

It is well recognized that patients with bilateral retinoblastoma undergoing radiotherapy are highly susceptible to developing second primary malignancies, particularly osteosarcoma and soft tissue sarcomas.1 Rhabdomyosarcoma is a rare secondary malignancy in these children.2–9 Herein, we report a clinicopathologic correlation of rhabdomyosarcoma of the temporalis muscle as a radiation-induced second neoplasm following retinoblastoma therapy, with an unusually rapid presentation.
Case Report
A male Puerto Rican child was diagnosed with familial, advanced, bilateral retinoblastoma (RB, group Vb) at 3 months of age. The patient was managed with 8 cycles of vincristine (0.05 mg/kg) and carboplatin (560 mg/m2) systemic chemoreduction therapy. Due to recurrent disease development 3 months later, at 11 months of age, the patient received external beam radiation therapy (EBRT) of 45 Gy to both orbits. At age 2 years 11 months, (2 years following radiotherapy and 2 years 8 months after RB diagnosis), the ocular tumor was felt to be stable; however, the patient had developed a rapidly increasing left temporal mass in the field of radiation, and lymphadenopathy on physical exam.
Radiographic imaging revealed a “grape-like” solid mass in the left temporalis muscle involving both infra and suprazygomatic components, as well as enlarged and abnormal lymph nodes in the posterior aspect of the parotid gland (Figure 1).
Figure 1.
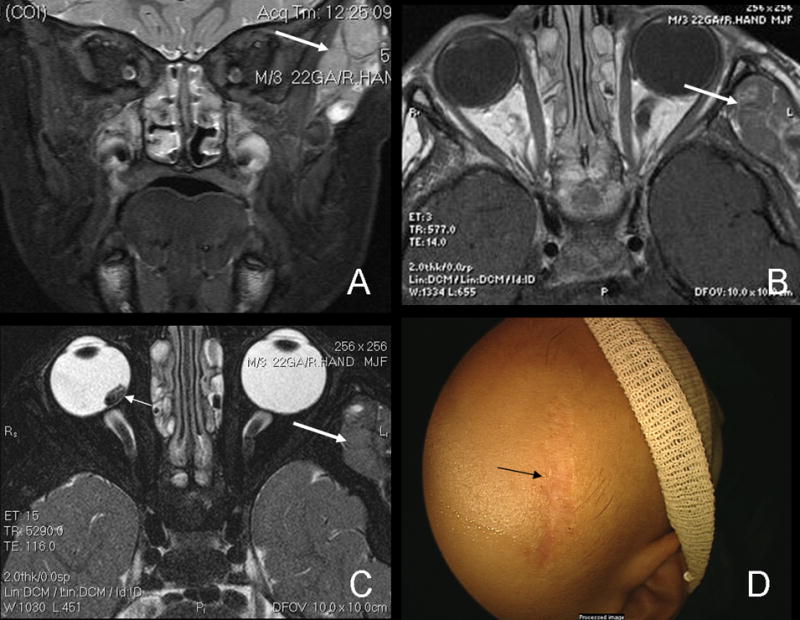
Short inversion-time inversion-recovery (STIR) MRI of temporalis mass (A). The heterogeneous, well circumscribed mass which is hyperintense to muscle can also be seen on T1-weighted images (B) as well as T2 images (C), large arrows. There is no radiological evidence of extension into the globe or bones. The residual retinoblastoma tumor can be seen on the T2 image as well (small arrow, C). Post-chemotherapy for rhabdomyosarcoma, the mass has resolved, leaving only the biopsy scar (D).
The patient’s biopsy revealed morphologic features consistent with rhabdomyosarcoma (Figure 2). Pathologic analysis of hematoxylin and eosin (H&E) stained sections revealed small, round, blue neoplastic cells with scanty cytoplasm, sometimes in nests, forming a pseudoalveolar pattern (Figure 2). Nuclei were uniform. No Flexner-Wintersteiner or other rosette-like structures were identified. Special immunostains demonstrated that the neoplastic cells were positive for myogenin and myo-D1 (Figure 2), and negative for CD99, TdT and synaptophysin, ruling-out Ewing’s sarcoma/primitive neuroectodermal tumors (PNET), lymphoma, and retinoblastoma.
Figure 2.
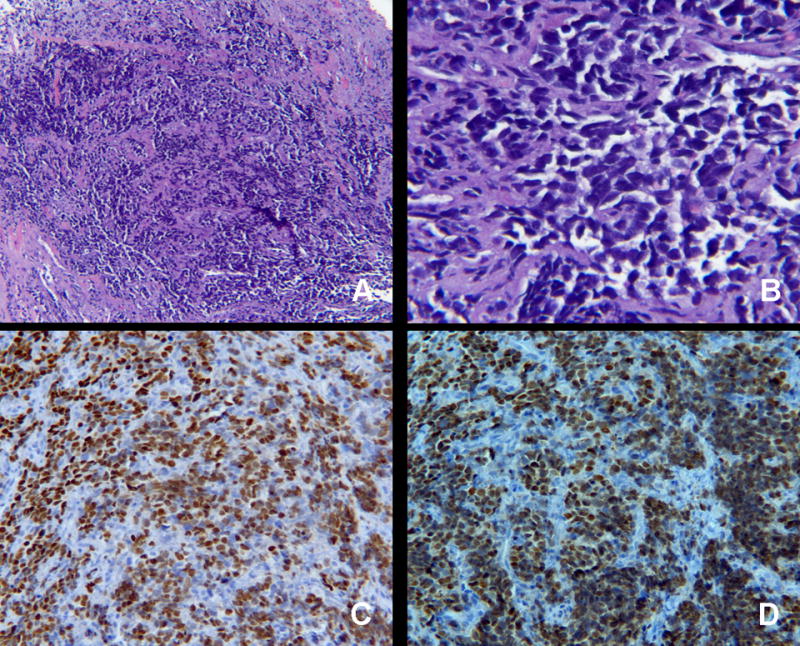
Histopathology of left temporalis mass. H&E is shown demonstrating sheets of round, homogenous blue cells at 40 × magnification (A) and 400× magnification (B). The neoplastic cells are positive for rhabdomyosarcoma markers myogein (C) and myod-D1 (D), magnification 200×.
The patient’s family moved and shortly presented to our ocular oncology service. He is continuing the low-risk COG rhabdomyosarcoma chemotherapy protocol and has had complete resolution of the temporal mass (Figure 1). He continues to undergo examinations under anesthesia and laser therapy for advanced bilateral retinoblastoma (Figure 3).
Figure 3.
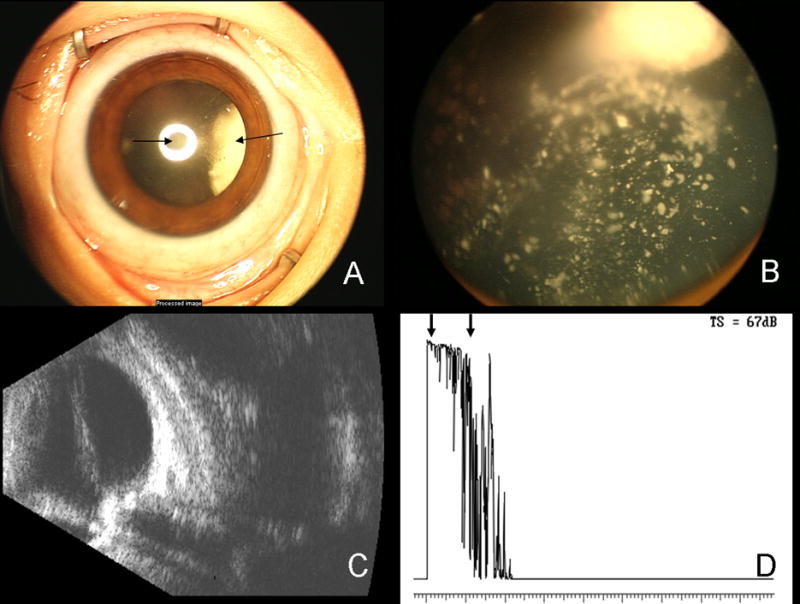
Ret-Cam photograph of the patient’s right eye with advanced retinoblastoma (A). A posterior subcapsular cataract, induced by radiation, can be seen (arrow, center) as well as the white tumor around 3:00 (arrow, right). Ret-Cam photograph of the fundus of the right eye shows the advanced retinoblastoma, including inactive calcific vitreous seeds (B). B-scan ultrasound of the right eye demonstrates the shadowing generated by the calcific RB tumors (C). Diagnostic A-scan demonstrates high internal reflectivity of the calcific tumor (arrows, D).
DISCUSSION
Rhabdomyosarcoma is a rare second cancer in hereditary retinoblastoma patients. In the largest series of soft tissue sarcomas in survivors of hereditary RB, only 8 out of 963 patients developed rhabdomyosarcoma.7
The case presented herein is unusual for the appearance of a solid malignancy in the field of radiation, only 2 years following EBRT, and 2 years 8 months after diagnosis of RB. In a review of historical cases of retinoblastoma by Hasegawa et al., the mean length of time to develop secondary rhabdomyosarcoma tumors was 6 years 6 months, with a range of 1 year 3 months to 15 years 6 months after the diagnosis of RB.2 In the largest series of soft tissue sarcomas in survivors of hereditary RB, 12% (8/69 soft tissue sarcoma patients) were diagnosed with rhabdomyosarcoma, with 4 cases presenting between 1–9 years, 3 cases presenting 10–19 years, and 1 case presenting more than 30 years after the RB diagnosis.7 All cases had received radiotherapy for RB.
Further evaluation of the data from Kleinerman et al.7 revealed that the mean and median ages from RB to rhabdomyosarcoma diagnosis were 12.6 and 9.5 years, respectively (+/− 12.0 years standard deviation, Table 1). The age at presentation of the second cancer ranged from age 3 to 42 years. In a Dutch series of 263 hereditary RB patients, rhabdomyosarcoma was diagnosed in three patients at ages 8 and 10 after EBRT, and at age 9 after EBRT in combination with unknown type of chemotherapy.9
Table 1.
Comparison of current case with data from Kleinerman et al. and Moll et al.
| Patient Number | Gender | Age RB | Age Rhabdomyosarcoma | Year of Radiation Treatment | Rhabdomyosarcoma Location |
|---|---|---|---|---|---|
| 1 ref. 7 | M | 5 y | 12y | 1934 | Temporal region |
| 2 ref. 7 | F | 1y | 42y | 1948 | Ethmoid sinus |
| 3 ref. 7 | M | <1y | 14y | 1949 | Temporal muscle |
| 4 ref. 7 | M | 1y | 8y | 1950 | Lower eye lid |
| 5 ref. 7 | F | <1y | 12y | 1971 | Cheek |
| 6 ref. 7 | M | <1y | 13y | 1975 | Temporal parotid |
| 7 ref. 7 | F | <1y | 9y | 1976 | Leptomeninges |
| 8 ref. 7 | M | <1y | 3y | 1983 | Temporal region |
| 9 ref. 9 | M | <1 y | 9y | 1968 | Face |
| 10 ref. 9 | F | < 1 y | 8y | 1984 | Zygoma |
| 11 ref. 9 | F | <1 y | 10y | 1987 | Os lacrimalis |
| Current Case | M | <1 y | 2y 11 mo | 2003 | Temporalis muscle |
The case we present herein was diagnosed with rhabdomyosarcoma 2 years and 8 months after RB, and the earliest case of the Kleinerman et al. series was diagnosed 3 years and 3 months after RB. Both of these early onset cases had shorter latency intervals than all of the other rhabdomyosarcoma cases reported from both the Kleinerman and Moll studies. (Table 1).
Considerations for the early onset of rhabdomyosarcoma in our case include genetic predisposition, the combination of radiotherapy with chemotherapy, or both. The predisposition to soft tissue sarcomas in RB patients has been attributed to a germline mutation in the RB gene10 and the appearance of a solid tumor in less than 3 years after retinoblastoma suggests this possibility. In two large series of childhood cancer patients (not including retinoblastoma), the risk of a second primary soft tissue sarcoma was increased with radiotherapy in one study11 and with both high-dose radiotherapy and alkylating agents in the other study.12 However, neither of these studies included soft tissue sarcomas occurring less than 3 years after the first cancer. It is also possible that the chemotherapy in this case acted as a radiation sensitizer that could have contributed to the early appearance of his rhabdomyosarcoma.
Future studies should evaluate hereditary RB cases carefully for any links between chemotherapy-promoted, radiation-induced second cancers, particularly for rhabdomyosarcoma.
Acknowledgments
We would like to thank Dr. Dan S. Gombos for helpful comments.
References
- 1.Abramson DH, Ellsworth RM, Kitchin FD, Tung G. Second nonocular tumors in retinoblastoma survivors. Are they radiation-induced? Ophthalmology. 1984;91:1351–5. doi: 10.1016/s0161-6420(84)34127-6. [DOI] [PubMed] [Google Scholar]
- 2.Hasegawa T, Matsuno Y, Niki T, Hirohashi S, Shimoda T, Takayama J, Watanabe C, Kaneko A, Sano T, Sato M, Suzuki J. Second primary rhabdomyosarcomas in patients with bilateral retinoblastoma: a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1998;22:1351–60. doi: 10.1097/00000478-199811000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Dickman PS, Barmada M, Gollin SM, Blatt J. Malignancy after retinoblastoma: secondary cancer or recurrence? Hum Pathol. 1997;28:200–5. doi: 10.1016/s0046-8177(97)90107-6. [DOI] [PubMed] [Google Scholar]
- 4.Chemello PD, Nelson CL, Tomich CE, Sadove AM. Embryonal rhabdomyosarcoma arising in the masseter muscle as a second malignant neoplasm. J Oral Maxillofac Surg. 1988;46:899–905. doi: 10.1016/0278-2391(88)90060-2. [DOI] [PubMed] [Google Scholar]
- 5.Tefft M, Vawter GF, Mitus A. Second primary neoplasms in children. Am J Roentgenol Radium Ther Nucl Med. 1968;103:800–22. doi: 10.2214/ajr.103.4.800. [DOI] [PubMed] [Google Scholar]
- 6.Brookes CN, van Velzen D. Rhabdomyosarcoma, presenting as a facial swelling in a child. A case report and review of the literature. Br J Oral Maxillofac Surg. 1990;28:117–21. doi: 10.1016/0266-4356(90)90137-a. [DOI] [PubMed] [Google Scholar]
- 7.Kleinerman RA, Tucker MA, Abramson DH, Seddon JM, Tarone RE, Fraumeni JF., Jr Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24–31. doi: 10.1093/jnci/djk002. [DOI] [PubMed] [Google Scholar]
- 8.Aerts I, Paquement H, Aerts I, Pacquement H, Doz F, Mosseri V, Desjardins L, Sastre X, Michon J, Rodriquez, Schlienger P, Zucker JM, Quintana E. Outcome of second malignancies after retinoblastoma: a retrospective analysis of 25 patients treated at the Institut Curie. Eur J Cancer. 2004;40:1522–9. doi: 10.1016/j.ejca.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 9.Moll AC, Imhof SM, Schouten-Van Meeteren AYN, Kuik DJ, Hofman P, Boers M. Second primary tumors in hereditary retinoblastoma: A register-based study, 1945–1997. Ophthalmology. 2001;108:1109–1114. doi: 10.1016/s0161-6420(01)00562-0. [DOI] [PubMed] [Google Scholar]
- 10.Friend SH, Horowitz JM, Gerber MR, Wang XF, Bogenmann E, Li FP, Weinberg RA. Deletions of a DNA sequence in retinoblastomas and mesenschymal tumors: Organization of the sequence and its encoded protein. Proc Natl Acad Sci U S A. 1987;84:9059–63. doi: 10.1073/pnas.84.24.9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkinson JC, Winter DL, Marsden HB, Stovall MA, Stevens MCG, Stiller CA, Hawkins MM. A study of soft tissue sarcomas after childhood cancer in Britain. Brit J Cancer. 2007;97:695–99. doi: 10.1038/sj.bjc.6603908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henderson TO, Whitton J, Stovall M, Mertens AC, Mitby P, Friedman D, Strong LC, Hammond S, Neglia JP, Meadows AT, Robison L, Diller L. Secondary sarcomas in childhood cancer survivors: A report from the childhood cancer survivor study. JNCI. 2007;99:300–8. doi: 10.1093/jnci/djk052. [DOI] [PMC free article] [PubMed] [Google Scholar]