.Hasegawa M, Arai T, Akiyama H, et al. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain. 2007;130(pt 5):1386–1394. doi: 10.1093/brain/awm065. [DOI] [PubMed] [Google Scholar]
- 17.Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61(5):435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pickering-Brown SM, Baker M, Gass J, et al. Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain. 2006;129(pt 11):3124–3126. doi: 10.1093/brain/awl289. [DOI] [PubMed] [Google Scholar]
- 19.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 20.Leverenz JB, Yu CE, Montine TJ, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130(pt 5):1360–1374. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- 21.Van Deerlin VM, Wood EM, Moore P, et al. Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol. 2007;64(8):1148–1153. doi: 10.1001/archneur.64.8.1148. [DOI] [PubMed] [Google Scholar]
- 22.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15(1):7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 23.Huey ED, Grafman J, Wassermann EM, et al. Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann Neurol. 2006;60(3):374–380. doi: 10.1002/ana.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesulam M, Johnson N, Krefft TA, et al. Progranulin mutations in primary progressive aphasia: the PPA1 and PPA3 families. Arch Neurol. 2007;64(1):43–47. doi: 10.1001/archneur.64.1.43. [DOI] [PubMed] [Google Scholar]
- 25.Spina S, Murrell JR, Huey ED, et al. Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology. 2007;68(11):820–827. doi: 10.1212/01.wnl.0000254460.31273.2d. [DOI] [PubMed] [Google Scholar]
- 26.Davion S, Johnson N, Weintraub S, et al. Clinicopathologic correlation in PGRN mutations. Neurology. 2007;69(11):1113–1121. doi: 10.1212/01.wnl.0000267701.58488.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruni AC, Momeni P, Bernardi L, et al. Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology. 2007;69(2):140–147. doi: 10.1212/01.wnl.0000265220.64396.b4. [DOI] [PubMed] [Google Scholar]
- 28.Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat. 2008;29(12):1373–1386. doi: 10.1002/humu.20785. [DOI] [PubMed] [Google Scholar]
- 29.Behrens MI, Mukherjee O, Tu PH, et al. Neuropathologic heterogeneity in HDDD1: a familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord. 2007;21(1):1–7. doi: 10.1097/WAD.0b013e31803083f2. [DOI] [PubMed] [Google Scholar]
- 30.Bronner IF, Rizzu P, Seelaar H, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet. 2007;15(3):369–374. doi: 10.1038/sj.ejhg.5201772. [DOI] [PubMed] [Google Scholar]
- 31.Schymick JC, Yang Y, Andersen PM, et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis–frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 2007;78(7):754–756. doi: 10.1136/jnnp.2006.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Zee J, Le Ber I, Maurer-Stroh S, et al. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat. 2007;28(4):416. doi: 10.1002/humu.9484. doi:10.1002/humu.9484. [DOI] [PubMed] [Google Scholar]
- 33.D’Souza I, Schellenberg GD. Tau exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J Biol Chem. 2002;277(29):26587–26599. doi: 10.1074/jbc.M203794200. [DOI] [PubMed] [Google Scholar]
- 34.Mukherjee O, Pastor P, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60(3):314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benussi L, Binetti G, Sina E, et al. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging. 2008;29(3):427–435. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 36.Boeve BF, Baker M, Dickson DW, et al. Frontotemporal dementia and parkinsonism associated with the IVS1+1G®A mutation in progranulin: a clinicopathologic study. Brain. 2006;129(pt 11):3103–3114. doi: 10.1093/brain/awl268. [DOI] [PubMed] [Google Scholar]
- 37.Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129(pt 11):3115–3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- 38.Snowden JS, Pickering-Brown SM, Mackenzie IR, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006;129(pt 11):3091–3102. doi: 10.1093/brain/awl267. [DOI] [PubMed] [Google Scholar]
- 39.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 40.Ferrer-Costa C, Orozco M, de la Cruz X. Sequence-based prediction of pathological mutations. Proteins. 2004;57(4):811–819. doi: 10.1002/prot.20252. [DOI] [PubMed] [Google Scholar]
- 41.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297(5583):1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 44.He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9(2):225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- 45.Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuroinflammation. 2007;4:7. doi: 10.1186/1742-2094-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48(7):999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- 47.Chiba S, Suzuki M, Yamanouchi K, Nishihara M. Involvement of granulin in estrogen-induced neurogenesis in the adult rat hippocampus. J Reprod Dev. 2007;53(2):297–307. doi: 10.1262/jrd.18108. [DOI] [PubMed] [Google Scholar]
- 48.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5(2):89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 49.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3(4):285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 50.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17(4):419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 51.Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. 2004;5(5):389–396. doi: 10.1038/nrg1327. [DOI] [PubMed] [Google Scholar]
- 52.Shen LX, Basilion JP, Stanton VP., Jr. Single-nucleotide polymorphisms can cause different structural folds of mRNA. Proc Natl Acad Sci U S A. 1999;96(14):7871–7876. doi: 10.1073/pnas.96.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frittitta L, Ercolino T, Bozzali M, et al. A cluster of three single nucleotide polymorphisms in the 3′-untranslated region of human glycoprotein PC-1 gene stabilizes PC-1 mRNA and is associated with increased PC-1 protein content and insulin resistance-related abnormalities. Diabetes. 2001;50(8):1952–1955. doi: 10.2337/diabetes.50.8.1952. [DOI] [PubMed] [Google Scholar]
- 54.Muro AF, Caputi M, Pariyarath R, Pagani F, Buratti E, Baralle FE. Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol Cell Biol. 1999;19(4):2657–2671. doi: 10.1128/mcb.19.4.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.