

Abstract
A series of alkyne containing type II inhibitors with potent inhibitory activity of T315I Bcr-Abl has been identified. The most active compound 4 exhibits an EC50 of less than 1 nM against wild-type Bcr-Abl and an EC50 of 10 nM against T315I mutant but is broadly active against a number of other kinases.
The success of Bcr-Abl inhibitor imatinib (Gleevec®, Novartis Pharma AG) for the treatment of Chronic Myelogenous Leukemia (CML) has provided the paradigm for targeting dominant oncogenes with small molecules.1,2 Imatinib resistance is rare in chronic phase patients, however for patients with blast crisis phase CML or Philadelphia chromosome-positive CML, resistance is common after an initial response in the first year.3,4 To address these relapses, two more potent ATP-site directed agents: nilotinib (AMN107)5 and dasatinib (BMS-354825)6 have been approved as the second-line therapy. Although both compounds inhibit most of the mutations that induce resistance to imatinib, neither compound is capable of inhibiting the so-called ‘gatekeeper’ T315I mutation.7 Because of the clinical importance of this mutation, there has been intense interest in the synthesis of novel inhibitors that are able to circumvent this mutation.
Recently, several compounds from the Type-II class8 that recognize the “DFG-out” conformation have been reported to inhibit T315I. These include cyclic urea compound 14,9 BGG463,10 AP24163,11 DSA series compounds,12 HG-7-85-0113 and AP2453414. A co-crystal structure of T315I with AP24534, an imidazo[1,2b]-pyridazine-based multi-targeted inhibitor demonstrates how this compound can circumvent a larger residue at the gatekeeper reside.14
In our efforts to identify new molecular scaffolds that could target T315I mutant of Bcr-Abl, we recently reported the discovery of HG-7-85-01, a small molecule type II inhibitor that inhibits the proliferation of cells expressing the major imatinib-resistant gatekeeper mutants, BCR-ABL-T315I, Kit-T670I, PDGFRα-T674M/I, as well as Src-T341M/I.13 HG-7-85-01 was designed as a hybrid between the type I inhibitor dasatinib and the type II inhibitor, nilotinib. Specifically, a superposition of the Abl-bound conformation of dasatinib (PDB code: 2GQG)15 and nilotinib (PDB code: 3CS9)5 guided the choice of how to connect the aminothiazole hinge-interacting motif of dasatinib with the N-(3-(trifluoromethyl)phenyl)-benzamide substructure of nilotinib, which is known to be responsible for inducing the “DFG-out” flip that is characteristic of type II kinase inhibitors. Our results demonstrate that it is possible to design a Type-II inhibitor that can circumvent the T315I Bcr-Abl “gatekeeper” mutation by bridging the ATP and allosteric binding site using a linker segment that can accommodate a larger gatekeeper residue. Here we report on our efforts applying this strategy to synthesize type II inhibitor using an alkyne as a linear linkage segment that can traverse a larger gatekeeper residue. A number of compounds from this series exhibit highly potent activities against both wild-type and T315I mutant of Bcr-Abl.
Molecular modeling suggested that the triple-bond linkage should be used to connect the toluene moiety of imatinib/nilotinib with a variety of heterocycles that would be capable of forming hydrogen bonding interactions with the kinase hinge region (Figure 1). This scaffold is exemplified by structures I and II. Concise synthetic routes were developed to prepare I and II (Scheme 1 and 2). Sonogashira coupling16 is used as the key reaction in both synthetic routes. Scheme 1 shows the details of synthesis of compound 3, starting with the amide condensation of freshly prepared 3-iodo-4-methylbenzoyl chloride with 4-((4-ethyl-piperazin-1-yl)methyl)-3-(trifluoromethyl)benzenamine to afford the iodo-intermediate 1. Alkyne intermediate 2 is obtained using a Sonogashira coupling of intermediate 1 with ethynyltrimethylsilane followed by deprotection of the TMS group. The final product 3 is obtained using another Sonogashira coupling of 2 with 3-iodopyridine. Compounds 4 to 9 were synthesized analogously using different heteroaromatic iodides or bromides in the final coupling step.
Figure 1.

Scaffold design strategy.
Scheme 1.

Synthetic route of 3.a
a Reagents and conditions: (a) SOCl2, reflux, 1h; (b) 4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)-benzenamine, DIEA, CH2Cl2, 0 °C to RT, 56% over two steps; (c) ethynyltrimethylsilane, Pd(PPh3)4, CuI, DIEA, DMF, RT, 62%; (d) TBAF, THF, RT, 72%; (e) 3-iodopyridine, Pd(PPh3)4, CuI, DIEA, DMF, 50 °C, 72%.
Scheme 2.

Synthetic route of 12.a
a Reagents and conditions: (a) ethynyltrimethylsilane, Pd(PPh3)4, CuI, DIEA, DMF, 50 °C, 55%; (b) TBAF, THF, RT, 77%; (c) 1, Pd(PPh3)4, CuI, DIEA, DMF, RT, 82%.
Synthesis of 12 was accomplished by introduction of ethynyl group to 5-bromo-1H-pyrrolo[2,3-b]pyridine followed by coupling with iodo-intermediate 1 (Scheme 2). Compounds 13-20 were obtained following this synthetic route.
To assess the cellular activity of the compounds, we tested them against parental, wild-type and T315I Bcr-Abl transformed Ba/F3 cells. Wild-type Ba/F3 cell proliferate only in the presence of interlukin-3 (IL-3) while Ba/F3 cells transformed with oncogenic kinases such as Bcr-Abl become capable of growing in the absence of IL-3 and provides a robust and commonly used assay for selective kinase inhibition.17
The first compound we synthesized 3 exhibited an EC50 of less than 1 nM on wild-type Bcr-Abl and an EC50 of 92 nM on T315I. The EC50 against parental Ba/F3 cells was 1.59 uM demonstrating that the antiproliferative activity was derived from on-target inhibition of Bcr-Abl. Encouraged by this initial result, a small set of compounds were synthesized to investigate the SAR and validate our design strategy (Table 1). Introduction of substituted amino group to the pyridine 6-position (4 and 5) resulted in an approximate 8-fold improvement relative to 3 against T315I mutant Bcr-Abl. This might be attributed to the introduction one additional hydrogen bond to the kinase hinge from the amino-group. While a carbonyl group as is present in Sorafenib at the 6-position resulted in a compound 6 that exhibited an 6-fold less potent EC50 against both wild-type and T315I Bcr-Abl. Replacing the pyridine head with pyrimidine and pyrazine resulted in approximately equipotent compounds 7 and 8 against wild-type but decreased potency on T315I mutant. The identification of highly potent compounds 3-5 clearly validates our design strategy. The results also demonstrate that T315I Bcr-Abl is less potently inhibited relative to wild-type by this inhibitor series.
Table 1.
SAR of mono-heterocyclic head alkynyl analogs.
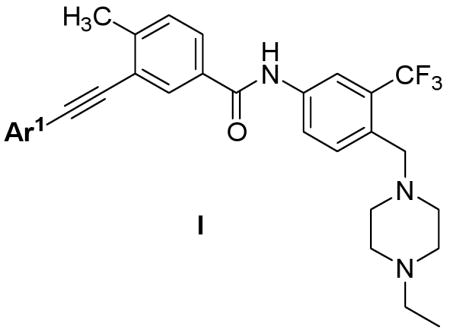 | ||||
|---|---|---|---|---|
| Compds | Ar1 | Cellular antiproliferative activity (EC50, μM) | ||
| Bcr-Abl (wt)a | Bcr-Abl (T315I)b | BaF3c | ||
| 3 | 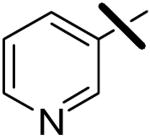 |
<0.001 | 0.092 | 1.59 |
| 4 | 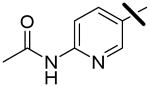 |
<0.001 | 0.010 | 2.05 |
| 5 | 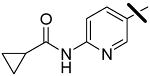 |
<0.001 | 0.013 | 1.80 |
| 6 | 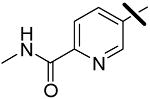 |
0.041 | 2.58 | 3.98 |
| 7 | 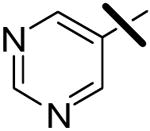 |
<0.001 | 0.99 | 2.19 |
| 8 | 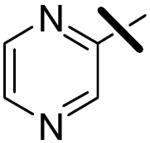 |
0.006 | 3.03 | 2.49 |
| 9 | 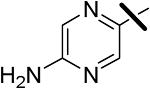 |
<0.001 | 0.165 | 0.86 |
Cellular antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
Cellular antiproliferative activity (EC50, μM) on mutant Bcr-Abl-T315I-Ba/F3.
Cytotoxicity (EC50, μM) on wt-Ba/F3.
We next investigate the effects of using 6-5 and 6-6 fused heterocyclic rings such as 7-aza-indole, imidazopyridine, pyridopyrazine and benzofuran as hinge-interacting motifs (Table 2). Most of the resulting compounds exhibited EC50 values of less than two-digit nanomolar against wild-type Bcr-Abl, but only compounds 12 and 14 exhibited EC50 values of less than 100 nM against T315I. A comparison of potencies of compounds 12 and 17 demonstrate that the presence of the toluene methyl group is an important structural element for achieving potent inhibition against both wild-type and T315I Bcr-abl. An analysis of the Abl bound conformations of imatinib, nilotinib and AP24534 suggests that the methyl group favors the twisted conformation required for high affinity binding.5,14,18 The orientation of amide found in nilotinib (12) is favored over the reverse amide orientation found in imatinib (20).
Table 2.
SAR of bi-heterocyclic head alkynyl analogs.
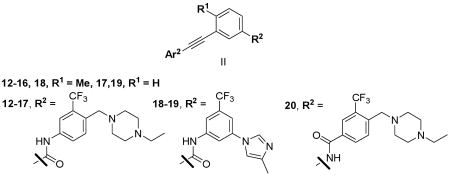 | ||||
|---|---|---|---|---|
| Compds | Ar1 | Cellular antiproliferative activity (EC50, μM) | ||
| Bcr-Abl (wt)a | Bcr-Abl (T315I)b | BaF3c | ||
| 12 | 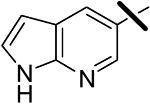 |
<0.001 | 0.080 | 1.00 |
| 13 | 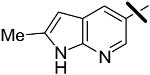 |
0.026 | 0.56 | 1.00 |
| 14 | 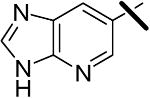 |
<0.001 | 0.014 | 0.060 |
| 15 | 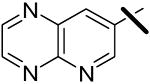 |
0.037 | 5.52 | 3.32 |
| 16 | 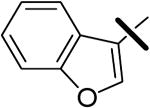 |
0.24 | 8.37 | 6.76 |
| 17 | 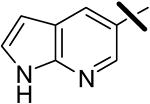 |
0.011 | 0.22 | >10.0 |
| 18 | 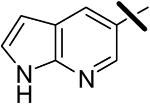 |
0.037 | 0.47 | >10.0 |
| 19 | 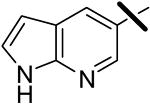 |
0.015 | 0.30 | 2.83 |
| 20 | 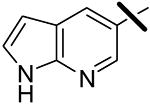 |
0.070 | 2.63 | 7.86 |
Cellular antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
Cellular antiproliferative activity (EC50, μM) on mutant Bcr-Abl-T315I-Ba/F3.
Cytotoxicity (EC50, μM) on wt-Ba/F3.
The selectivity of this scaffold was assessed using KINOMEscan™ (Ambit Biosciences, San Diego, CA),19 a high-throughput method for screening kinase inhibitors against a panel of 442 kinases. Compounds 3 and 12, were screened at concentration of 10 μM. This analysis revealed that the compounds possessed an extremely broad selectivity profile with compounds 3 and 12 exhibiting 193 and 203 kinases with ambit score of less than 10% of the DMSO control respectively. The kinase hits for 12 with ambit score less 0.1% of the DMSO control were highlighted in spot tree (Figure 2, please see supplemental material for full screening data of 12 and 3). The potently targeted kinases were mainly from the TK, TKL, STE and CMGC groups. The selectivity scores19 for 3 (S10 = 0.427) and 12 (S10 = 0.449) indicate that these compounds are considerably less specific relative to compounds such as HG-7-85-01 (S10 = 0.056)13. As compounds 3 and 12 are highly rigid structures they are most likely only to bind with high affinity to the ‘DFG-out’ conformation as confirmed by the recent co-structure of AP24534 with T315I Bcr-Abl14. These kinase profiling results demonstrate that a very large number of kinases can be potently targeted in this conformation and inhibitors 3 and 12 represent starting points for the design of multitargeted inhibitors with the potential to target diverse combinations of kinase targets.
Figure 2.
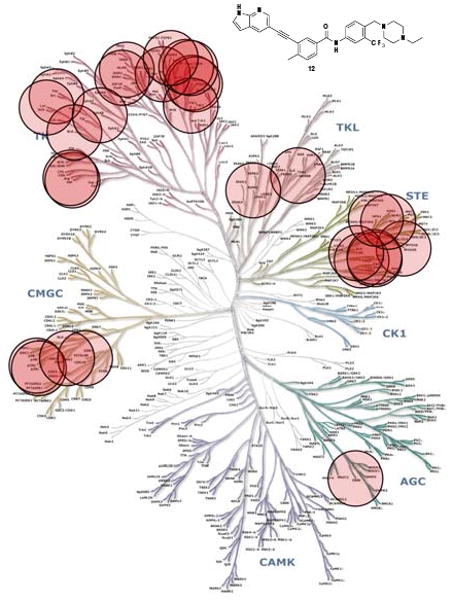
KINOMEscan™ profiling of 12. Compounds were screened at a concentration10 μM against 442 kinases and the most potently bound kinases are indicated by red circles and include: ABL, ARG, BLK, PITSLRE, CRIK, FMS/CSFR, DDR1, EphA8, FGFR4, HPK1, JNK2, KIT, LOK, LYN, GCK, MUSK, MYO3B, p38α, p38β, PDGFRα, PDGFRβ, RET, RIPK1, SRC, TAO1, TAO2, TAO3 and TIE2.
In summary, we have used a structure-based design approach to design new type II scaffold using an alkyne as a linker segment between a heterocyclic hinge interacting motif and a trifluoromethylphenylamide motif that binds to the pocket created by the ‘DFG-out’ conformation. The compounds exhibit very potent cellular activity against wild-type and T315I Bcr-Abl. Despite being extremely promiscuous kinase inhibitors, compounds such as 3 and 12 are not non-specifically cytotoxic and exhibit up to 1000-fold selectivity for Bcr-Abl dependent cellular growth. Further medicinal chemistry efforts are in progress to develop analogs from this compound series whose multitargeted inhibition profile is tailored for optimal activity against particular cancer genotypes.
Supplementary Material
Acknowledgments
This work was supported by the NIH, R01 CA130876-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Nat Med. 1996;2:561. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. New Engl J Med. 2001;344:1031. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 3.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, Tura S, Fischer T, Deininger MW, Schiffer CA, Baccarani M, Gratwohl A, Hochhaus A, Hoelzer D, Fernandes-Reese S, Gathmann I, Capdeville R, O'Brien SG. Blood. 2002;100:1965. doi: 10.1182/blood-2001-12-0181. [DOI] [PubMed] [Google Scholar]
- 4.Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MWN, Fischer T, O'Brien SG, Stone RM, Gambacorti-Passerini CB, Russell NH, Reiffers JJ, Shea TC, Chapuis B, Coutre S, Tura S, Morra E, Larson RA, Saven A, Peschel C, Gratwohl A, Mandelli F, Ben-Am M, Gathmann I, Capdeville R, Paquette RL, Druker BJ. Blood. 2002;99:3530. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 5.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Cancer Cell. 2005;7:129. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Science. 2004;305:399. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 7.O'Hare T, Walters DK, Deininger MW, Druker BJ. Cancer Cell. 2005;7:117. doi: 10.1016/j.ccr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Gray NS. Nat Chem Biol. 2006;2:358. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 9.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Nat Struct Mol Biol. 2008;15:1109. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manley PW, Bruggen J, Floersheimer A, Furet P, Jensen MR, Mestan J, Pissot C, Cowan-Jacob S. Chimia. 2008;62:579. [Google Scholar]
- 11.Huang WS, Zhu X, Wang Y, Azam M, Wen D, Sundaramoorthi R, Thomas RM, Liu S, Banda G, Lentini SP, Das S, Xu Q, Keats J, Wang F, Wardwell S, Ning Y, Snodgrass JT, Broudy MI, Russian K, Daley GQ, Iuliucci J, Dalgarno DC, Clackson T, Sawyer TK, Shakespeare WC. J Med Chem. 2009;52:4743. doi: 10.1021/jm900166t. [DOI] [PubMed] [Google Scholar]
- 12.Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, Maly DJ. Cancer Res. 2009;69:2384. doi: 10.1158/0008-5472.CAN-08-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisberg E, Choi HG, Ray A, Barrett R, Zhang J, Sim T, Zhou W, Seeliger M, Cameron M, Azam M, Mayeda M, Moreno D, Kung AL, Janne PA, Khosravi-Far R, Melo JV, Manley P, Adamia S, Wu C, Gray NS, Griffin JD. Blood. 2010 doi: 10.1182/blood-2009-11-251751. PMID: 20299508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA, 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. Cancer Cell. 2009;16:401. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FY, Borzillerri R, Lombardo LJ, Xie D, Zhang Y, Klei HE. Cancer Res. 2006;66:5790. doi: 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- 16.Chinchilla R, Najera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 17.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, Beigi R, Xia G, Harig RA, Asatryan H, Yan SF, Zhou Y, Gu XJ, Saadat A, Zhou V, King FJ, Shaw CM, Su AI, Downs R, Gray NS, Schultz PG, Warmuth M, Caldwell JS. Proc Natl Acad Sci U S A. 2006;103:3153. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Science. 2000;289:1938. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 19.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. Nat Biotechnol. 2008;26:127. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.