

Abstract
Alzheimer's disease (AD) is a debilitating neurodegenerative disorder affecting millions of elderly individuals worldwide. Advances in the genetics of AD have led to new levels of understanding and treatment opportunities. Here, we used a systems biology approach based on weighted gene coexpression network analysis to determine transcriptional networks in AD. This method permits a higher order depiction of gene expression relationships and identifies modules of coexpressed genes that are functionally related, rather than producing massive gene lists. Using this framework, we characterized the transcriptional network in AD, identifying 12 distinct modules related to synaptic and metabolic processes, immune response, and white matter, nine of which were related to disease progression. We further examined the association of gene expression changes with progression of AD and normal aging, and were able to compare functional modules of genes defined in both conditions. Two biologically relevant modules were conserved between AD and aging, one related to mitochondrial processes such as energy metabolism, and the other related to synaptic plasticity. We also identified several genes that were central, or hub, genes in both aging and AD, including the highly abundant signaling molecule 14.3.3 ζ (YWHAZ), whose role in AD and aging is uncharacterized. Finally, we found that presenilin 1 (PSEN1) is highly coexpressed with canonical myelin proteins, suggesting a role for PSEN1 in aspects of glial-neuronal interactions related to neurodegenerative processes.
Keywords: microarray, presenilin, systems biology, WGCNA, Alzheimer's disease, aging
Introduction
Since the introduction of microarray technology over 10 years ago (Schena et al., 1995; Lockhart et al., 1996), many studies have been performed to determine the mechanisms behind various neurodegenerative disorders, including frontotemporal dementia (Karsten et al., 2006) and Alzheimer's disease (AD) (Loring et al., 2001; Colangelo et al., 2002; Blalock et al., 2004; Small et al., 2005; Emilsson et al., 2006). However, comparing gene lists from different studies often lacks a functional foundation and requires new methods to allow cross-study integration. Thus, despite the fact that AD is the most common and well studied neurological disorder of the elderly (Drachman, 2006), no coherent picture of AD pathological interactions has emerged from microarray studies. Additionally, many technical issues, such as small sample sizes and postmortem artifacts, have further reduced power (Vawter et al., 2002; Mirnics and Pevsner, 2004).
Here, we apply weighted gene coexpression network analysis (WGCNA) (Zhang and Horvath, 2005; Horvath et al., 2006; Oldham et al., 2006), a method that organizes gene expression data into a functionally relevant framework, to explore the pathophysiology of AD from a systems perspective. We demonstrate the ability of WGCNA to capture the underlying transcriptome organization inherent in a study of the CA1 region of the hippocampus in AD (Blalock et al., 2004), yielding modules of coexpressed genes involved in common disease-related processes. Nine modules are related to disease progression, including two groupings that correspond to synaptic function and metabolic processes. Within these modules, we are also able to identify the most central genes in the network (“hub genes”), such as voltage-dependent anion channel 1 (VDAC1), which are predicted to play prominent roles in the disease process.
WGCNA also creates a functional basis on which to compare different microarray studies by organizing gene lists into relevant transcriptional modules based on their coexpression relationships. So, we also applied WGCNA to determine whether there were any shared processes revealed by gene expression changes observed during the progression of AD and in normal aging (Lu et al., 2004). Although AD has many features that clearly distinguish it from normal aging, whether there are areas of biological overlap with normal aging remains an important issue (Smith et al., 1991; Price and Morris, 1999; Drachman, 2006; Keller, 2006). Remarkably, despite the differences between the samples used in the two studies analyzed here, we find significantly overlapping aspects of network organization, highlighting common biological changes that result from the progression of both AD and aging. From these networks, we also identify key hub genes that are in central positions in both studies and relate them to their underlying biological processes. Although the roles of some of these genes, such as cyclin-dependent kinase 5 (CDK5) and presenilin 1 (PSEN1), are well characterized in AD, WGCNA also identifies many novel genes in the context of AD pathology, providing a basis for further study.
Materials and Methods
Data acquisition.
Microarray data sets from two separate studies were used in this analysis: (1) microarrays assessing gene expression from the CA1 region of the hippocampus from 31 individuals, comprising nine controls, seven with incipient AD, eight with moderate AD, and seven with severe AD, as defined by the MiniMental State Examination (MMSE) score and neurofibrillary tangle (NFT) burden [(Blalock et al., 2004) henceforth, “the AD study”], and (2) 30 microarrays representing a study of the effects of aging on frontal lobe gene expression of individuals who died of natural causes between the ages of 26 and 106 [(Lu et al., 2004) henceforth, “the aging study”]. The raw data (.cel files) were obtained from the Geo DataSets database (http://www.ncbi.nlm.nih.gov/geo/) and directly from the authors (Lu et al., Harvard Medical School, Boston, MA). Experimental assays are described in full in the initial publications (Blalock et al., 2004; Lu et al., 2004). The AD study used Affymetrix HG-U133A chips containing 22,283 probe sets, and the aging study used HG-U95A chips with 12,625 probe sets.
Data processing.
Both datasets were processed identically for consistency. All preprocessing was performed in R (http://cran.r-project.org/), a freely available programming language, using microarray-specific packages available through Bioconductor (http://www.bioconductor.org/). We used the “expresso” function in the “affy” library of Bioconductor, including methods for background correction (mas), normalization (quantile), perfect match probe correction (mas), and expression summary (medianpolish) based on the study by Choe et al. (2005). Details about the significance of each step have been described previously (Huber et al., 2005).
Outlier subjects were determined calculating the un-normalized correlation matrix between subjects using the Pearson correlation (Oldham et al., 2006). Subjects were clustered based on their dissimilarity, and any arrays with average intersubject correlation <2 (AD study) or 3 (aging study) SDs (ς) below the mean were removed. This process was repeated, until no arrays needed to be removed (Oldham et al., 2006). In all, three arrays were removed from the AD study (4.3ς, 2.61ς, and 2.08ς below mean) and two were removed from the aging study (3.76ς and 3.27ς below mean). Data were processed using “expresso” as described above, this time including quantile normalization. Any probe sets that were called “present” in three or fewer arrays using the “mas5calls” function in the “affy” library were considered unreliable and removed from further analysis. Control probe sets and those not associated with known genes were also removed from further analysis. These steps left 12,073 and 6741 probe sets remaining for the AD and aging studies, respectively.
Determining variably expressed genes.
We first identified the genes with variable expression patterns across conditions (variable genes), both to simplify computation and to eliminate genes that do not change and therefore do not contribute to the correlation matrix (Zhang and Horvath, 2005; Oldham et al., 2006). We performed two types of network analyses: one in which the analysis was agnostic to any clinical or pathophysiological information (“unsupervised”), and a second in which clinical information was used to guide gene selection (“supervised”). We used a supervised analysis because this allowed us to compare networks related to the progression of aging (age as clinical variable) directly to the progression of dementia (as measured by MMSE score and NFT burden). For the unsupervised AD analysis, we selected the top 5000 most variable genes, as measured by coefficient of variance (Oldham et al., 2006). For the supervised AD study, we applied the following filters to identify a relatively inclusive set of genes: (1) Bayes ANOVA test (p < 0.05) (Baldi and Long, 2001) comparing all four groups (control, incipient AD, moderate AD, and severe AD) identified 1122 probe sets; (2) t test (p < 0.05) comparing controls and any other group identified 2010 probe sets; (3) Pearson's correlation (p < 0.05) of gene expression values with dementia stage as determined by NFT burden or MMSE score identified 3024 probe sets. Merging these gene lists together resulted in a total of 3890 variable genes for the AD supervised study. Any probe sets and associated genes for which any of the above three conditions were true are described as “correlated with AD.” We also performed a supervised analysis on the aging study, where we identified 1505 probe sets whose expression correlated significantly with age of subject as measured by Pearson correlation (p < 0.05). We did not perform an unsupervised aging analysis because the purpose of this study was to characterize normal aging only in the context of AD progression. False discovery rates [(cutoff p value) × (number of probe sets tested)/(total number of positives)] for the AD and aging studies, respectively, were 34% and 22%, so these sets of genes represent inclusive gene lists, which are appropriate as starting points for additional analysis.
Network analysis.
WGCNA was performed on variably expressed genes in each study as previously described (Zhang and Horvath, 2005; Horvath et al., 2006; Oldham et al., 2006). After finalizing the gene list, the correlation matrix was obtained by calculating the Pearson correlations between all variable probe sets across all subjects. Next, the adjacency matrix was calculated by raising the absolute values of the correlation matrix to a power (β = 6 for the unsupervised AD analysis, β = 8 for the supervised AD analysis, and β = 7 for the supervised aging analysis) (Zhang and Horvath, 2005). For computational reasons, the gene list was then further restricted by omitting probe sets with very low connectivity (k), the summation of connection strengths for each gene with all other genes. In the unsupervised AD analysis, probe sets with k/kmax < 0.10 were omitted, whereas in the AD and aging supervised analyses, the cutoff value was k/kmax < 0.05, leaving 2628, 2157, and 1334 probe sets remaining in the three respective analyses. Topological overlap (TO), a biologically meaningful measure of node similarity (how close the neighbors of gene 1 are to the neighbors of gene 2), was then calculated as described previously (Zhang and Horvath, 2005; Oldham et al., 2006). Next, the probe sets were hierarchically clustered using 1-TO as the distance measure and modules were determined by choosing a height cutoff for the resulting dendrogram (see Fig. 4a, top trace) or by using a dynamic tree-cutting algorithm (see Fig. 1a, top trace) (http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/).
Figure 4.
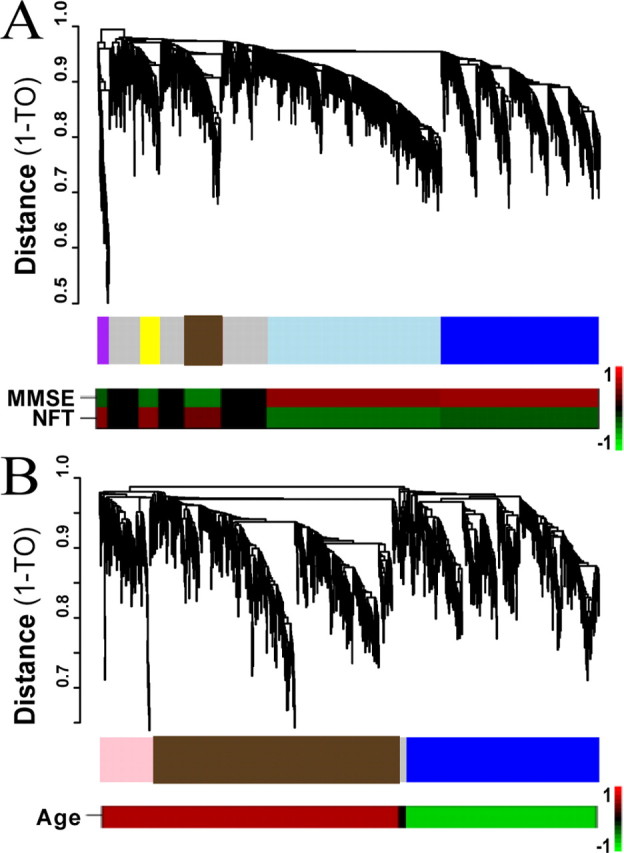
Direct comparison of AD and aging by supervised network analysis. A, B, Cluster dendrogram of genes corresponds to the AD gene expression network (A, top trace) and to the aging gene expression network (B, top trace). The y-axis corresponds to topological distance (1-TO). A height cutoff was used to characterize modules to allow for a large number of genes per module. Middle traces, The modules in each study are color-coded such that modules with significant overlap between studies (A, AD; B, aging) share a module color. Bottom traces, Heat maps corresponding to the correlation between each ME and the relevant phenotypic measures [MMSE score and NFT burden in the AD study (A) and age in the aging study (B)]. The color scale bar to the right of the bottom trace represents the Pearson correlation ranging from −1 (green) to 1 (red).
Figure 1.
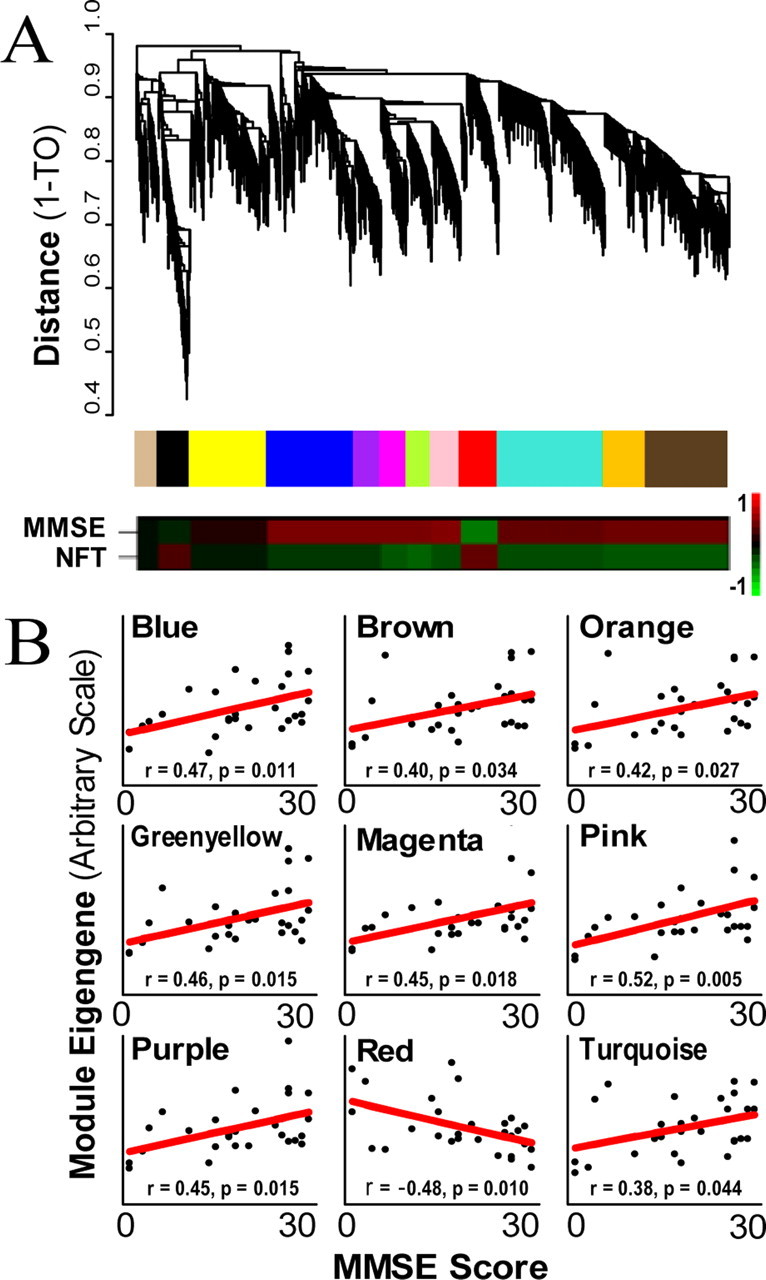
Clustering by topological overlap reveals modules of genes that are characterized by distinct expression patterns. A, Top trace, Cluster dendrogram of genes in the unsupervised AD study groups genes into distinct modules. The y-axis corresponds to distance determined by the extent of topological overlap (1-TO). Dynamic tree cutting was used to identify the most parsimonious module definitions (Materials and Methods), generally dividing modules at significant branch points in the dendrogram. Middle trace, The genes in each of the 12 modules are color-coded. Bottom trace, Heat maps corresponding to the correlation between each ME and both MMSE score and NFT burden. The color scale bar to the right of the bottom trace represents the Pearson correlation ranging from −1 (green) to 1 (red). B, MMSE score (x-axis) plotted vs module eigengene (y-axis) for all nine modules that are significantly correlated with MMSE score. Each point represents a single subject and the line is the line of best fit determined by linear regression across subjects. Only the red module shows negative correlation, indicating an increase in gene expression with AD.
Once modules were identified, the module eigengene (ME; i.e., first principal component of the expression values across subjects) was calculated using all probe sets in each module. The MEs were then correlated to relevant clinical traits using the Pearson correlation. Within-module connectivity (kin) for each probe set was determined by summing the connectivities of that probe set with each other probe set in that module. Networks were graphically depicted using the program VisANT (Hu et al., 2004; Oldham et al., 2006). Unless otherwise noted, these “network depictions” show only the top 250 reciprocal within-module gene–gene interactions (“connections”) with the strongest TO. The genes were colored based on the module color and labeled as a “hub” if they had at least 15 connections depicted.
MultiTOM analysis.
The Multinode Topological Overlap Measure for Gene Neighborhood Analysis (MultiTOM) software was also used to create “local networks” for genes of interest (Li and Horvath, 2007). MultiTOM takes an expression array and a “seed” [probe set(s) of interest] as input, defining this seed as the initial module. The probe set in the expression array with the highest TO to the seed is then added to the module, forming a larger module. This process is repeated recursively until the module contains a specified number of probe sets (typically 60), after which its network depictions are graphed (as described above), with orange or gray assigned as the module color. Unlike the network analyses, these local networks were not restricted to variably expressed genes; any probe set that was present with an associated gene symbol could form part of a MultiTOM module.
Comparative analysis.
We performed comparative analyses of the two studies by limiting the analysis to genes with probe sets on both arrays, choosing first the probe set with the highest kin followed by the probe set with the highest correlation to phenotype when multiple probe sets for a single gene were present. This approach identified 4827 overlapping genes between the two studies. We tested whether the number of genes correlated with both AD and aging was significantly larger than expected by chance using a hypergeometric distribution. Similar analyses were done comparing only genes changing in parallel or opposite directions in AD and aging, as well as genes in overlapping modules between the two studies.
Functional categorization of genes.
Group analyses were performed on all generated gene lists using Expression Analysis Systematic Explorer (EASE) (Hosack et al., 2003) to functionally categorize genes in an unbiased manner. Modules were characterized based on the gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), or SwissProt terms associated with their gene list that had the lowest EASE scores (similar to a p value), as long as such scores were significant (p < 0.05). When EASE failed to provide such categories, WebGestalt (http://bioinfo.vanderbilt.edu/webgestalt/) was used for module categorization. Smaller gene lists (<50 probe sets) were analyzed using Chilibot, a literature-mining tool that creates biological networks based on co-occurrence of terms in PubMed abstracts (Chen and Sharp, 2004). Gene lists were also compared with terms related to clinical traits of interest (such as “Alzheimer's disease”) to explore the potential involvement of a given gene in that process. Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Redwood City, CA) was applied to a subset of modules to augment the functional annotation.
Results
Network analysis of AD hippocampus
To circumvent the problems associated with tissue and clinical heterogeneity and directly address the issue of regional vulnerability, Blalock et al. (2004) performed a microarray study of one of the earliest and most consistently affected brain regions in AD, the CA1 region of the hippocampus, using specimens from subjects with relatively well defined clinical and pathological stages of AD. After preprocessing this data ourselves using R and removing three outlier arrays (Materials and Methods), these samples represent eight controls, as well as six incipient, eight moderate, and six severely afflicted AD patients, as determined by NFT burden and MMSE score (Blalock et al., 2004). Because CA1 is a highly vulnerable region of the medial temporal lobe affected fairly early in the progression of AD (Braak and Braak, 1991), these samples represent snapshots at a maximal range of NFT burden. To complement traditional differential expression analyses, we performed WGCNA on these samples, as groups of coexpressed genes are biologically related, and such network analyses may shed light on underlying functional processes in a manner complementary to standard differential expression analyses (Horvath et al., 2006). WGCNA starts by calculating a matrix containing all pairwise Pearson correlations between all genes. The Pearson correlations are used to calculate TO, a more robust, and biologically meaningful measure of gene coexpression that takes into account the shared neighbors of each gene pair in the network (Ravasz et al., 2002; Zhang and Horvath, 2005). TO was calculated between each gene and every other gene, and groups of genes with high TO were then identified by hierarchical clustering to define modules (Materials and Methods) (Zhang and Horvath, 2005).
We first used WGCNA to analyze the 5000 most variable genes determined by their coefficient of variance, rather than any sample characteristics such as disease or control status. Because we did not filter by any clinical or pathological features, and genes therefore were not selected based on such top-down information, we refer to this analysis as “unsupervised.” Twelve modules of genes with high TO were identified (Fig. 1a) (a full list of genes by module appears in supplemental Table 1, available at www.jneurosci.org as supplemental material). Genes in each module share expression patterns that are more similar to one another than to the expression patterns of genes in other modules. We were able to biologically characterize most modules using EASE, a program that checks for an over-representation of genes with specific GO, KEGG, and SwissProt terms relative to a reference list (Hosack et al., 2003). In all modules except black and yellow, the most significant category identified by EASE was a process previously associated with AD. These categories included synaptic transmission and extracellular transport, mitochondrial and metabolic functions, and myelination (Table 1) (Mesulam, 1999; Arendt, 2000; Blass et al., 2002; Bartzokis, 2004; Beal, 2005). These observations are consistent with the notion that modules represent groups of biologically related genes, as has been demonstrated in several systems (Jeong et al., 2001; Barabasi and Oltvai, 2004; Horvath et al., 2006; Oldham et al., 2006).
Table 1.
Top EASE category associated with each module
| Module | Gene category | EASE score |
|---|---|---|
| Blue | Metal ion transport (glycoprotein) | 1.06 × 10−5 |
| Magenta | Transmission of nerve impulse | 8.98 × 10−5 |
| Pink | Synaptic transmission | 2.03 × 10−5 |
| Purple | Synaptic transmission | 3.46 × 10−5 |
| Green yellow | Ion transport (calcium transport) | 3.61 × 10−4 |
| Brown | Mitochondrion | 7.96 × 10−23 |
| Orange | RNA processing | 4.24 × 10−3 |
| Red | Transferase (oncogenesis) | 3.61 × 10−2 |
| TurquoiseI | Intracellular transport (cytoskeleton) | 7.58 × 10−4 |
| Yellow | Antigen processing (ribosome) | 4.79 × 10−5 |
| Black | Heat shock protein activity | 1.41 × 10−24 |
| Tan | Myelin (plasma membrane) | 5.69 × 10−5 |
EASE categories include GO, KEGG, and SwissProt categories. For some modules, the second most significant EASE category is included in parentheses. p values in bold are significant (p < 0.05) after accounting for multiple comparisons. At least one of the presented EASE categories for each module contains 10 or more genes, in an attempt to provide an overall categorization of the modules rather than characterizing small subsets of the modules.
Modular organization and relationship to underlying biological processes
Although the expression patterns in each module were different, many of the modules shared similar GO categorizations, suggesting that some modules may be functionally related. To determine how related each module was to every other and to phenotypic assessments of AD, we performed a principal component analysis (PCA) to obtain the module eigengene (ME; the first PC of the expression values across subjects) for each module. PCA simplifies a data set by reducing the dimensionality of the data, sorting the PCs based on the amount of variance each component explains (supplemental Table 2, available at www.jneurosci.org as supplemental material). In this case, the ME represents the main trend of gene expression for a module, and modules with genes sharing similar expression will also have similar MEs. We plotted each module in two-dimensional space using the first two PCs (Fig. 2), which grouped modules with identical or highly related significant EASE categories (Table 1). Five modules were involved in synaptic processes (blue, green yellow, magenta, pink, and purple), four in metabolic processes (brown, orange, red, and turquoise), and two in injury response (black and yellow). This analysis also identified a single module (tan) containing myelin-related genes. Because the definition of a module is subjective such groupings were not unexpected, and in fact suggest that our results are robust with respect to module size. There was also a strong relationship between module function (as measured by EASE analysis) and disease progression, with modules in the synaptic and metabolic groups correlated with MMSE score, a cognitive indicator of AD progression (Fig. 1b) (Folstein et al., 1975). However, most genes in the immune response group do not correlate with MMSE score, and are likely variable because of postmortem effects or other immune responses unrelated to AD. These results demonstrate that unsupervised analyses can identify groups of genes not only with shared biological functions, but also showing significant correlation to a pathological measure of disease progression and clinical phenotype in AD.
Figure 2.

Multidimensional scaling (MDS) plot of top two PCs of each module reveals clear functional groupings. The first two PCs of each module in the unsupervised AD analysis, PC1 (x-axis) and PC2 (y-axis), were plotted against one another as a quantitative measure of module similarity, using the same scaling for both axes. Each colored point corresponds to a module presented in Figure 1, using the same color depiction. The modules cluster into four distinct groups that can be functionally annotated using the EASE categories from Table 1, resulting in the following descriptive group titles: the “synaptic” group (blue, magenta, pink, purple, green yellow), the “metabolic” group (brown, green, turquoise, red), and the “immune response” group (yellow, black). The first PC of each module in the “synaptic” and “metabolic” groups correlates with MMSE score, whereas the first PC of the red module is also correlated with NFT burden.
The red module, which was the only module whose ME correlated positively with AD progression (Fig. 1b), was not readily characterized using EASE. Because this module was relatively small, we decided to perform more direct assessments of gene interactions. Using Chilibot (Chen and Sharp, 2004), we found that many of these genes (45 of 165) shared literature co-occurrences (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that underlying structure may exist in this module that was not identified using EASE. To provide added confidence, we used the more powerful IPA (Ingenuity Systems), which identified a significant number of genes involved in the mitogen-activated protein kinase kinase kinase (MAPKKK) cascade (p < 10−4). Similarly, using WebGestalt (Zhang et al., 2005), we found a large number of genes in this module related to MAPK signaling (p < 0.01). Inspection of this module found more uncharacterized genes than expected by chance (p < 0.05), suggesting that genes within this module would make interesting candidate genes for follow up studies, because many of the genes characterize hypothetical proteins that have not been previously associated with AD.
Identification of key hub genes in AD hippocampus
Evidence suggests that a gene's network position has significant functional implications, with more centralized genes in the network (“hub genes” or “hubs”) more likely to be vital to proper cellular function than peripheral genes (nodes). For example, hubs have previously been shown to play important roles in yeast protein networks (Jeong et al., 2001) and in glioblastoma gene networks (Horvath et al., 2006), where hubs have been shown to be therapeutic targets. Network depictions for the AD study were created by graphing the top 250 gene–gene interactions based on TO, defining a hub as any gene involved in at least 15 of the these interactions (for selected network depictions, see Fig. 3). The brown, “mitochondrial” module has three hub genes transcribing mitochondrial membrane proteins involved in ion transport (VDAC1, VDAC3, and ATP5F1) (Fig. 3a). At least two hub genes in both the pink (WDR7 and SYNJ1) and purple (STXBP1 and SNAP91) “synaptic” modules are directly involved in synaptic vesicle fusion and endocytosis (Fig. 3b,c). Other hub genes within these modules are also likely to play roles in synaptic transmission: CACNB2 is a subunit of voltage-dependent calcium channels, whereas GLRB is a subunit of the glycine receptor, a neurotransmitter-gated ion channel. All of these hub genes are present in modules that are significantly correlated with MMSE score (supplemental Table 2, available at www.jneurosci.org as supplemental material), likely reflecting mitochondrial dysfunction and synaptic loss, which are known consequences of AD (Arendt, 2000; Beal, 2005). Finally, the central positions of several unknown genes within the red module (for example, FLJ14346 and LOC152719) (Fig. 3d), suggest that these genes may play important roles in cell signaling pathways such as the MAPKKK cascade, or in other unknown processes upregulated with AD progression, a result that would never have been uncovered using traditional microarray analysis methods.
Figure 3.

Network depictions of selected modules allow visualization of intramodular connections and hub genes. A, The brown module contains a significant cohort of mitochondrial genes (p < 10−22), including three mitochondrial membrane proteins as hubs (VDAC1, VDAC3, and ATP5F1). B, C, Both the pink (B) and purple (C) modules contain genes primarily related to synaptic transmission, including four hub genes, WDR7, SYNJ1, STXBP1, and SNAP91. D, The red module contains hubs of largely unknown function, but that are connected with genes involved in important signaling pathways, such as the MAP kinase cascade. For each network depiction, orange lines indicate positively correlated genes, whereas black lines indicate negatively correlated genes. Large, labeled nodes (genes) represent hub genes with at least 15 connections of the 250 displayed in each plot. The length of each line and the position of each node were arbitrarily chosen by VisANT to highlight network structure.
Comparative network analysis of AD and aging studies
The previous unsupervised analysis did not use knowledge of sample characteristics or disease pathophysiology, and therefore provided an unbiased view of transcriptome organization in AD. However, using a supervised analysis, we provide a complementary view in which additional phenotypic information ensures that a more inclusive set of condition-related genes is used to construct the network. Here, we filtered genes to include those most correlated with MMSE score and NFT burden, as well as including genes that differ significantly in expression between control subjects and any AD group, so as to enrich our analysis with genes related to broad aspects of disease progression (Braak and Braak, 1991; Mouton et al., 1998; Giannakopoulos et al., 2003), allowing direct comparison to other data sets. In effect, by using supervised analyses we can improve our signal by filtering out genes that are not directly correlated with the pathological progression of AD.
From this analysis, we identified five large gene coexpression modules (Fig. 4a, top two traces), all showing significant correlation with AD as measured by NFT score (purple), MMSE score (blue), or both (brown, light blue, and yellow). The blue and light blue modules show decreased expression with AD progression and the remaining modules consist of genes whose expression is increasing (Fig. 4a, bottom trace, supplemental Table 2, available at www.jneurosci.org as supplemental material). Furthermore, all five modules in this analysis show significant gene overlap with the modules identified in the unsupervised analysis (supplemental Table 3, available at www.jneurosci.org as supplemental material), demonstrating that both network analyses group genes by function in a robust manner. For example, all five modules in the “synaptic group” show >20% overlap (four showing >50% overlap) with the blue module in the supervised analysis, suggesting that the new blue module from the supervised analysis is a synaptic module. Similar overlap can be found between three unsupervised modules in the “metabolic group” and the light blue supervised module, which has significant EASE categories associated with energy metabolism, ion transport, and mitochondria. Thus, regardless of the method used to select genes, modules corresponding both to the mitochondrion and to synaptic function show decreased expression with AD progression, further suggesting that these processes are disrupted in AD. Furthermore, use of a supervised analysis, where all genes are correlated with AD progression and all samples are age-matched, allows us to compare transcriptional patterns with a disease-free study of aging, because in both cases we know that the genes selected are related to progression of the relevant process.
To approach the question of which aspects of gene expression in AD overlap with normal aging, or whether AD is essentially a unique process, we compared the supervised AD network to a supervised aging study of comparable size and design (Lu et al., 2004). Because the point of this analysis was to compare normal aging with AD, not to characterize normal aging in isolation, an unsupervised aging analysis was not necessary. Unfortunately, two precisely matched data sets with appropriate clinical and pathological data were not available, so we compared data from the only two studies with high quality, publicly available data: the AD study above and a study of normal aging in the frontal lobes of disease-free individuals between the ages of 26 and 106 (Lu et al., 2004). To our knowledge, this aging study is the only one of its type; thus, we could not match brain regions with the AD study. Despite the different brain regions in the two studies, by limiting our analysis to only those genes correlated with AD or aging, our results would identify general cellular processes in common between AD and normal aging should they exist, rather than region-specific changes. Furthermore, in both studies, progression of the key aspects of the phenotype (MMSE score age or NFT burden for AD; for normal aging) could be directly correlated with expression for all genes. Such a correlation with essential metrics of phenotype progression was essential for interpretation of overlapping processes identified in this analysis. However, we cannot separate true biological differences in AD and aging from technical confounding factors, and therefore it is not possible to obtain firm conclusions about distinctions between AD and aging gene expression in this analysis.
After selecting probe sets correlated with age (p < 0.05), we identified three modules of highly coexpressed genes (Fig. 4b, top two traces), two of which included genes with expression predominately increasing with age, the third containing mostly genes decreasing in expression with age (Fig. 4b, bottom trace). As with the AD study, each of these modules was comprised of genes involved in related biological processes. For example, the blue module, which contains genes downregulated with aging, has significant EASE categories associated with nucleotide binding, signal transduction, and metabolism (supplemental Table 4, available at www.jneurosci.org as supplemental material).
To determine whether similar processes are shared between AD and aging, we compared the genes in each module between the two networks. Strikingly, we observed that three module pairs had significantly more overlapping genes than expected by chance (Fig. 5, left column). Furthermore, two of these module pairs, the blue AD and aging modules and the light blue AD and the blue aging modules, contained overlapping GO biological process categories in addition to specific genes (Fig. 5, right). Finally, among the 328 genes that were placed in modules in both analyses, eight genes (p = 0.08) were identified as hub genes (kin > 0.6) in both analyses, seven of which fell into the three overlapping modules pairs, again indicating similar processes in AD and aging (Table 2). In short, whether measuring by number of genes, GO category, or number of hub genes, the blue (“synaptic”) and light blue (“mitochondrial”) modules in the supervised AD analysis significantly overlap with the blue aging module.
Figure 5.

Modules from AD analysis overlap significantly with modules from aging analysis, as measured by both gene number and gene ontology categories. A, Blue AD and blue aging modules. B, Light blue AD and blue aging modules. C, Brown AD and brown aging modules. Left trace, Number of genes overlapping between modules in the AD and aging studies. *p = 0.001; **p < 10−9. p values were obtained using a hypergeometric distribution. Right trace, Top GO biological process categories for overlapping modules in the two studies. Italicized categories are GO molecular function. p values in bold are significant (p < 0.05) after accounting for multiple comparisons. Because there were no GO categories significant in both brown modules, categories with nearly significant p values in both AD and aging are included.
Table 2.
Hub genes common to both AD and aging
| Gene symbol | AD |
Aging |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Probe set | Assay | p value | Module | Kin | Probe set | p value | Module | Kin | Gene description | |
| ATP5A1 | 213738_s_at | MMSE | 4.17 × 10−2 | Light blue | 0.67 | 40096_at | 7.44 × 10−5 | Blue | 0.91 | ATP synthase alpha chain, mitochondrial |
| ATP6V1G2 | 214762_at | ANOVA | 1.52 × 10−3 | Light blue | 0.85 | 33033_at | 1.50 × 10−4 | Blue | 0.64 | Vacuolar ATP synthase subunit G 2 |
| RAB6A | 210406_s_at | MMSE | 3.05 × 10−2 | Light blue | 0.76 | 622_at | 1.58 × 10−4 | Blue | 0.86 | RAB6A, member RAS oncogene family |
| YWHAZ | 200638_s_at | MMSE | 3.44 × 10−3 | Light blue | 0.97 | 34642_at | 1.68 × 10−6 | Blue | 0.96 | 14-3-3 protein zeta/delta |
| CDK5 | 204247_s_at | MMSE | 3.13 × 10−3 | Blue | 0.63 | 1206_at | 2.28 × 10−4 | Blue | 0.64 | Cyclin-dependent kinase 5 |
| PRKCB1 | 207957_s_at | ANOVA | 2.88 × 10−4 | Blue | 0.71 | 1217_g_at | 1.66 × 10−5 | Blue | 0.77 | Protein kinase C, beta 1 |
| DICER1 | 213229_at | CvsM | 1.78 × 10−2 | Brown | 0.75 | 38764_at | 1.33 × 10−2 | Brown | 0.64 | Dicer1, Dcr-1 homolog (Drosophila) |
| LARP4 | 212714_at | NFT | 6.72 × 10−3 | Purple | 0.64 | 35180_at | 2.72 × 10−2 | Brown | 0.73 | La ribonucleoprotein domain family member 4 |
These genes are highly connected in each data set and are highly correlated with the respective phenotype of aging or AD progression; both DICER1 and LARP4 increase expression with increasing AD progression and aging, whereas all the other hubs decrease expression with both phenotypes. The method of measuring probe set significance in AD that led to the lowest p value is presented in the AD assay column (with its associated p value). The aging p value represents the probe set's correlation with age. For both studies, kin represents the intramodular connectivity, with kin > 0.6 considered significant. In the CvsM assay, the control and moderate AD samples were compared using a t test.
Several of these hub genes have known functions that suggest plausible connections between AD and normal aging. For example, cyclin-dependent kinase 5 (CDK5) is one of the major tau kinases, and accumulation of pathologically phosphorylated tau is a hallmark of AD (Geschwind, 2003; Cruz and Tsai, 2004). Another potential AD- and aging-related gene is YWHAZ (14.3.3 ζ), one of the most highly connected genes in both studies (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). YWHAZ is a member of the highly conserved family of 14.3.3 genes involved in cell signaling, regulation of cell cycle progression, cytoskeletal structure, and transcription (Aitken, 2006), making up ∼1% of the cytosolic protein content in the brain (Arendt, 2000). In both aging and AD, decreased expression of this key linker gene was observed. All overlapping genes in these modules, their intramodular connectivities, and their correlations to phenotype are included in supplemental Table 5 (available at www.jneurosci.org as supplemental material).
Local network analyses of AD-related genes
Next, we compared the local networks of known AD genes in the AD study with their local networks in the aging study using MultiTOM (Materials and Methods) (Li and Horvath, 2007), which constructs modules by starting with a seed gene and recursively adding the next gene with the highest TO to the current module. Presenilin 1 (PSEN1) is a hub gene in its local network of 60 genes in the AD study, but is not highly connected with its nearest 60 neighbors in the aging study, suggesting that PSEN1 function in CA1 during AD may be different from PSEN1 function in the prefrontal cortex during normal aging (Fig. 6). Although this effect may reflect differences in brain regions, microarrays, or AD progression, a changed role for PSEN1 in the AD network is consistent with its established role in the disease. Interestingly, the organization and composition of the local network involving PSEN1 is similar in AD and aging. Autotaxin (ENPP2) is a hub gene in both local networks, and five other genes are also observed in the local network of PSEN1 in both studies (CD9, FRMD4B, GPR37, LIPA, and ZNF536), constituting significantly higher overlap than expected by chance (p < 10−8). These local networks are likely related to myelination processes, as PSEN1 is highly coexpressed with canonical oligodendrocyte markers, including myelin associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG) and transferrin (TF) in the AD study, and plasmolipin (PLLP) and myelin proteolipid protein (PLP1) in the aging study (Fig. 6). As additional evidence that these overlapping networks are myelin-related, in a core list of 50 “oligodendrocyte-related genes” compiled in our lab using multiple human brain data sets (M. C. Oldham and J. A. Miller, unpublished observations), 20 overlapped with the local module of PSEN1 in AD (p < 10−36), whereas 18 overlapped with the aging PSEN1 module (p < 10−26). These results provide a new set of evidence supporting the hypothesis that demyelination and oligodendrocyte dysfunction may play a role in AD progression (Braak and Braak, 1991; Bartzokis, 2004).
Figure 6.

PSEN1 modules in AD and aging. A, B, VisANT was used to create network depictions as in Figure 3 for the AD network (A), where PSEN1 is a hub, and aging network (B), where PSEN1 is not a hub. The local networks of PSEN1 are similar in AD and aging, however, as seven genes (including the hubs ENPP2 and LIPA) overlap between both PSEN1 local networks (hypergeometric probability; p < 10−8). Both hub genes (large nodes) and canonical oligodendrocyte genes are labeled, and all lines represent positive gene–gene correlations. As with Figure 3, node positions and line lengths are chosen to highlight network structure and do not have any biological meaning.
Comparative gene expression analysis of AD and aging genes
We also performed a comparative analysis between AD and aging that follows the traditional approach of creating lists of genes that are differentially expressed between two groups. In this case, comparing multiple phenotypes allowed for the statistical analysis of gene overlap between the two studies, regardless of the microarray platforms used. Of the 4826 genes in common between studies, 2010 were significantly correlated with AD progression (Materials and Methods) (902 increased, 1108 decreased), whereas 1221 were significantly correlated with aging (738 increased, 483 decreased). Because the samples in the AD study were age matched, any gene expression changes in the AD study were caused by changes above and beyond normal aging. Not only did we find significantly more overlapping genes between the two studies than expected by chance (558 vs 508; p = 4 × 10−4) (for a list of all overlapping genes, see supplemental Table 6, available at www.jneurosci.org as supplemental material), but also more genes than expected showed parallel expression changes with advancing age and dementia (Fig. 7) (p < 10−22). Likewise, significantly fewer genes than expected changed in opposite directions (p < 10−15). GO and IPA suggest that genes upregulated with both AD and aging are related to transcription and cell death, whereas genes downregulated with both AD and aging are involved in neurotransmission and signaling pathways (supplemental Table 6, available at www.jneurosci.org as supplemental material), consistent with previous data (Vernadakis, 1985; Arendt, 2004) and with our network analyses.
Figure 7.

Overlapping genes in AD and aging show parallel changes. Among genes with probe sets that are present in both studies, there are more genes significantly correlated with both AD and aging (∼48% of possible overlapping genes) than expected by chance (590 vs 526; p = 4 × 10−4), and most of these genes increase or decrease with both AD and aging. Both the over-representation of genes with parallel changes (p < 10−22) and the under-representation of genes with opposite changes (p < 10−15) are highly significant. p values were obtained using a hypergeometric distribution (for a list of these genes, see supplemental Table 6, available at www.jneurosci.org as supplemental material).
These results become even more impressive when only the genes in overlapping modules between the supervised AD and aging studies are considered (Fig. 5, supplemental Table 5, available at www.jneurosci.org as supplemental material); of 160 such genes, all but three (98%) show parallel expression changes with advancing age and dementia, and only one of these is negatively correlated with its respective ME. Thus, traditional differential expression analyses provide support that similar transcriptional changes occur in AD and normal aging; however, WGCNA organizes this information into a framework that reflects underlying biological relationships (Horvath et al., 2006), allowing biologically relevant changes to be partially separated from changes attributable to inherent variability. Together, these analyses lend support to the idea that several processes known to occur in both normal aging and AD (including mitochondrial and synaptic dysfunction and cell death) result in similar transcriptional changes and thus may have similar underlying mechanisms.
Discussion
Moving from gene lists to function remains a challenge in most microarray studies, especially when the focus is on gene-by-gene analysis of differential expression, which relies on known biological categorizations such as GO to provide notions of biological structure. Network analysis moves beyond single gene investigation to provide a systems level understanding of the relationships between members of a network (Ravasz et al., 2002; Barabasi and Oltvai, 2004). WGCNA in particular provides a framework based on the underlying transcriptome organization measured in a given study, and allows identification of hub genes that play central roles in the biological system in question (Horvath et al., 2006; Oldham et al., 2006). There is increasing evidence linking gene and protein coexpression to functional relationships. For example, a previous meta-study of 60 diverse data sets across many human tissues found that many genes share similar coexpression patterns in multiple data sets, and that these coexpressed genes tend to have overlapping GO terms (Lee et al., 2004). Similar results have been found in studies of cancer tissue (Mischel et al., 2004; Horvath et al., 2006), yeast protein networks (Jeong et al., 2001), and Escherichia coli (Ravasz et al., 2002).
Several lines of evidence suggest that the above networks are biologically significant. First, WGCNA sorts variable genes by biological or molecular function, delineating genes into modules with highly significant and specific EASE categorizations (Table 1), many of them correlated with AD progression (Fig. 1). It is reassuring that many of the resulting EASE categorizations, such as synaptic function, are processes specific to the CNS, because we would expect modules to be enriched for such genes. However, the glial or neuronal specificity of these genes alone would not account for the fact that expression within these modules is strongly related to both progression of AD and aging. Second, hub genes from many modules are key players in the biological processes suggested by EASE. For example, VDAC1, a hub in the brown (mitochondrial) unsupervised AD module (Fig. 3a), is an important mitochondrial membrane protein involved in ATP regulation and apoptosis that has isoforms known to show decreased expression in the temporal lobe of AD patients (Yoo et al., 2001). Finally, multiple network modules are conserved between AD and aging, including the mitochondrial and synaptic modules from Figure 5, and the PSEN1 local networks (Fig. 6), which contain numerous canonical myelin-related genes.
Gene discovery: identification of hub genes as candidate AD-related genes
Determining whether the high connectivities of hubs in this study are attributable to a functional role of these genes in disrupting essential cellular processes, or whether they instead mark organelle dysfunction and neuronal loss induced through other means, will require further study. In either case, hubs of modules correlated with AD progression play key roles in processes disrupted in AD, and understanding the function of such genes may lead to a better understanding of AD progression. For example, the role of YWHAZ, an abundant signaling protein involved in many essential cellular processes (Aitken, 2006) that shows high correlations to both AD and aging (Table 2, supplemental Fig. 2, available at www.jneurosci.org as supplemental material) warrants follow up study. This network analysis also identified CDK5 as a reproducible hub in both data sets. CDK5/p25 is known to participate in the pathological phosphorylation of tau (Geschwind, 2003) and cause neurodegeneration when overexpressed in mice (Cruz et al., 2003). Furthermore, emerging evidence suggests that CDK5/p25 dysregulation may also lead to aberrant amyloid precursor protein (APP) processing and accumulation of forebrain β-amyloid (Cruz et al., 2006). These data strongly suggest that this pathway may be crucial for AD pathophysiology, providing a potential leverage point for understanding the role of environmental and other genetic factors related to tau phosphorylation in the transition between normal aging and AD.
Neurodegeneration of CA1 is another hallmark pathology of AD. Thus, neuronally expressed genes, such as synaptic markers, decreasing in expression with advancing AD could be partially explained by neuronal loss. It is less likely that such changes are directly responsible for genes positively correlated with AD. Thus, the over-representation of uncharacterized genes, and especially hubs, in the unsupervised red module is particularly interesting (Fig. 3d). This module likely represents distinct biological processes, given the large number of genes with shared literature co-occurrences (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), as well as the over-representation of MAPK signaling genes (see Results). Furthermore, this module falls within the metabolic group, suggesting that the unknown hubs in this module (LOC152719, FLJ14346, and FLJ12151) may play important roles in processes leading to or caused by mitochondrial and metabolic dysfunction.
PSEN1 module suggests a role for oligodendrocyte dysfunction in AD
Mutations in the presenilins account for over half of the early onset familial AD cases. PSEN1 is one component of the gamma secretase complex, which cleaves APP, preventing buildup of toxic moieties of β-amyloid peptide (Borchelt et al., 1996; Cai et al., 2003). Of all the genes involved in APP cleavage or β-amyloid production in the AD study, only APBB2, an APP-binding protein involved in cholesterol metabolism (Carter, 2007), was a member of the PSEN1 coexpression network, suggesting multiple roles of PSEN1 in the adult brain. The coexpression of PSEN1 with known myelin-associated genes in both studies, and its high connectivity in the AD study, suggests that PSEN1 may play a role in oligodendrocyte dysfunction or demyelination in AD. Pak et al. (2003) found that oligodendrocytes in PSEN1 mutant knock-in mice were more prone to damage by demyelinating agents and death by glutamate and β-amyloid toxicity. These results fit the hypothesis that AD progression follows a trajectory opposite in time to cortical myelination (Braak and Braak, 1996), because the later-made oligodendrocytes progressively myelinate a greater number of axonal segments, making them more vulnerable to AD risk factors such as head injury and high cholesterol (Bartzokis, 2004).
Mitochondria and synaptic dysfunction in AD
The unsupervised network analysis of AD provides an interesting perspective on mitochondrial and synaptic dysfunction. Because both mitochondria and synapses fail with increasing age and disease progression, and because oxidative damage is one of the earliest pathological changes seen in AD (Nunomura et al., 2001), it has been suggested that mitochondrial dysfunction is the underlying cause for disease pathology (Beal, 2005; Lin and Beal, 2006). Although the expression profiles for the mitochondrial and synaptic modules look relatively similar, they can clearly be separated by their PCs (Fig. 2, the brown point falls in the “metabolic group,” not the “synaptic group”). This separation suggests that mitochondrial and synaptic genes are likely involved in distinct, yet related processes. Such results cannot determine causal relationships, yet the idea that mitochondrial dysfunction may in part lead to neuroplasticity failure, as suggested by Beal et al. (Beal, 2005; Lin and Beal, 2006), remains intriguing.
Common features shared between AD and aging
A fundamental question in both AD and aging research is whether AD is an extreme form of aging, or whether it is an entirely distinct process (Finch and Cohen, 1997; Drachman, 2006; Keller, 2006; Pereira et al., 2007). Because our data sets involve distinct individuals, microarray platforms, and brain regions, it is unclear which network differences identified between studies are attributable to these potential confounders. These same issues, however, make any similarities between the studies even more surprising, given the generally poor repeatability of microarray experiments. Even in experimental replications, it is relatively uncommon to find lists of differentially expressed genes with significant overlap between microarray studies (Kuo et al., 2002). For example, of the 29 differentially expressed genes identified in a similar, older study of AD in CA1 (Colangelo et al., 2002) also present in the Blalock et al. (2004) AD study, only eight correlated with MMSE or NFT, and six of these genes changed in opposite directions between the studies. Thus, our current finding of highly significant overlap between two entirely different conditions is remarkable, and likely biologically meaningful.
Our analysis uncovered evidence of significant overlap between AD and aging at a molecular level, identifying core biological processes and genes they share. Because samples from the AD study were taken from age-matched subjects at varying states of cognitive decline (Blalock et al., 2004), all variable genes represent genes upregulated or downregulated in AD separate from what would be seen in normal aging. For example, the downregulation of synapse-related genes in both studies suggests that synaptic transmission is disrupted with normal aging, and then further disrupted with AD. Not only do specific genes show parallel expression changes in AD and aging, but many also cluster into modules within a transcriptional coexpression network (Fig. 5), with shared GO categories generally related to synaptic and mitochondrial function. These data support the notion that AD and aging share common pathophysiological processes. Given these results, a comprehensive analysis of both conditions in tandem, for example using the same tissues and microarray platforms across both age and AD progression, would be quite powerful. Such direct comparison would complement these analyses, permitting quantitative assessment of biological differences, as well as the similarities, between aging and AD.
Footnotes
This work was supported by Neurobehavioral Genetics Training Grant T32MH073526-01A1 (J.A.M., D.H.G.); National Institutes of Health, National Research Service Award F31 AG031649 from the National Institute on Aging (J.A.M.); and National Institutes of Health–National Institute on Aging Grant R01 AG26938 (D.H.G.). We thank Tao Lu for providing the raw data from the aging paper and Eric Blalock for valuable discussions and assistance with preprocessing data from the AD study.
References
- Aitken A. 14-3-3 proteins: a historic overview. Semin Cancer Biol. 2006;16:162–172. doi: 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Arendt T. Alzheimer's disease as a loss of differentiation control in a subset of neurons that retain immature features in the adult brain. Neurobiol Aging. 2000;21:783–796. doi: 10.1016/s0197-4580(00)00216-5. [DOI] [PubMed] [Google Scholar]
- Arendt T. Neurodegeneration and plasticity. Int J Dev Neurosci. 2004;22:507–514. doi: 10.1016/j.ijdevneu.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Baldi P, Long A. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Barabasi A, Oltvai Z. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer's disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. author reply 49–62. [DOI] [PubMed] [Google Scholar]
- Beal M. Mitochondria take left stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Blalock E, Geddes J, Chen K, Porter N, Markesbery W, Landfield P. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass J, Gibson G, Hoyer S. The role of the metabolic lesion in Alzheimer's disease. J Alzheimers Dis. 2002;4:225–232. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- Borchelt D, Thinakaran G, Eckman C, Lee M, Davenport F, Ratovitsky T, Prada C, Kim G, Seekins S, Yager D, Slunt H, Wang R, Seeger M, Levey A, Gandy S, Copeland N, Jenkins N, Price D, Younkin S, Sisodia S. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol (Berl) 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- Cai D, Leem J, Greenfield J, Wang P, Kim B, Wang R, Lopes K, Kim S, Zheng H, Greengard P, Sisodia S, Thinakaran G, Xu H. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- Carter CJ. Convergence of genes implicated in Alzheimer's disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem Int. 2007;50:12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Chen H, Sharp B. Content-rich biological network constructed by mining PubMed abstracts. BMC Bioinformatics. 2004;5:147. doi: 10.1186/1471-2105-5-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S, Boutros M, Michelson A, Church G, Halfon M. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 2005;6:R16. doi: 10.1186/gb-2005-6-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colangelo V, Schurr J, Ball M, Pelaez R, Bazan N, Lukiw W. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- Cruz J, Tsai L. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Cruz J, Tseng H, Goldman J, Shih H, Tsai L. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Cruz J, Kim D, Moy L, Dobbin M, Sun X, Bronson R, Tsai L. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006;26:10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman D. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006;67:1340–1352. doi: 10.1212/01.wnl.0000240127.89601.83. [DOI] [PubMed] [Google Scholar]
- Emilsson L, Saetre P, Jazin E. Alzheimer's disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol Dis. 2006;21:618–625. doi: 10.1016/j.nbd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Finch C, Cohen D. Aging, metabolism, and Alzheimer disease: review and hypotheses. Exp Neurol. 1997;143:82–102. doi: 10.1006/exnr.1996.6339. [DOI] [PubMed] [Google Scholar]
- Folstein M, Folstein S, McHugh P. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Geschwind D. Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann F, Bussière T, Bouras C, Kövari E, Perl D, Morrison J, Gold G, Hof P. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, Lee Y, Scheck AC, Liau LM, Wu H, Geschwind DH, Febbo PG, Kornblum HI, Cloughesy TF, Nelson SF, Mischel PS. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci USA. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosack D, Dennis G, Sherman B, Lane H, Lempicki R. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, DeLisi C. VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004;5:17. doi: 10.1186/1471-2105-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Irizarry R, Gentleman R, Carey VJ, Dudoit S. New York: Springer; 2005. Bioinformatics and computational biology solutions using R and bioconductor (statistics for biology and health) [Google Scholar]
- Jeong H, Mason S, Barabasi A, Oltvai Z. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Karsten S, Sang T, Gehman L, Chatterjee S, Liu J, Lawless G, Sengupta S, Berry R, Pomakian J, Oh H, Schulz C, Hui K, Wiedau-Pazos M, Vinters H, Binder L, Geschwind D, Jackson G. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Keller J. Age-related neuropathology, cognitive decline, and Alzheimer's disease. Ageing Res Rev. 2006;5:1–13. doi: 10.1016/j.arr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Kuo W, Jenssen T, Butte A, Ohno-Machado L, Kohane I. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics. 2002;18:405–412. doi: 10.1093/bioinformatics/18.3.405. [DOI] [PubMed] [Google Scholar]
- Lee H, Hsu A, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004;14:1085–1094. doi: 10.1101/gr.1910904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Horvath S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics. 2007;23:222–231. doi: 10.1093/bioinformatics/btl581. [DOI] [PubMed] [Google Scholar]
- Lin M, Beal F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lockhart D, Dong H, Byrne M, Follettie M, Gallo M, Chee M, Mittmann M, Wang C, Kobayashi M, Horton H, Brown E. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Loring J, Wen X, Lee J, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer's disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao S-Y, Li C, Kohane I, Chan J, Yankner B. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Mesulam M. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Pevsner J. Progress in the use of microarray technology to study the neurobiology of disease. Nat Neurosci. 2004;7:434–439. doi: 10.1038/nn1230. [DOI] [PubMed] [Google Scholar]
- Mischel P, Cloughesy T, Nelson S. DNA-microarray analysis of brain cancer: molecular classification for therapy. Nat Rev Neurosci. 2004;5:782–792. doi: 10.1038/nrn1518. [DOI] [PubMed] [Google Scholar]
- Mouton P, Martin L, Calhoun M, Dal Forno G, Price D. Cognitive decline strongly correlates with cortical atrophy in Alzheimer's dementia. Neurobiol Aging. 1998;19:371–377. doi: 10.1016/s0197-4580(98)00080-3. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj E, Jones P, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood C, Petersen R, Smith M. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Oldham M, Horvath S, Geschwind D. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci USA. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak K, Chan S, Mattson M. Presenilin-1 mutation sensitizes oligo-dendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. Neuromolecular Med. 2003;3:53–64. doi: 10.1385/NMM:3:1:53. [DOI] [PubMed] [Google Scholar]
- Pereira A, Wu W, Small S. Imaging-guided microarray: isolating molecular profiles that dissociate Alzheimer's disease from normal aging. Ann NY Acad Sci. 2007;1097:225–238. doi: 10.1196/annals.1379.005. [DOI] [PubMed] [Google Scholar]
- Price J, Morris J. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Ravasz E, Somera A, Mongru D, Oltvai Z, Barabasi A. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis R, Brown P. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim T-W. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Smith C, Carney J, Starke-Reed P, Oliver C, Stadtman E, Floyd R, Markesbery W. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter M, Crook J, Hyde T, Kleinman J, Weinberger D, Becker K, Freed W. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr Res. 2002;58:11–20. doi: 10.1016/s0920-9964(01)00377-2. [DOI] [PubMed] [Google Scholar]
- Vernadakis A. The aging brain. Clin Geriatr Med. 1985;1:61–94. [PubMed] [Google Scholar]
- Yoo B, Fountoulakis M, Cairns N, Lubec G. Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer's disease and Down syndrome. Electrophoresis. 2001;22:172–179. doi: 10.1002/1522-2683(200101)22:1<172::AID-ELPS172>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article 17. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]