

Abstract
Enterovirus infection in newborn infants is a significant cause of aseptic meningitis and encephalitis. Using a neonatal mouse model, we previously determined that coxsackievirus B3 (CVB3) preferentially targets proliferating neural stem cells located in the subventricular zone within 24 h after infection. At later time points, immature neuroblasts, and eventually mature neurons, were infected as determined by expression of high levels of viral protein. Here, we show that blood-derived Mac3+ mononuclear cells were rapidly recruited to the CNS within 12 h after intracranial infection with CVB3. These cells displayed a myeloid-like morphology, were of a peripheral origin based on green fluorescent protein (GFP)-tagged adoptive cell transplant examination, and were highly susceptible to CVB3 infection during their migration into the CNS. Serial immunofluorescence images suggested that the myeloid cells enter the CNS via the choroid plexus, and that they may be infected during their extravasation and passage through the choroid plexus epithelium; these infected myeloid cells ultimately penetrate into the parenchyma of the brain. Before their migration through the ependymal cell layer, a subset of these infected myeloid cells expressed detectable levels of nestin, a marker for neural stem and progenitor cells. As these nestin+ myeloid cells infected with CVB3 migrated through the ependymal cell layer, they revealed distinct morphological characteristics typical of type B neural stem cells. The recruitment of these novel myeloid cells may be specifically set in motion by the induction of a unique chemokine profile in the CNS induced very early after CVB3 infection, which includes upregulation of CCL12. We propose that intracranial CVB3 infection may lead to the recruitment of nestin+ myeloid cells into the CNS which might represent an intrinsic host CNS repair response. In turn, the proliferative and metabolic status of recruited myeloid cells may render them attractive targets for CVB3 infection. Moreover, the migratory ability of these myeloid cells may point to a productive method of virus dissemination within the CNS.
Introduction
Enterovirus infections have been previously associated with a wide range of neurological disorders in a clinical setting, including persistent encephalitis (Berger et al., 2006), white matter damage (Verboon-Maciolek et al., 2006), and acute disseminated encephalomyelitis (Saitoh et al., 2004). Coxsackievirus B3 (CVB3) infections are relatively common and cause a number of human enterovirus-associated diseases, including pancreatitis, myocarditis, and aseptic meningitis (Whitton et al., 2005). Newborns in particular are highly susceptible to infection and disease (Romero, 2008). We have previously described the ability of CVB3 to infect neural stem cells in the neonatal CNS, induce apoptosis within infected neurons, and establish a persistent infection (Feuer et al., 2003, 2005, 2009). The choroid plexus was previously shown to be an early site of CVB3 replication (Feuer et al., 2003). However, a close examination of the infected choroid plexus at the cellular level has not previously been undertaken.
The choroid plexus remains a poorly understood organ in the CNS which has recently been shown to harbor previously unidentified host functions (Emerich et al., 2005). One of the critical functions of the choroid plexus is to regulate the production of CSF in the CNS. The choroid plexus also forms the blood–CSF barrier in the CNS (Ransohoff et al., 2003). The blood–CSF barrier is distinct from the blood–brain barrier (BBB) in that entry is controlled by the tight junctions of the choroid plexus cuboidal epithelium, as opposed to the endothelial cell layer comprising the BBB. Furthermore, the blood–CSF barrier may be an important entry point for activated immune cells (Ransohoff et al., 2003). Certain substances, such as transthyretin, may be actively transported by the choroid plexus into the CSF (Dickson et al., 1986). However, many other functions have recently been described for the choroid plexus, including the production of growth factors (Shingo et al., 2003) and an active involvement in neurogenesis (Falk and Frisén, 2002).
Here, we more closely determined the involvement of the choroid plexus during the early stages of CVB3 infection in the neonatal CNS. The choroid plexus epithelial cells appeared to be spared from CVB3 infection. Instead, infected cells morphologically similar to myeloid cells were found clustered throughout the choroid plexus tissue and within the lateral ventricle. By analyzing serial immunofluorescence sections by microscopy, we carefully characterized the phenotype of myeloid cells undergoing extravasation through the columnar epithelium of the choroid plexus and eventually entering the parenchyma of the neonatal CNS. These cells were seen in abundance only after infection, suggesting that initial CVB3 infection in the CNS may induce chemoattractant molecules into the surrounding area. Furthermore, many recruited myeloid cells expressed nestin, a marker for neural stem and progenitor cells (Lendahl et al., 1990). We also examined the early induction of chemokines which might be responsible for the recruitment of these unique myeloid cells, and their possible fate as they enter the parenchyma of the neonatal CNS. We hypothesize that CVB3 may induce the migration of these unique myeloid cells by the upregulation of chemokines, including CCL12, to aid virus dissemination within the neonatal CNS.
Materials and Methods
Isolation and production of a recombinant coxsackievirus.
The generation of a recombinant coxsackievirus expressing eGFP has been described previously (Feuer, 2002). Briefly, the CVB3 infectious clone (pH3) (obtained from Dr. Kirk Knowlton at University of California at San Diego) was engineered to contain a unique SfiI site which facilitates the insertion of any foreign sequence into the CVB3 genome (pMKS1). For the recombinant CVB3 expressing dsRED (dsRED-CVB3), dsRED gene was amplified from a dsRED expression plasmid (Clontech Laboratories) using dsRED sequence-specific primers with flanking Sfi1 sequences. The PCR product was cloned into pMKS1. Following transfection of HeLa RW cells with dsRED-CVB3 plasmid, infectious virus was generated. All virus stocks were grown on HeLa RW cells maintained in DMEM (Cellgro) supplemented with 10% fetal bovine serum. Virus titrations were performed as described previously.
Mice and viral inoculations.
Mouse experimentation conformed to the requirements of the San Diego State University Animal Research Committee and the National Institutes of Health. BALB/c and C57BL/6 mice were obtained from the Scripps Research Institute animal facilities or Harlan Sprague Dawley. Breeding pairs were checked every day, and 1-d-old pups were infected intracranially with 2 × 106 or 1 × 107 pfu eGFP-CVB3, 5 × 103 pfu wild-type CVB3 (WT-CVB3), 5 × 103 pfu lymphocytic choriomeningitis virus (LCMV), or mock-infected with DMEM. The procedure for intracranial (i.c.) inoculation of 1-d-old pups has been described previously (Feuer et al., 2003, 2005). Pups were killed at various time points between 12, 24, and 48 h postinfection (PI) by hypothermia/CO2, followed by immediate decapitation. The brains were fixed by immersion in 10% neutral-buffered formalin for >4 h, paraffin-embedded, and immunostained or stained with hematoxylin and eosin (H&E).
Adoptive transfer of GFP-expressing bone marrow-derived cells or newborn liver-derived cells into recipient animals infected with CVB3.
Adult bone marrow-derived cells (BMDCs) were isolated from the femurs of actin promoter-GFP transgenic mice and washed three times with PBS solution. Alternatively newborn livers from actin promoter-GFP transgenic mice were homogenized into a single cell suspension and washed three times with PBS. A total of ∼107–108 GFP-expressing donor cells were injected intrahepatically (i.h.) into 1- or 3-d-old C57BL/6 recipient mice (Massengale et al., 2005). Either simultaneously or 1 d later, recipient mice were infected intracranially with dsRED-CVB3 (2.5 × 104 pfu) or mock-infected with DMEM. On days 1, 2, and 6 PI, the brain and livers from recipient mice were harvested, paraffin-embedded, and inspected for the presence of peripheral GFP-expressing donor cells and virus protein expression (dsRED) by immunofluorescence microscopy.
In situ hybridization.
Our in situ hybridization procedure has been described previously (Feuer et al., 2003). Briefly, a 33P-labeled antisense RNA (421 bases) probe for the 5′ untranslated region of CVB3 was generated using the MAXIscript In vitro Transcription Kit (Ambion Inc.), as described by the manufacturer. In situ hybridization procedures were performed using the mRNAlocater In Situ Hybridization Kit (Ambion Inc.), as described by the manufacturer. The radio-labeled probe (107 cpm) was applied to deparaffinized sections, and the sample was sealed in a humidified chamber and incubated at 46°C for 18 h. After washing and RNase A treatment, slides were immersed in photographic emulsion, held at 4°C for 6 d, then developed. Finally, slides were stained with hematoxylin (1 min) and eosin (1.5 min), and mounted with Cytoseal (VWR International).
Myeloid cell quantification of the choroid plexus.
Oil images (100×) of H&E-stained brain sections were taken using a Zeiss Axiovert 200 inverted microscope with an attached Zeiss ICc3 Color Camera. Brain sections of BALB/c mice infected with eGFP-CVB3, and C57BL/6 mice infected with eGFP-CVB3, WT-CVB3, and LCMV at 12, 24, and 48 h postinfection were used. The brain sections with the most intact choroid plexus were used for imaging. Three images of the most representative regions were taken of each choroid plexus, using two animals per time point. Myeloid cells in and around the choroid plexus were counted based on morphology and location. Data were entered into GraphPad Prism 3.0 software, analyzed, and displayed graphically.
Immunofluorescence staining.
eGFP (from eGFP-CVB3 or GFP-expressing donor cells) or dsRED expression was observed in unstained paraffin-embedded sections overlaid with PBS. For immunofluorescence staining, paraffin-embedded sections (3 μm thickness) were deparaffinized with three washes in xylene and serial washes in 100%, 95%, 70% ethanol, followed by a wash in PBS and distilled water. Thereafter, high temperature antigen unmasking in 0.01 m citrate buffer, pH 6.0, was performed. The Mouse on Mouse kit (Vector Laboratories Inc.) was used for primary antibodies derived from mouse. Two main immunofluorescence staining protocols were used. For GFP (1:10, AB3080; Millipore Corporation; or 1:100, ab13970, Abcam), nestin (1:50, MAB353; Millipore Corporation), predilute Ki67 (neat, RTU-Ki67-MM1, Novocastra Laboratories) RC2 (1:50, Developmental Studies Hybridoma Bank, Iowa City, IA), doublecortin (1:100, AB5910; Millipore Bioscience Research Reagents), ZO-1 (1:100, MAB1520; Millipore Bioscience Research Reagents), CD11b/c (1:200, ab53187; Abcam Inc.), phospho-p44/42 (pERK1/2, Thr202/Tyr204) MAP Kinase (1:100, #9101, Cell Signaling Technology Inc.), LeX-CD15 (1:50, #559045 Clone MMA, BD Biosciences Inc.), Musashi (1:100, AB5977; Millipore Corporation), Iba1 (1:100 or 1:200, #016-20001; Wako Pure Chemical Industries, Ltd.), Mac-3 (1:20, #550292; BD Biosciences Inc.), prediluted Cyclin D1 (neat, #P236AA; Biocare Medical Inc.), and laminin (1:200, L 9393; Sigma-Aldrich Inc.) antibodies; sections were blocked with 10% normal goat serum for 30 min, primary antibody was put on overnight at 4°C, goat anti-rabbit, anti-rat, or anti-mouse antibody conjugated to biotin at 1:100, or bovine anti-chicken FITC (sc2700, Santa Cruz Biotechnologies) was applied for 30 min. Finally sections were incubated with streptavidin-Alexa Fluor 488 or streptavidin-Alexa Fluor 594 at 1:500 for 30 min, respectively. For CD105 (1:100, ab56298; Abcam Inc.), CD133 (1:200, ab19898; Abcam Inc.), CD34 (1:50, ab8158; Abcam Inc.), CD11b/c (1:200, ab53187; Abcam Inc.), CD73 (1:50, sc-14682; Santa Cruz Biotechnology Inc.), CCL12/MCP-5 (anti-MCP-5 (K19)-sc9718, Santa Cruz Biotechnology Inc.), c-Kit (1:20, AF1356; R&D Systems), Sca-1 (1:20, R&D Systems Inc.), B220 (1:30, 550286; BD Biosciences Inc.), mCAR (1:100, Santa Cruz Biotechnology Inc.), Tyramide Signal Amplication (TSA) Kit #25 (Invitrogen Inc.) was used according to manufacturer's protocol. Briefly, sections were quenched with 3% H2O2 for 1 h, blocked in 1% Blocking Solution for 1 h, incubated in primary antibody in 1% Blocking Solution overnight at 4°C, goat anti-rabbit antibody conjugated to biotin at 1:100 in 1% Blocking Solution was applied for 2 h, streptavidin-HRP was applied at 1:100 in 1% Blocking Solution for 1 h, and last sections were incubated with tyramide-594 at 1:100 in Amplification Buffer for 20 min. Specificity controls for immunostaining included sections stained in the absence of primary antibody, or in the presence of rabbit IgG control antibody at 0.1 μg/ml (Vector Laboratories, Inc), rat IgG1 K isotype control (1:100, Clone R3-34, #559072, BD Biosciences Inc.), or mouse IgG1-negative isotype control (1:20, Clone DD7, #CBL600; Millipore Bioscience Research Reagents). For detection of DNA/nuclei, sections were overlaid with Vectashield Mounting Medium with DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) (Vector Laboratories). Sections were observed by fluorescence microscopy (Zeiss Axiovert 200 inverted microscope) for GFP (green), the indicated cellular marker (red) and DAPI nuclear staining (blue). Green, red, and blue channel images were merged using AxioVision software.
Confocal microscope imaging.
Confocal images were captured using a Zeiss Laser Confocal Scanning Microscope (LCSM) running the latest Zen 2009 Zeiss software suite (Carl Zeiss Inc.). All serial optical 8 bit image sections [obtained with a 63× Plan-Aprochromat (1.4 numerical aperture) objective at 0.3 mm interval step slices] were imported and spatially reassembled using Imaris software (BitPlane Inc.) to generate a three-dimensional representation of the tissue of which single snapshots and movies were created. Maximum projected two-dimensional images were created in Image Pro Plus (Media Cybernetics Inc.), or ImageJ (National Institutes of Health, Bethesda, MD) software for the montage image panel presentation.
Quantitative real-time reverse transcriptase PCR for CVB3.
5′-untranslated (UTR) CVB3-specific primers (forward primer 5′-CACACTCCGATCAACAGTCA-3′; reverse primer 5′-GAACGCTTTCTCCTTCAACC-3′), and a FAM/TAMRA-labeled probe (5′-CGTGGCACACCAGCCATGTTT-3′) were used for the Taqman method of quantitative real-time reverse transcriptase (qRT)-PCR. A total of 1 μg of total RNA from the samples was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen Inc.) according to the procedure described by the manufacturer. Separate RT reactions were performed for the reverse primer or forward primer to quantitate the number of positive and negative-sense viral genomes, respectively. RT reactions were treated with 1 μl of Ribonuclease H (Invitrogen Inc.) to remove RNA complementary to the cDNA. PCR amplification was done using Platinum Quantitative PCR SuperMix-UDG ready to use cocktail (Invitrogen Inc.) containing all components except the amplification primers and FAM/TAMRA-labeled probe, as described by the manufacturer. Quantitative analysis of viral RNA was performed using a Bio-Rad iQ5 Real-Time PCR System in 96 well optical reaction plates heated to 50°C for 2 min to digest dUTP-containing contaminants, followed by 95°C for 2 min to deactivate UNG and activate Platinum TaqDNA polymerase. Forty cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 30 s were carried out. To normalize the amount of RNA in each sample, separate RT reactions using random primers instead of CVB3-specific primers were performed and amplified for 18S ribosomal RNA. The 18S ribosomal products were amplified with 18S forward (5′-CGGCTACCACATCCAAGGAA-3′) and reverse (5′-GCTGGAATTACCGCGGCT-3′) primers, and included a FAM/TAMRA (5′-TGCTGGCACCAGACTTGCCCTC-3′) probe. The standard curve was generated using in vitro transcribed pH3 CVB3 RNA. The standard curve was based on Ct values (cycle number at which relative fluorescence crossed the threshold) and Ct values from unknown samples (normalized for 18S ribosomal RNA signal) were compared with the standard curve to determine viral RNA copy numbers.
Quantitative real-time RT-PCR for LCMV.
LCMV glycoprotein (GP)-specific primers (forward primer 5′-CATTCACCTGGACTTTGTCAGACTC-3′; reverse primer 5′-GCAACTGCTGTG-TTCCCGAAAC-3′) and a FAM/TAMRA-labeled probe (5′-TTATTGCCTGACCAAATGGA-3′) were used for Taqman method of quantitative real-time RT-PCR. RT-PCR was performed as described for CVB3. Forty cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 30 s were carried out. To normalize the amount of RNA in each sample, separate RT reactions using random primers instead of LCMV-specific primers were performed and amplified for 18S ribosomal RNA. The 18S ribosomal products were amplified with 18S forward (5′-CGGCTACCACATCCAAGGAA-3′) and reverse (5′-GCTGGAATTACCGCGGCT-3′) primers, and included a FAM/TAMRA (5′-TGCTGGCACCAGACTTGCCCTC-3′) probe. The standard curve was generated using the LCMV pSG5-GP plasmid, a generous gift from Dr. Juan Carlos de la Torre (Department of Immunology and Microbial Sciences, The Scripps Research Institute). The LCMV GP gene was isolated from the plasmid using restriction enzyme digest (BglII and EcoRI) and gel purified. Isolated GP DNA was then quantified using a spectrophotometer, converted to copy number, and serially diluted (10-fold dilutions). The standard curve was based on Ct values; Ct values from unknown samples (normalized for 18S ribosomal RNA signal) were compared with the standard curve to determine viral RNA copy numbers.
Microarray analysis.
Total RNA was isolated from homogenized brain samples using the Qiagen RNeasy Kit. RNA was sent to the BIOGEM Microarray Core at University of California at San Diego and analyzed for quality. Samples were reverse transcribed to cDNA and then in vitro transcribed to cRNA with the incorporation of biotin. cRNA was hybridized to Illumina Whole Mouse Genome 6 Samples chips, which analyze ∼45,200 genes. Hybridized chips were incubated with Cy3, washed, and then read using the Illumina BeadArray Station. Normalized gene expression was used to examine differences and similarities in the following samples; GFP-CVB3, WT-CVB3, LCMV, and mock-infected C57BL/6 mice at 12 h postinfection. Relative chemokine/cytokine expression levels were identified via microarray analysis. More specifically, gene expression levels in the brain of eGFP-CVB3 or LCMV-infected neonatal mice were compared with mock-infected control mice. The fold change in gene expression levels for known chemokines compared with mock-infected control mice was analyzed to determine possible chemoattractant molecules associated with myeloid cell recruitment through the choroid plexus specifically following coxsackievirus infection.
ApopTag staining.
Detection of cells undergoing apoptosis was evaluated using the ApopTag Red In Situ Apoptosis Detection Kit (Millipore Inc.), as specified by the manufacturer. Briefly, paraffin-embedded sections were deparaffinized and pretreated with proteinase K (20 mg/ml) for 15 min. Equilibration buffer was added directly onto the specimen, after which terminal deoxynucleotidyl transferase (TdT) enzyme in reaction buffer was added for 1 h at 37°C. Sections were washed in working strength Stop/Wash buffer for 10 min. Prewarmed working strength anti-digoxigenin conjugate (rhodamine) was added to the sections and incubated at room temperature for 30 min. The samples were washed with PBS and observed by fluorescence microscopy, as described above.
Statistical analyses.
Statistical significance was determined using by one-way ANOVA with Newman–Keuls post hoc comparison. For myeloid cell quantification (see Fig. 5H), A 6 × 2 ANOVA with group (BALB/c–eGFP-CVB3, C57BL/6–eGFP-CVB3, C57BL/6–WTCVB3, C57BL/6–LCMV, C57BL/6–MOCK, and C57BL/6–CCL12) as a between-group variable and time (12 h, 24 h, 48 h) as a within-group variable, and average number of myeloid cells per animal as the dependent variable; revealed a significant main effect of group F(5,6) = 81.97, p < 0.001, a significant main effect of time F(2,12) = 17.59, p < 0.001, and a significant group × time interaction F(10,12) = 3.95, p < 0.05. A Newman–Keuls post hoc comparison test of the main effect of group revealed that the average number of myeloid cells per animal in BALB/c–eGFP-CVB3, C57BL/6–eGFP-CVB3, C57BL/6–WTCVB3, and C57BL/6–CCL12 was significantly higher (p < 0.05) from C57BL/6–MOCK, while C57BL/6–LCMV was the only group not significantly higher. In addition, there was no difference observed between the C57BL/6–WTCVB3 and C57BL/6–CCL12 groups. A Newman–Keuls post hoc comparison test of the main effect of time revealed that the average myeloid cells per animal was significantly higher (p < 0.05) at the 24 h time point compared with both the 12 and 24 h time points. A Newman–Keuls post hoc comparison test of the group × time interaction revealed that the average number of myeloid cells per animal in C57BL/6–eGFP-CVB3 was significantly higher (p < 0.05) from C57BL/6–MOCK and C57BL/6–LCMV at the 12 h, 24 h, and 48 h time points. For apoptosis quantification in the choroid plexus (see Fig. 11A), A one-way ANOVA with group (C57BL/6–MOCK, C57BL/6–eGFP-CVB3, and BALB/c–eGFP-CVB3) as a between-group variable, and ApopTag signal per choroid plexus as the dependent variable, revealed significant between-group differences F(2,8) = 4.84, p < 0.05. A Newman–Keuls post hoc comparison test revealed that ApopTag signal per choroid plexus was significantly higher (p < 0.05) in C57BL/6–eGFP-CVB3 and BALB/c–eGFP-CVB3 compared with C57BL/6–MOCK, with no difference between C57BL/6–eGFP-CVB3 and BALB/c–eGFP-CVB3.
Figure 5.

Myeloid cell infiltration is specific to coxsackievirus infection. A, B, D, G, Neonatal mice were monk-infected or infected (intracranially) with eGFP-CVB3, wild-type (wt) CVB3, or LCMV. The brains were harvested 12 h PI, and sections were stained by H&E. Large numbers of myeloid cells were observed in BALB/c mice (A), or C57BL/6 neonatal mice infected with eGFP-CVB3 (B), wt CVB3 (D), or CCL12-inoculated mice (G). C, Higher magnification of B identified myeloid cells migrating through the choroid plexus (black arrows). E, F, In contrast, infection of C57BL/6 mice with LCMV (E), or mock-infected C57BL/6 mice (F) showed little to no myeloid cell infiltration. H, Images (100.8× magnification) of H&E-stained brain sections were used to determine myeloid cell number based on their morphology and location near the choroid plexus. Using ANOVA with Newman–Keuls post hoc comparison, a significant difference was observed when comparing the C57BL/6–mock-infected group and either the BALB/c–eGFP-CVB3 group (p < 0.05), the C57BL/6–eGFP-CVB3 group (p < 0.05), the C57BL/6–WTCVB3 group (p < 0.001), or the C57BL/6 CCL12-inoculated group (p < 0.05). There was no significant difference between the C57BL/6–LCMV group and the C57BL/6–mock-infected group (p > 0.05). Error bars represent the SEM. I, J, Total RNA was isolated from the brain of infected samples and CVB3 RNA was quantified by qRT-PCR using primers and probe specific to the 5′-UTR, as described in Materials and Methods. K, Similarly, LCMV RNA was quantified by qRT-PCR using primers and probe specific for GP. A, B, D–G, 20× objective; C, 100.8× magnification.
Figure 11.

Induction of apoptosis in the choroid plexus and the possible outcome of CVB3-infected myeloid cells following their migration into the parenchyma of the CNS. A–G, Quantification of ApopTag signal in the choroid plexus of C57BL/6 mice infected with eGFP-CVB3, BALB/c mice infected with eGFP-CVB3, or mock-infected C57BL/6 mice 24 h PI. Infected mice showed a statistically significant increase in ApopTag levels (asterisks) compared with mock-infected mice (p < 0.05 using ANOVA with Newman–Keuls post hoc comparison). Representative images of ApopTag signal (red) and viral protein expressions (green) in the choroid plexus is shown at 20× and 63× magnification for infected (B, C, E, F) or mock-infected (D, G) mice. DAPI nuclei-stained cells are shown in blue. H, An illustration of the possible outcome of CVB3-infected myeloid cells is shown. Following inoculation into the lateral ventricle, initial eGFP-CVB3 infection may lead to CCL12 induction. Virions may bind to CAR expressed within the tight junctions of the choroid plexus epithelium. During myeloid cell recruitment, virions may infect myeloid cells upon extravasation across the epithelium. Eventually, CVB3-infected myeloid cells may undergo apoptosis near the choroid plexus (1) or after entering the parenchyma of the CNS (2). 3, Some infected myeloid cells may be phagocytosed by Iba1+ macrophages/microglia. 4, Alternatively, some infected myeloid cells may escape detection and assist in the establishment of persistent CVB3 infection. 5, Infectious virus may be released from myeloid cells upon entry through the ECL and subsequently infect type B and eventually progenitor cells, immature and mature neurons. 6, Finally, nestin+ myeloid cells responding to infection may contribute to CNS repair/neurogenesis.
Results
Myeloid cells migrating into the CNS are highly susceptible to infection
Previously, we determined the ability of CVB3 to target neurogenic regions of the CNS at early time points following infection. However, the choroid plexus was also highly susceptible to infection. Upon closer inspection, the choroid plexus epithelial cells appeared to be spared from CVB3 infection. Instead, H&E staining revealed the presence of cells with myeloid-like morphology which appeared to infiltrate through the vascular stroma (Fig. 1A, black arrows). These cells were seen in abundance only after infection, suggesting that initial CVB3 infection may induce chemoattractant molecules into the surrounding area. In addition, the presence of red blood cells external to the vascular regions of the choroid plexus suggested some hemorrhaging due to infection. In contrast, no infiltration of myeloid-like cells was observed for mock-infected control sections (Fig. 1B). Higher magnification of the infected choroid plexus revealed the extravasation of myeloid-like cells through the tight junctions of the choroidal epithelial cells (Fig. 1C, black arrows) and penetration through the ependymal cell layer (ECL) of the CNS (Fig. 1C, notched cyan arrow). These cells accounted entirely for the presence of viral signal in the choroid plexus and lateral ventricle, as shown by in situ hybridization using a CVB3-specific 5′-UTR probe (Fig. 1D, black signal), although viral signal was also identified (as published previously) near the ECL. In contrast, choroid plexus epithelial cells were generally spared from infection. Similar results were obtained when sections were inspected for viral protein expression (Fig. 1E, eGFP signal).
Figure 1.
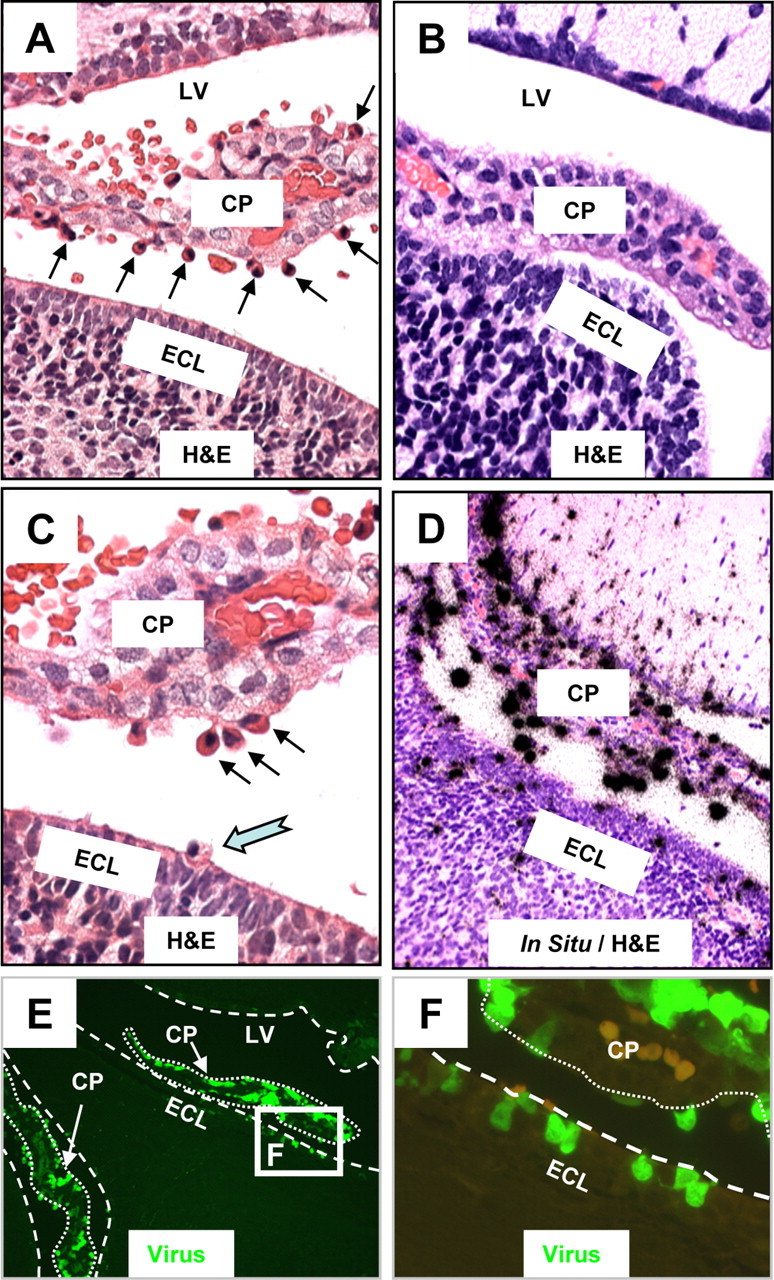
Myeloid cells migrating into the CNS are highly susceptible to CVB3 infection. The brains of mock and eGFP-CVB3-infected neonatal mice (intracranially) were harvested 24 h PI. Brain sections were cut sagittally, deparaffinized, observed by fluorescence microscopy for viral protein expression (eGFP), and stained by H&E (see Materials and Methods). The location of viral RNA was evaluated by in situ hybridization using a 5′-UTR probe specific for CVB3. A, CVB3 infection induced the recruitment of cells having a myeloid-like morphology (black arrows) which migrated from vascular stromal cores through the choroid plexus columnar epithelium. A number of these cells were found within the CSF of the ventricles. B, In contrast to infected mice, the choroid plexus of mock-infected mice revealed no apparent influx of myeloid-like cells near the choroid plexus epithelial cell layer. C, Higher magnification of the infected brain revealed the presence of myeloid-like cells adjacent to the epithelial cell layer of the choroid plexus (black arrows), many of which appeared to migrate through the ECL and into the parenchyma of the brain (cyan notched arrow). D, E, In situ hybridization (D) and fluorescence microscopy (E) demonstrated that these myeloid-like cells found adjacent to the choroid plexus supported robust viral replication (D, black dots) and expressed high levels of viral protein (E, green signal). F, Higher magnification of the white box in E showed multiple infected cells at different stages of diapedesis passing through the lumen of the choroid plexus (outlined with dotted white lines), along with the stepwise migration of these myeloid-like cells across the ECL (outlined with dashed white line). A few autofluorescing red blood cells (light orange) can be seen in the choroid plexus. A, B, D, 40× objective; C, 63× objective; E, 20× objective; F, 100.8× magnification. LV, lateral ventricle.
Infected myeloid cells extravasate through the choroid plexus and into the ependymal cell layer
Higher magnification of fluorescent microscopic images revealed the apparent stepwise migration of infected myeloid cells from the vascular stroma of the choroid plexus, through the epithelial cell layer, across the CSF of the lateral ventricle, and into the parenchyma of the brain (Fig. 1F, eGFP signal). These results suggest a route of entry for CVB3 through the blood–CSF barrier of the neonatal CNS using cells of the myeloid lineage, similar to what has been observed for HIV and the blood–brain barrier (Eugenin et al., 2006). Extravasation of infected myeloid cells through the choroid plexus (CP) columnar epithelium was observed in sections immunostained with laminin and zona occludens 1 (Z0–1) antibodies (Fig. 2A,B, respectively). In higher-magnification images, infected myeloid cells were observed in the process of extravasation through laminin+ (red) basement membranes outlining the capillaries of the choroid plexus (Fig. 2C, small gray arrows). Despite the early age of infected pups, ZO-1 staining (red) identified the tight junctions of the columnar epithelium which organizes the blood–CSF barrier in the CP (Fig. 2D). The blood–CSF barrier appeared to be predominantly intact, and infected myeloid cells could be seen trafficking through the columnar epithelium (Fig. 2D, small gray arrows). The activation status of infected myeloid cells was inspected by staining for Ki67 (cell cycle marker) and pERK1/2 (Fig. 2E,F, respectively). As expected, Ki67 staining was observed in neurogenic regions comprising the subventricular zone (SVZ). However, numerous infected myeloid cells also expressed high levels of Ki67, suggesting active proliferation. Intriguingly, the distribution of Ki67 was routinely cytoplasmic in infected myeloid cells (Fig. 2G, white arrows), as opposed to the normal nuclear distribution within uninfected cells found nearby (Fig. 2G, notched cyan arrows). Similar, the majority of infected myeloid cells expressed high levels of pERK1/2 in the cytoplasm, although other infected myeloid cells showed little to no expression (Fig. 2H, small white arrows). Both cytoplasmic and nuclear distribution of pERK1/2 has been described previously (Refojo et al., 2005). These results indicate that the majority of myeloid cells were undergoing proliferation or were highly activated, which might assist CVB3 replication. High expression levels of the CVB3 receptor [murine coxsackie and adenovirus receptor (mCAR)] was observed in regions within the tight junctions of the columnar epithelium (overlapping with ZO-1) and in the SVZ (Fig. 2I). In addition, detectable levels of mCAR were seen on infiltrating myeloid cells (Fig. 2J, white arrows).
Figure 2.
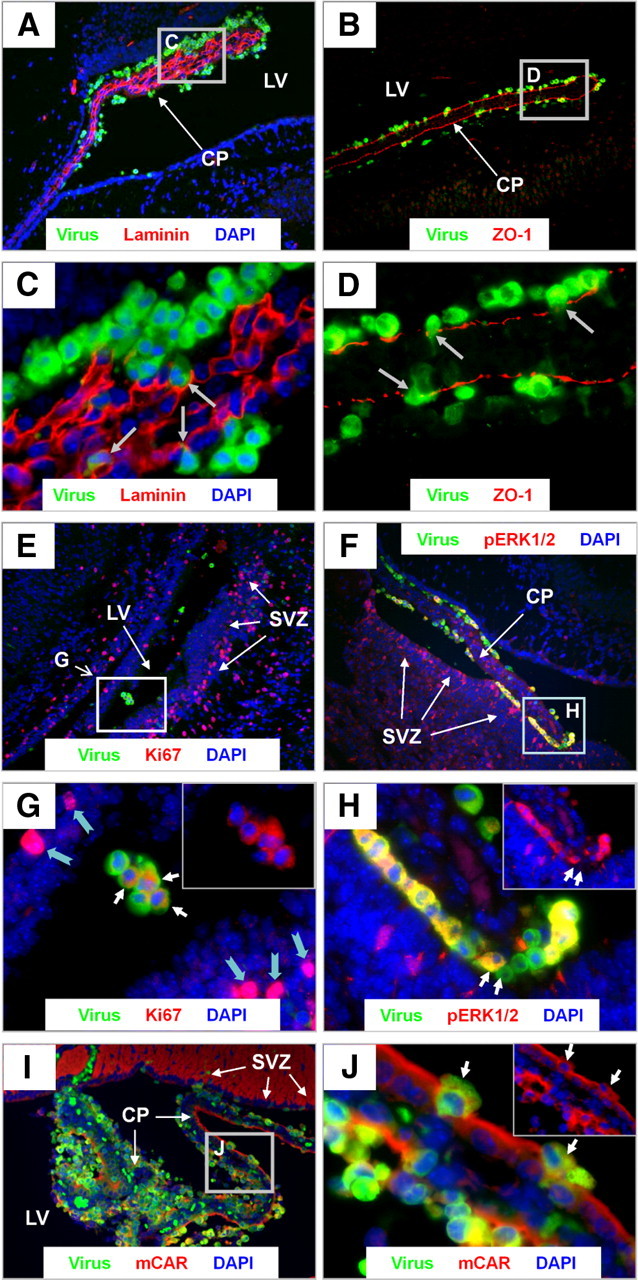
Infected myeloid cells extravasate through the choroid plexus and into the ependymal cell layer. Sagittal brain sections from eGFP–CVB3-infected pups (intracranially) were harvested 24 h PI, deparaffinized, and immunostained with antibodies against GFP, laminin, ZO-1, and mCAR. A, C, Laminin staining (red) which outlined the basement membrane revealed infected myeloid cells (green) trafficking through the vascular core of the choroid plexus. B, D, ZO-1 staining (red) showed infected myeloid cells (green) migrating through the tight junctions of the choroid plexus. E, Cellular proliferation assessed by Ki67 staining (red) revealed cellular proliferation in the subventricular zone and in myeloid cells within the lateral ventricle. G, Higher magnification of E showed a cytoplasmic localization for Ki67 (normally found in the nucleus) within infected cells (white arrows), and made clear in the inset image in the top right corner (red and blue channel only). In contrast, uninfected cells nearby showed a normal nuclear distribution for Ki67 (notched cyan arrows). F, The majority of infected myeloid cells also expressed high levels of pERK1/2 (red). H, Higher magnification of F revealed both pERK1/2+ and pERK1/2−-infected myeloid cells (white arrows). The pERK1/2 cytoplasmic staining pattern is illuminated by the inset image in the top right corner (red and blue channel only). I, mCAR expression (red) was observed within the tight junctions of the choroid plexus epithelium and in the subventricular zone. J, Higher magnification illustrated detectable mCAR expression levels on infected myeloid cells (white arrows). The mCAR staining pattern is illuminated by the inset image in the top right corner (red and blue channel only). A, B, E, F, I, 20× objective; C, D, G, H, J, 100.8× magnification. LV, Lateral ventricle.
We more carefully inspected the process of myeloid cell extravasation by confocal microscopy and IMARIS 3D analysis (Fig. 3). The progressive infiltration of infected myeloid cells through the basement membrane was observed in laminin (red)-stained sections of the choroid plexus at 12 and 24 h PI. Confocal microscopy revealed the progressive entry of infected myeloid cells through the tight junctions of the choroid plexus columnar epithelium. In addition, a 3D IMARIS movie representation was created at 12 and 24 h to inspect infected myeloid cell entry (supplemental Movies 1 and 2, respectively, available at www.jneurosci.org as supplemental material). Higher magnification (Fig. 3, zoom 1) revealed that myeloid cells expressed high levels of viral protein (green) only upon entry through the basement membrane. No eGFP+ myeloid cells were seen within the capillaries of the choroid plexus, indicating that infection of these cells occurred during myeloid cell extravasation (Fig. 3, gray-scale images of each color channel). IMARIS 3D analysis with a diminishing laminin label more clearly outlined the entry of infected myeloid cells through the basement membrane of the choroid plexus (Fig. 3, zoom IMARIS 3D with diminishing laminin label). Higher-magnification images showed the process of diapedesis of infected myeloid cells into the surrounding lateral ventricle (Fig. 3, zoom 2—white arrows).
Figure 3.

Progressive extravasation of infected myeloid cells through the basement membrane as determined by confocal microscopy and Imaris 3D analysis. Neonatal mice were infected (intracranially) with eGFP-CVB3, and the brains were harvested 12 and 24 h PI. The kinetics of myeloid cell migration through the basement membrane (outlined by laminin staining in red) was observed by confocal microscopy and IMARIS 3D analysis at 12 h (top) and 24 h PI (bottom). Low magnification of immunofluorescence images showed infected myeloid cell migration (green) through the tight junctions of the choroid plexus epithelium. Gray-scale images of all three colors (virus, green; laminin, red, DAPI, blue) at 12 and 24 h PI revealed the intensity and organization of the myeloid cell infiltration in greater detail. To better visualize myeloid cell entry, IMARIS 3D with diminishing laminin label (diminishing red) was performed on a higher magnification (zoom 1) for both 12 and 24 h PI images. Higher magnification of zoom 1 revealed the extravasation of infected myeloid cells through the basement membrane (zoom 2, white arrows). Original images were obtained with a 63× Plan-Aprochromat objective at 0.3 mm interval step slices (as described in Materials and Methods).
Myeloid cells may be recruited through the blood–CSF barrier by a unique set of chemokines which include CCL12 expression within the choroid plexus
RNA viruses are known to upregulate key chemokines, cytokines and type I interferon response genes (Takeuchi and Akira, 2009). We expected that novel chemoattractant factors may be upregulated to induce myeloid cell recruitment in the neonatal CNS specifically in response to CVB3 infection. Such chemoattractant molecules may also have a direct role on the recruitment of bone marrow stem cells into damaged tissues (Dar et al., 2006). We used Illumina BeadArray Technology (MouseWG-6 v2 Expression Beadchips) and the BeadChip BioMedical Genomics Microarray Facility (Department of Medicine, UCSD) to identify novel genes upregulated in the neonatal CNS during nestin+ myeloid recruitment. RNA was isolated from mock-infected neonatal mice, or neonatal mice infected with eGFP-CVB3, wtCVB3 and LCMV at 12 h PI. By comparing CVB3-infected, LCMV-infected and mock-infected animals using Illumina's genome-wide gene expression profiling, we were able to analyze gene expression alterations for nearly 46,000 transcripts. A number of chemokines were shown to be upregulated following CVB3 infection compared with mock-infected control mice (Fig. 4A). Chemokines upregulated in response to eGFP-CVB3 infection included CCL7, CCL9, and CCL4. In particular, extremely high levels of CCL12 were induced following eGFP-CVB3 infection. However, cytokines and interferon response gene expression were not significantly different between the treatments, except for a moderate increase in IRF-1 and OAS1 g for eGFP-CVB3-infected samples (data not shown). Next, we wanted to determine the location and level of CCL12 protein in the neonatal CNS by immunofluorescence staining. CCL12 (red) was highly expressed in eGFP-CVB3-infected samples as early as 12 h PI (Fig. 4B). Furthermore, higher-magnification images indicated that protein expression was localized to the to the choroid plexus and ependymal cell layer (Fig. 4D). In contrast, little or no CCL12 signal was observed in mock-infected samples (Fig. 4C,E).
Figure 4.
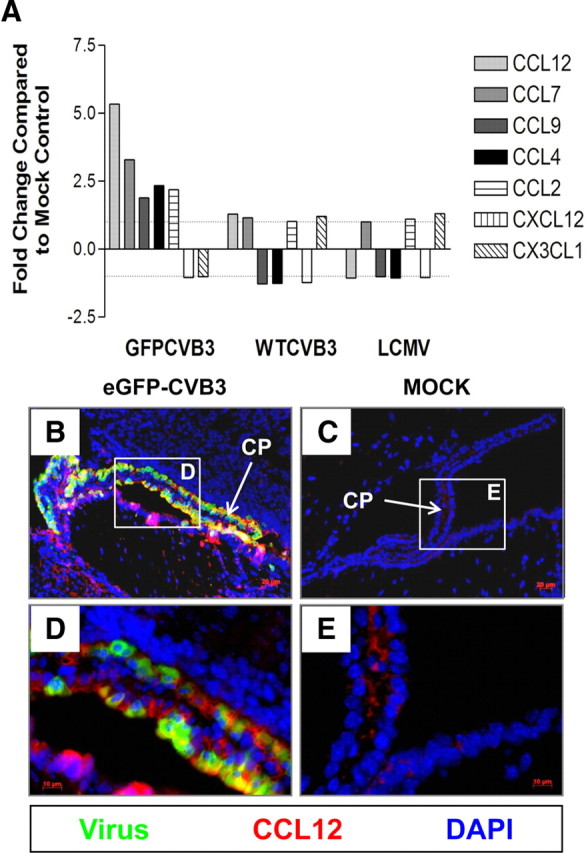
Myeloid cells may be recruited through the blood–CSF barrier by a unique set of chemokines, specifically CCL12 expressed by the choroid plexus. Total RNA was isolated from infected and mock-infected brain samples 12 h PI. cRNA generated from isolated RNA was hybridized to Illumina whole mouse genome chips (n = 2). A, Analysis of chemokine gene expression via microarray analysis showed the induction of particular chemokines in response to eGFP-CVB3 infection, including CCL12, CCL7, CCL9, and CCL4. Given that CCL12 was the most highly upregulated chemokine, immunostaining was performed using a CCL12 antibody on infected or mock-infected brain to evaluate the degree and location of chemokine protein expression. B, D, At 12 h PI, high levels of CCL12 (red) was expressed in the choroid plexus near or within eGFP-CVB3-infected myeloid cells (green), as shown in representative images. C, E, Little to no CCL12 was expressed in mock-infected control samples. DAPI nuclei-stained cells are shown in blue.
Myeloid cell infiltration is specific to coxsackievirus infection
To determine whether myeloid cell infiltration was specific to CVB3 infection, an additional RNA virus, LCMV, was analyzed concurrently with CVB3-infected samples at 12, 24 and 48 h PI. Myeloid cell infiltration was observed in the choroid plexus of both neonatal BALB/c and C57BL/6 mice infected with GFP-CVB3 (Fig. 5A,B, respectively). Higher magnification, showed the characteristic myeloid cell infiltration (Fig. 5C, black arrows). A relatively low inoculum (5 × 103 pfu, i.c.) of wild-type CVB3 (wt CVB3) also led to a limited induction of myeloid cell infiltration (Fig. 5D). In contrast, little or no myeloid cell infiltration was observed for either LCMV or mock-infected neonatal C57BL/6 mice (Fig. 5E,F, respectively). Also, intracranial administration of CCL12 was found to induce the recruitment of myeloid cells independent of CVB3 infection, albeit at a lower level (Fig. 5G).
Myeloid cell recruitment was quantified by counting the number of cells present in high-magnification images in three independent regions of the choroid plexus for two animals per treatment (Fig. 5H). Using this quantitative methodology, both neonatal BALB/c and C57BL/6 mice infected with eGFP-CVB3 induced high numbers of myeloid cells when compared with mock-infected control mice (p < 0.05 using ANOVA with Newman–Keuls post hoc comparison). A lower number of myeloid cells was observed in neonatal mice given a substantially lower inoculum of wtCVB3 or intracranially inoculation of CCL12 chemokine, compared with the mice infected with a high dose of eGFP-CVB3. The small increase in the number of myeloid cells for either wtCVB3 or CCL12 inoculation remained significantly greater than that observed in mock-infected control mice (p < 0.05), and graphical results showed a trend toward greater numbers of myeloid cells over three independent time points (12, 24 and 48 h PI) in wtCVB3-infected or CCL12 inoculated mice, compared with mock-infected control mice. In addition, no significant difference was observed between LCMV and mock-infected control mice (p > 0.05). The few myeloid-like cells observed in the choroid plexus of LCMV-infected and mock-infected mice might represent resident epiplexus cells (Ransohoff et al., 2003).
Next, we performed qRT-PCR on RNA isolated from infected CNS tissue to examine viral load in these samples over time, as described previously (Feuer et al., 2009). The quantity of eGFP-CVB3 and wtCVB3 genomic RNA increased over a period of 48 h PI (Fig. 5I,J, respectively). Higher viral copy numbers were observed for eGFP-CVB3 compared with wtCVB3, most likely reflecting the greater initial starting inoculum for eGFP-CVB3 infected mice (1 × 107 pfu versus 5 × 103 pfu, respectively). Using qRT-PCR, an increase in viral load in the neonatal CNS over time was also observed for LCMV (Fig. 5K).
Infected myeloid cells near the choroid plexus express both neural stem/progenitor cell and myeloid-lineage markers
Remarkably, immunofluorescence data revealed that many CVB3-infected myeloid cells in the choroid plexus (eGFP) expressed nestin (red), a marker which identifies neural stem and progenitor cells (Lendahl et al., 1990) (Fig. 6A,C). As expected, the majority of nestin staining was observed within the SVZ. Surprisingly, numerous infected myeloid cells located within or near the choroid plexus expressed high levels of nestin. These infected myeloid cells also expressed high levels of Mac3 (red), an antigen normally found on myeloid-lineage cells (Fig. 6B,D) (Ho and Springer, 1983). Furthermore, numerous Mac3+ cells were found within the SVZ, suggesting their eventual entry into the parenchyma of the brain. Isotype control staining for both nestin and Mac3 demonstrated the specificity of our immunofluorescence methodology in these sections (Fig. 6E,F, respectively). Higher magnification of sections in Figure 6A revealed nestin+ cells morphologically similar to that of myeloid cells located adjacent, yet distinct, from the columnar epithelium of the CP and the ependymal cell layer (Fig. 7A,C,E). These nestin+ myeloid cells were not only distinct from the columnar epithelium of the choroid plexus and the ependymal cell layer, but exhibited morphology similar to that of type B neural stem cells with cellular protrusions penetrating the ECL and contacting the lateral ventricle. Identical sections used for fluorescence microscopy as described above were stained by H&E to reveal infected cells having characteristic myeloid morphology and located adjacent to the choroid plexus or within the lateral ventricle (Fig. 7B,D,F, black arrows). Interestingly, many infected cells expressed nestin in a polarized manner within discrete cellular regions in the apparent direction of cell movement. A few infected myeloid cells in other sections expressed nestin near ciliated structures or cellular processes extending into the ependymal cell layer, made apparent after H&E staining of the same section (Fig. 7G,H, notched cyan arrows in inset images). This unique staining pattern suggests that functionally, nestin (an intermediate filament) may play a direct role in cell migration, which might fit well with what is known about intermediate filaments, cell motility (Helfand et al., 2003) and neuronal progenitor cell progression (Doetsch et al., 1999). We also observed a number of infected myeloid cells near the choroid plexus or within the ependymal cell layer expressing a radial glial marker (Fig. 7I,J) or a neuronal precursor cell marker (Fig. 7K,L).
Figure 6.
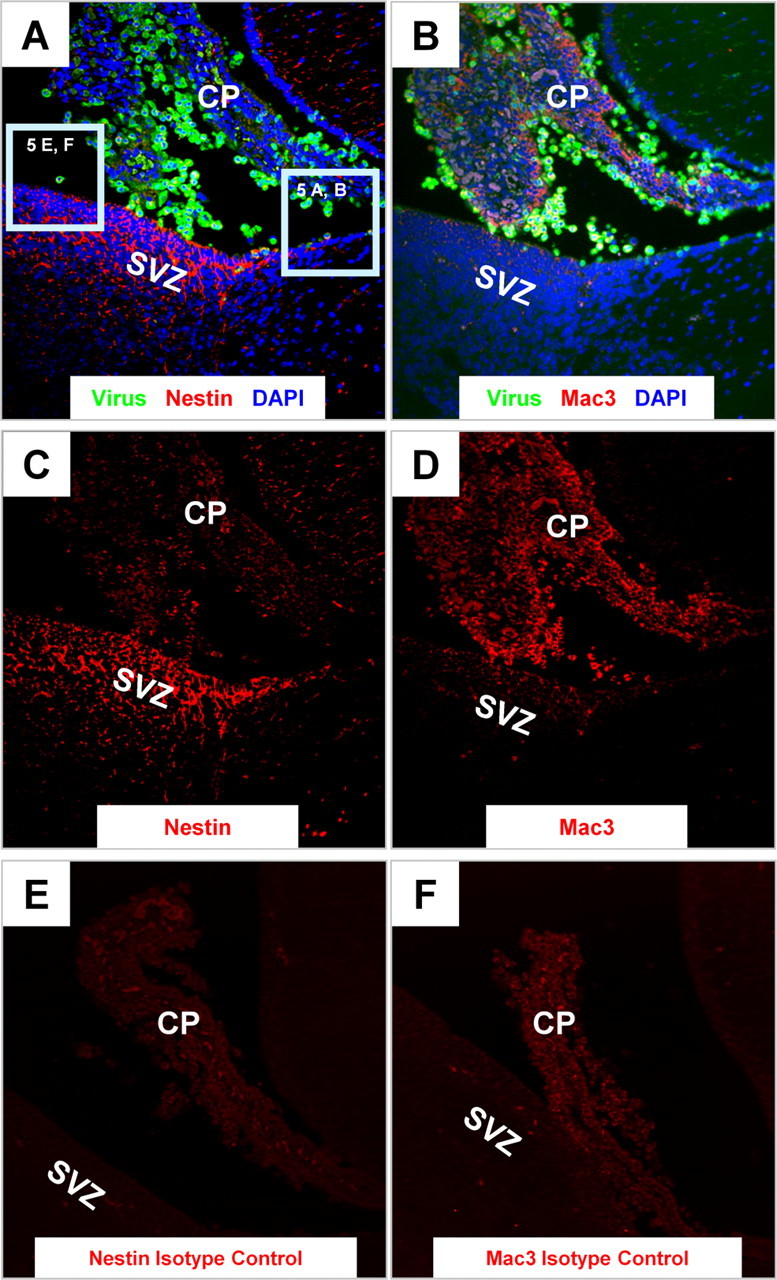
Infected myeloid cells near the choroid plexus express both neural stem/progenitor cell and myeloid-lineage markers. Sagittal brain sections from infected pups were harvested 24 h PI, deparaffinized, and immunostained with antibodies against nestin and Mac3. A, C, As expected, nestin staining (red) was evident in the SVZ. However, detectable levels of nestin expression were also seen within infected cells (green) adjacent to the choroid plexus and circulating within the CSF of the lateral ventricle. B, D, The majority of infected cells within the lateral ventricle expressed Mac-3 (a bone marrow myeloid-lineage marker) indicated by the high levels of expression (red) within the lateral ventricle. E, F, No signal was observed in parallel sections immunostained using a mouse IgG isotype control antibody for nestin (E) or a rat isotype control antibody for Mac3 (F). A–F, 20× objective.
Figure 7.
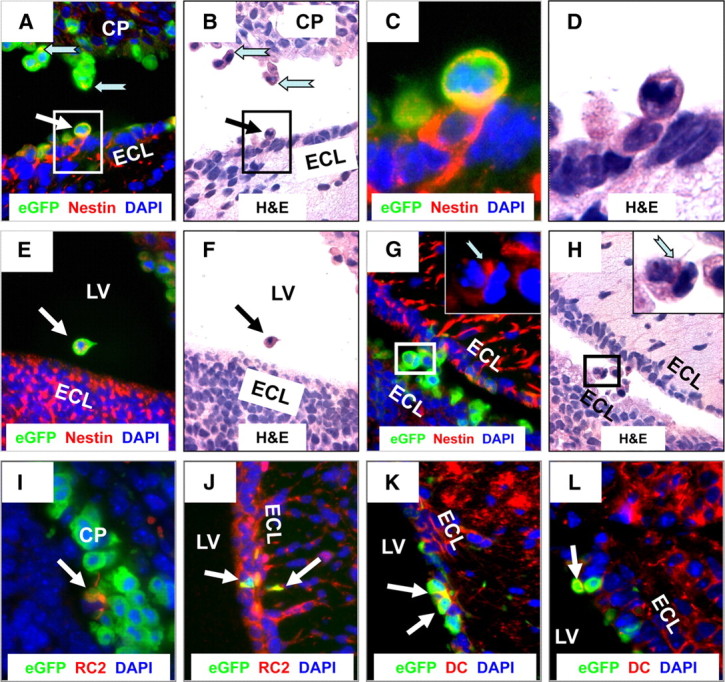
Infected myeloid cells in the ventricle express nestin and have the morphology of type B stem cells as they migrate though the ECL and into the parenchyma of the brain Sagittal brain sections from infected pups were harvested 24 h PI, deparaffinized, and immunostained with antibodies against nestin, RC2, and doublecortin (DC). A, E, Infected myeloid cells near the choroid plexus (A) (cyan notched arrows), adjacent to the ECL (white arrow), and within the lateral ventricle (E) (white arrow) expressed high levels of nestin. C, High magnification of A illustrated the cytoplasmic and extended cellular processes, typical of nestin staining, extending within the ECL. B, D, F, Identical sections were stained by H&E to reveal the myeloid-like morphology of these nestin+ cells (black arrow) in different regions of the lateral ventricle. G, H, A small number of infected myeloid cells (G) in transit through the LV and moving toward the SVZ expressed nestin (red, cyan notched arrow in high-magnification inset) in cytoplasmic locations adjacent to extended cellular processes, as visualized in the identical section stained by H&E in H (cyan notched arrow in high-magnification inset). Nestin expression appears to be localized in the direction of movement. I, J, A few infected myeloid cells (white arrows) near the choroid plexus and within the ECL expressed RC2, a marker for radial glial cells. K, L, Some myeloid cells (white arrows) adjacent to the ECL also expressed doublecortin, a marker for neuronal precursor cells. LV, lateral ventricle. A, B, E–H, J–L, 63× objective; C, D, 100× objective with a further ∼2-fold magnification; I, 100× objective.
Macrophages/microglia engulfed CVB3-infected myeloid cells entering the CNS
Within 12 and 24 h PI, Iba1+ macrophages/microglia were seen responding to CVB3 infection (Fig. 8). Iba1+ cells were observed near the choroid plexus and within the subventricular zone (Fig. 8A). In contrast to Mac3 staining results, infected myeloid cells in the choroid plexus did not express Iba1, a marker for activated monocytes/microglia/macrophages (Ito et al., 2001). Instead, Iba1+ macrophages/microglia were seen engulfing multiple infected myeloid cells (Fig. 8B–D, white arrows). Macrophage/microglia engulfment of CVB3-infected cells in the CNS has been described previously, although at later time points (Feuer et al., 2009). Higher magnification revealed the presence of Iba1+ gitter cells (macrophage/microglia cells that are globular and swollen following phagocytosis of CNS debris) engulfing virus-infected cells in some sections (Fig. 8B). Similar results illustrating the phagocytosis of multiple infected-myeloid cells in the CNS were also seen 24 h PI following either Iba1 (Fig. 8E,G, white arrows) or CD11b immunofluorescence staining (Fig. 8F,H, white arrows).
Figure 8.
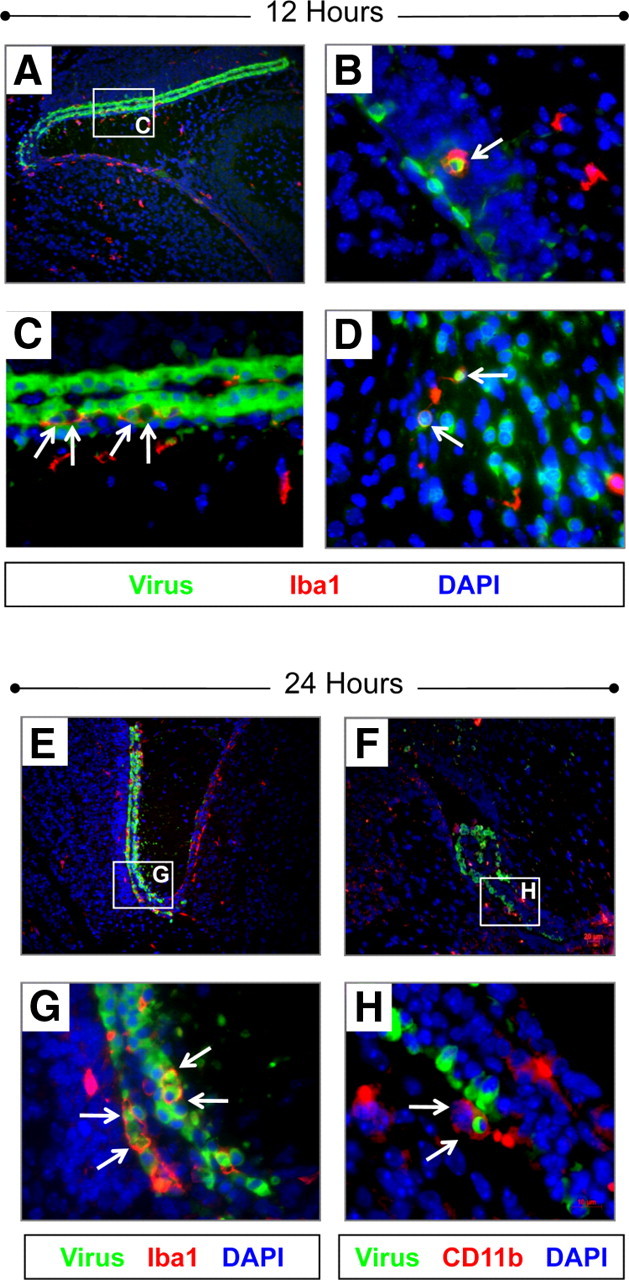
Iba1+ and CD11b+ macrophages/microglia engulfed CVB3-infected myeloid cells entering the CNS. Sagittal brain sections from infected pups were harvested 12 and 24 h PI, deparaffinized and immunostained with antibodies against Iba1 and CD11b. A, Iba1+ expression (red) was observed near the choroid plexus and SVZ at 12 h PI. B, High magnification of an Iba1+ macrophage (red) reminiscent of a “gitter cell” (white arrow) that appears to engulf an infected myeloid cell (green) near the SVZ a 12 h PI. C, Higher magnification of A showed numerous Iba1+ cells (red) engulfing multiple CVB3-infected myeloid cells (green) near the choroid plexus (white arrows) at 12 h PI. D, High magnification of an Iba1+ cell engulfing two infected cells (white arrows) within the rostral migratory stream at 12 h PI. E, G, At 24 h PI, phagocytosis of infected myeloid cells (green) near the choroid plexus (white arrows) was more pronounced, with multiple cells seen becoming engulfed by Iba1+ cells (red). F, H, Phagocytosis of infected myeloid cells by CD11b+ macrophages/microglia were also observed near the choroid plexus (white arrows). DAPI nuclei-stained cells are shown in blue. A, E, F, 20× objective; B–D, G, H, 100× objective.
Progressive increase in Iba1+ macrophages/microglia and engulfment of CVB3-infected myeloid cells undergoing apoptosis
We observed a progressive increase in Iba1+ macrophages/microglia within the choroid plexus over 48 h PI (Fig. 9). In contrast to the appearance of infected myeloid cells (green) as soon as 12 h PI, the kinetics of macrophage/microglia migration to the choroid plexus was delayed. Very few Iba1+ macrophages/microglia (red) were seen at 12 h PI (Fig. 9A,B). At 24 h PI, the number of Iba1+ macrophages/microglia increased at a point in time when infected myeloid cells appeared within the SVZ (Fig. 9C,D). By 48 h PI, the integrity of the choroid plexus appeared compromised, and Iba1+ macrophages/microglia were found in high numbers (Fig. 9E,F). Intriguingly, Iba1+ macrophages/microglia actively engulfed the few remaining infected myeloid cells within the choroid plexus (Fig. 9G). As shown by ApoTome analysis or “grid projection” which allows for optical sectioning on a motorized Zeiss Axio Observer fluorescence microscope, Iba1+ macrophages/microglia could be visualized engulfing multiple infected myeloid cells (Fig. 9H,z-stack image, white arrows; also shown in supplemental Movie 3, available at www.jneurosci.org as supplemental material). An x,z view (top part of image) and a y,z view (right-hand part of image) in Figure 9H show the internalization of infected myeloid cells by Iba1+ macrophages/microglia. Additionally, the engulfment of apoptotic bodies by Iba1+ macrophages/microglia in parallel sections was clearly visible at 48 h PI, as shown by ApoTome analysis (Fig. 9I,J, z-stack image, white arrows; also shown in supplemental Movie 4, available at www.jneurosci.org as supplemental material). An x,z view (top part of image) and a y,z view (right-hand part of image) in Figure 9J illustrates the engulfment of apoptotic bodies by Iba1+ macrophages/microglia.
Figure 9.
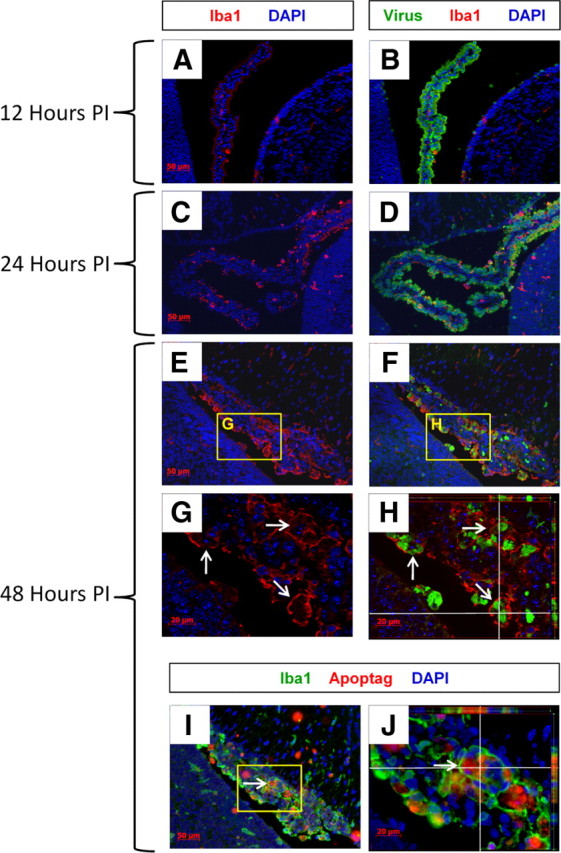
Progressive increase in Iba1+ macrophages/microglia and engulfment of CVB3-infected myeloid cells undergoing apoptosis. Sagittal brain sections from infected pups were harvested 12, 24, and 48 h PI, deparaffinized and immunostained with antibodies against Iba1 and viral protein (GFP) or identified for apoptotic activity using ApopTag. A, B, Infected myeloid cells (green) were seen in high numbers in the choroid plexus as soon as 12 h PI. In contrast, only low numbers of Iba1+ macrophages/microglia (red) were seen in the choroid plexus and SVZ at 12 h PI. C, D, By 24 h PI, a substantial increase in Iba1+ macrophages/microglia was seen in the choroid plexus. E, F, After 48 h, Iba1+ macrophages/microglia were observed at high numbers in the choroid plexus at a time when the number of infected myeloid cells substantially decreased. G, H, Higher magnification showed Iba1+ macrophages/microglia which actively phagocytosed the remaining infected myeloid cells (white arrows) in the choroid plexus. I, J, Infected myeloid cells underwent apoptosis, and the apoptotic bodies were engulfed by Iba1+ macrophages/microglia (white arrow). Z-stack images obtained using ApoTome Imaging System for 3 orthogonal section views—x,y, x,z, and y,z, were included in H and J, as shown in the “Cut View” (white lines) using AxioVision software. Z-stack images for H showed Iba1+ macrophages/microglia (red) surrounding infected myeloid cells (green). Z-stack images for J showed Iba1+ macrophages/microglia (green) surrounding apoptotic bodies (red) in parallel cuts. All images were acquired as five 1 μm z-stacks taken with a Zeiss Axiovert 200 inverted microscope with ApoTome Imaging System. A–F, I, 20× objective; G, H, J, 63× objective.
Expression of molecular markers by CVB3-infected myeloid cells in the CNS
A summation of antibodies used for immunofluorescence staining to identify recruited myeloid cells responding to CVB3 infection at 12 and 24 h PI is shown in Table 1. In general, antibodies were used to determine neural and hematopoietic stem cell, cell cycle/activation, and myeloid/monocyte/macrophage markers. Infected myeloid cells consistently expressed cell cycle/activation markers. Also as described above, these cells expressed relatively high levels of nestin and Mac3. In contrast, most infected myeloid cells failed to express high levels of other informative neural stem/progenitor cell or hematopoietic cell markers.
Table 1.
Expression of molecular markers by myeloid cells recruited into the CNS
| Neural stem cell markers | Hematopoietic/mesenchymal stem cell markers | Cell cycle/activation markers | Monocyte/macrophage markers |
|---|---|---|---|
| Nestin | CD105 | Ki67 (cytoplasmic) | Mac3 |
| ++ | +/− | +++ | +++ |
| RC2 | c-kit | Cyclin D1 (cytoplasmic) | Iba1 |
| +/− | − | +++ | − |
| Doublecortin | CD34 | pERK1/2 | B220 |
| +/− | − | +++ | +/− |
| Musashi | CD133 | CD11b/c | |
| − | − | − | |
| Lex (CD15) | Sca-1 | ||
| − | − | ||
| NG2 | CD73 | ||
| − | − |
The results of immunofluorescence staining for neural stem and progenitor cell markers, hematopoietic/mesenchymal stem cell markers, cell cycle/activation markers, and monocyte/macrophage markers on recruited myeloid cells 24 h PI are described. The relative level of expression for each marker is represented in a range of high expression levels (+++) to little or no expression (−). Recruited myeloid cells expressed high levels of cell cycle/activation markers. However, few cell lineage or neural stem and progenitor cell markers (except for nestin and mac3) were expressed on recruited myeloid cells.
Adoptively transferred donor cells from actin promoter-GFP transgenic mice inoculated intrahepatically follow a similar path to the CNS following CVB3 infection
We wished to reveal the peripheral origin of infected myeloid cells responding to CVB3 in the CNS. Therefore, adult BMDCs, or alternatively, newborn liver-derived cells from actin promoter-GFP transgenic mice were inoculated intrahepatically into 1- or 3-d-old recipient C57BL/6 mice, as described previously (Massengale et al., 2005). These recipient mice were subsequently infected (intracranially) with a recombinant coxsackievirus expressing dsRED protein either simultaneously, or 24 h following the intrahepatic inoculation of GFP-expressing donor cells (Fig. 10A). On days 1, 2 and 6 postinoculation, the brains of recipient mice were inspected for GFP+ myeloid cells near the choroid plexus and within the parenchyma similar to those observed in Figure 1. Surprisingly, the inoculation of newborn liver-derived donor cells (the site of hematopoiesis in newborn mice) routinely led to greater engraftment in the recipient liver, compared with adult BMDCs (Fig. 10B,C). Intriguingly, the appearance of numerous GFP+ myeloid-like cells within the brain was evident as soon as 24 h postinoculation (POI) in simultaneously infected mice, or alternatively, as soon as 48 h POI in mice infected 1 d after intrahepatic inoculation (Fig. 10D). GFP+ donor cells with myeloid morphology, similar to infected myeloid cells seen in Figure 6, were observed adjacent to the ECL, within the SVZ, near the choroid plexus, and within the olfactory bulb (Fig. 10F,H,I, respectively). Furthermore, mock-infected animals failed to display an influx of GFP+ donor cells (Fig. 10E); although a few GFP+ perivascular macrophage cells were seen near blood vessels in the brain parenchyma (data not shown). By day 6 POI, GFP+ donor cells, including a few cells with neuronal-like homology, were observed within the cortex (Fig. 10J,K). The majority of GFP+ donor cells did not express high levels of viral protein, and the degree of infection (as judged by dsRED expression) appeared not to be as robust as with eGFP-CVB3. We attribute this discrepancy due to the relatively low initial viral inoculum (2.5 × 104 pfu, i.c.) reflective of our marginal dsRED-CVB3 working stock, and a greater technical difficulty in observing dsRED expression compared with eGFP (fluorescence requires dsRED tetramer formation, and dsRED protein has a tendency to aggregate). Our recombinant CVB3 expressing dsRED may also be of lower viral fitness compared with eGFP-CVB3 (data not shown), perhaps due to the greater toxicity of dsRED in eukaryotic cells. Also at later stages of infection (day 6 POI), viral protein levels may be waning in surviving cells. Nonetheless, these data suggest a peripheral origin of myeloid cells in the choroid plexus and CNS responding to CVB3 infection.
Figure 10.

Recruitment of intrahepatically inoculated GFP-expressing donor cells into the CNS following infection with dsRED-CVB3. BMDCs or newborn liver-derived cells were isolated from actin promoter-GFP transgenic mice. A, GFP-expressing cells were inoculated intrahepatically into 1- or 3-d-old recipient C57BL/6 mice. Recipient mice were intracranially infected either simultaneously, or 24 h later, with dsRED-CVB3. The brains and liver of recipient mice were harvested 1, 2, and 6 d postinoculation. Recipient livers showed varying numbers of engrafted GFP-expressing donor cells. B, C, The most successful engraftment occurred in recipient mice inoculated intrahepatically with newborn liver cells (green), although the majority of recipient mice showed some level of successful engraftment. D, As soon as 24 h postinfection, GFP-expressing donor cells were observed in the CNS (representative animal shown). E, In contrast, no GFP-expressing donor cells were observed in the CNS in mock-infected mice. F, G, Higher magnification of D showed donor cells near the lateral ventricle and subventricular zone (white arrows). H, I, GFP-expressing donor cells were also observed within the choroid plexus and olfactory bulb (white arrows). J, By day 6 postinfection, GFP-expressing donor cells were observed in the entorhinal cortex (white and blue arrows). Also, viral protein expression (red) was observed in the entorhinal cortex, although no colocalization with donor was seen. K, Higher magnification of J identified GFP-expressing donor cells in the entorhinal cortex with neuronal-like processes (white arrow). B–E, 20× objective; F–J, 63× objective; K, 63× objective with a further ∼3-fold magnification.
Induction of apoptosis in the choroid plexus following CVB3 infection
We determined the ability of eGFP-CVB3 to induce apoptosis within infected myeloid cells by TUNEL (ApopTag staining). The amount of ApopTag signal was quantified on a per section basis and compared with mock-infected control samples (Fig. 11A). For both C57BL/6 and BALB/c mice, high levels of ApopTag signal was observed within infected myeloid cells adjacent to the choroid plexus 24 h PI. Little to no ApopTag signal was seen in C57BL/6 mock-infected control sections. The ApopTag signal levels were statistically significant (p < 0.05) using ANOVA with Newman–Keuls post hoc comparison for infected C57BL/6 and BALB/c mice, compared with mock-infected control samples. Representative fluorescent microscopic images of infected C57BL/6 and BALB/c neonatal mice revealed high levels of ApopTag signal (red) in myeloid cells adjacent to the choroid plexus (Fig. 11B,C, respectively). Higher magnification showed ApopTag signal predominantly within infected (eGFP+) myeloid cells (Fig. 11E,F). In contrast, little to no ApopTag signal was observed in mock-infected mice (Fig. 11D,G).
Possible outcome of CVB3-infected myeloid cells following their migration into the parenchyma of the CNS
Our present model suggests that upon inoculation, eGFP-CVB3 virions may provisionally bind to the tight junctions of the choroid plexus epithelial cells (Fig. 11H). The induction of key chemokines specific to CVB3 infection, including CCL12, may play a role in the recruitment of unique myeloid cells through the choroid plexus. Upon extravasation through the tight junctions of the choroid plexus epithelial cells, eGFP-CVB3 virions may target these highly activated myeloid cells, thereby leading to their infection and rapid expression of viral proteins (including eGFP). Infected myeloid cells may eventually enter the lateral ventricle and travel through the ependymal cell layer. Presently, our ability to follow infected myeloid cells through the ependymal cell layer into the parenchyma of the CNS is limited. However, our existing data indicate several possible outcomes for these cells as they migrate into the parenchyma. (1) Infected myeloid cells may undergo apoptosis and be engulfed by Iba1+ macrophages/microglia before entering the parenchyma. (2) Infected myeloid cells may reach the parenchyma of the brain and undergo virus-induced apoptosis shortly thereafter. (3) Iba1+ macrophages/microglia may engulf infected myeloid cells within the parenchyma. (4) Infected myeloid cells may remain for extended periods of time within the CNS and eventually contribute to CVB3 persistence, as described previously (Feuer et al., 2009). Additionally, these cells may assist in the dissemination of CVB3 deep within the CNS. (5) The production of infectious virus by these cells may contribute to the infection of type B neural stem cells, progenitor cells, or neurons; as shown previously (Feuer et al., 2003, 2005). (6) Finally, given their ability to express neural stem cell and progenitor cell markers, infected myeloid cells may exhibit long term effects in the brain. These effects may, either indirectly or directly contribute to neurogenesis and/or CNS repair. The outcome of such a scenario might be the eventual generation of infected neurons which eventually migrate into the cortex and olfactory bulb and contribute to viral persistence. Given the plasticity of cells in a developing neonate and the unique immunological environment produced following CVB3 infection, such an outcome might be a possibility.
Discussion
Our recent studies on coxsackievirus B3 (CVB3) tropism and pathogenesis in our neonatal mouse model of CNS infection have allowed us to make some intriguing observations. First, using a recombinant CVB3 expressing GFP, we determined that this virus preferentially infects proliferating neural stem cells (NSCs) in the neonatal CNS (Feuer et al., 2003, 2005; Whitton et al., 2005) and may persist in the CNS for months after initial infection (Feuer et al., 2009). Second, we now describe the early recruitment of nestin+ (marker for neural stem and progenitor cells) myeloid cells through the blood–CSF barrier shortly after infection, and before the establishment of infection in neurogenic regions of the brain (shown diagrammatically in Fig. 11H). These cells are Mac3+ upon entry, but then appear to increase their nestin expression as they move into the lateral ventricle. Finally, these peripherally recruited myeloid cells were highly susceptible to infection upon their entry across the tight junctions of the CP epithelial cells that provide the framework for the blood–CSF barrier.
The choroid plexus, an invagination of modified ependymal cells forming epithelium, may be considered a unique microenvironment and has been implicated in regulating neurogenesis (Krizhanovsky and Ben-Arie, 2006) and the immune response in the CNS (Reboldi et al., 2009). The recruitment of myeloid-like cells appears to be CVB3-specific, given that an unrelated RNA virus [lymphocytic choriomeningitis virus (LCMV)], failed to stimulate myeloid cell recruitment through the blood–CSF barrier. Also, a unique spectrum of chemoattractant molecules was expressed specifically following eGFP-CVB3 infection. In particular, CCL12, a chemokine known to induce the recruitment of monocytes, was highly expressed in regions of the choroid plexus and the ependymal cell layer. Furthermore, intracranial inoculation of CCL12 into neonatal mice independently induced the recruitment of myeloid cells through the choroid plexus, albeit at a lower level compared with eGFP-CVB3 infection (Fig. 5G,H). Also, it remains to be determined whether myeloid cells recruited following CCL12 inoculation are identical to those induced following CVB3 infection. Injection of a single chemokine may not completely mimic the microenvironment induced by CVB3 necessary to attract these novel myeloid cells. Perhaps the injection of a “chemokine cocktail” might more accurately recreate the immune response induced following CVB3 infection.
The temporal distribution of these infected cells suggests their extravasation through the vascular stromal cores of the choroid plexus columnar epithelium, across the CSF of the ventricles, and the continued migration across the ependymal cell layer into the parenchyma of the brain. Although these cells may be contributing to the immune response against CVB3 infection in the CNS, few signs of inflammation other than reactive microgliosis were evident in the brain parenchyma shortly after infection (Feuer et al., 2009). Furthermore, these myeloid cells failed to express Iba1, a marker for activation microglia/macrophages. This suggested to us that infected myeloid cells seen in the choroid plexus do not differentiate into microglia or macrophages in the neonatal CNS. Also, these cells did not have long dendrites typical of dendritic cells and failed to express dendritic cell markers. Dendritic cells have been previously shown to be resistant to coxsackievirus infection (Kramer et al., 2008).
Intriguingly, Iba1+ macrophages/microglia which did not express nestin were observed actively engulfing numerous infected myeloid cells. Serial sections demonstrated two distinct cell populations (infected myeloid cells and Iba1+ macrophages/microglia) having different migration kinetics entering the choroid plexus following CVB3 infection. Furthermore, the progression of cell migration captured by immunofluorescence microscopy highlighted the transient nature of infected myeloid cells, and illustrated their rapid movement away from the choroid plexus and into the parenchyma of the brain between 24 and 48 h PI. Also, the few remaining infected myeloid cells in the choroid plexus at 48 h PI underwent apoptosis, and these apoptotic bodies were subsequently phagocytosed by the large number of Iba1+ macrophages/microglia infiltrating into the choroid plexus at this time point following infection.
Additional immunostaining revealed that the majority of myeloid cells in the choroid plexus expressed high levels of phospho-p44/42 MAP kinase (pERK1/2) or Ki67 (marker for cell proliferation), which may explain the susceptibility of these cells to infection (Feuer et al., 2002, 2004; Esfandiarei et al., 2004; Feuer and Whitton, 2008). Nestin staining within infected myeloid cells was frequently localized to leading cellular processes in the direction of the ependymal cell layer. Although expression of nestin has been repeatedly used to identify migratory neural stem and progenitor cells, little is known regarding the function of nestin. Recent data suggest that nestin may participate in the phosphorylation of microtubules and direct cell migration and movement (Helfand et al., 2003; Kleeberger et al., 2007).
The extravasation of infected myeloid cells through the choroid plexus columnar epithelium was observed by immunostaining of sections with laminin and ZO-1 antibodies and by confocal microscopy with IMARIS 3D analysis. Collectively, staining for laminin and ZO-1 identifies two key structural components of the choroid plexus, basement membranes and tight junctions. Laminin is a ubiquitous connective tissue glycoprotein and a major component of basement membranes. Laminin helped to identify the endothelial cell layer of the choroid plexus and permitted us to observe the diapedesis of cells from the blood capillaries of the vascular stromal core. Infected myeloid cells were observed in the process of extravasation through laminin+ regions. Despite the early age of infected pups (12, 24, and 48 h postbirth), ZO-1 staining outlined the tight junctions of the columnar epithelium organizing the blood–CSF barrier in the choroid plexus. Infected myeloid cells could be seen trafficking through the columnar epithelium. The great majority of infected cells expressed high levels of viral protein during or after extravasation, indicating that infection may have taken place upon entry of these cells through the blood–CSF barrier. As suggested by our own CAR immunostaining, we hypothesize that binding of CVB3 virions to CAR located within the tight junctions (Nagai et al., 2003) may assist in virus dissemination following the recruitment and diapedesis of target cells through the tight junctions of the choroidal epithelium. These results also indicate that the morphology of the choroid plexus is maintained during myeloid cell infiltration up to 24 h PI, as mock infected samples had similar laminin and ZO-1 expression (data not shown).
As these novel cells enter the parenchyma of the brain, they exhibit morphological similarities to type B NSCs that normally reside within the SVZ and display typical cellular processes that characteristically penetrate through the ECL contacting the lateral ventricles (Alvarez-Buylla et al., 2002). Some myeloid cells may eventually undergo apoptosis, or become phagocytosed by resident macrophages/microglia. We suggest that CVB3 infection, and the subsequent damage to the neonatal CNS, may induce myeloid cell recruitment through the blood–CSF barrier to repair the brain following injury, such as after an infection. Moreover, CVB3 may exploit this inherent repair process in the neonatal CNS by intensifying myeloid cell recruitment for the purpose of maximizing virus dissemination into the brain. Early CVB3 infection and subsequent chemokine expression may induce the recruitment of myeloid cells and the eventual systemic distribution of virus into the CNS and other tissues, as described for murine cytomegalovirus by the Mocarski group (Noda et al., 2006). Aseptic meningitis caused by coxsackievirus infection in newborns may be exacerbated by myeloid cell recruitment since these cells may provide an effective method for viral dissemination throughout the neonatal CNS. The recruitment of mobile cells, and their subsequent susceptibility to infection, may be a common theme among viruses in their drive to disseminate through the host. The benefits to viral spread using these cells within the host are manyfold in this scenario. (1) Virions might be protected from neutralizing antibodies normally found in high titers in the sera. (2) Mobility of migrating cells might provide virions with rapid transportation to normally inaccessible organs (for example, the CNS). (3) Many different genera of viruses require activated or proliferating host cells for maximal viral replication, and the metabolic activity of migratory cells may make them attractive targets for viral infection.
Myeloid cells have not been previously identified as supporting neurogenesis, although some primary hematopoietic cells and CD14+ monocytes express nestin and other stem cell markers under certain culture conditions (Vitry et al., 2003; Chen et al., 2005). Recent studies have described the ability of CD14+ multipotent monocytes having the ability to differentiate in vitro into bone, skeletal muscle (Kuwana et al., 2003), cardiac muscle (Kodama et al., 2005), and most recently the neuronal lineage (Kodama et al., 2006). Others have described the close association of perivascular macrophages with regions of neurogenesis within the SVZ (Mercier et al., 2002). However, the role of these cells during neurogenesis has not been evaluated. Bone marrow-derived cells have been shown to migrate into the CNS in response to injury and help the repair process (Brazelton et al., 2000). A recent study suggests that neuronal progenitor cells may have a cell marker, LeX (ssea-1), found on monocytes (Capela and Temple, 2002). The possibility remains that progenitor cells may be induced to migrate from the blood only after expression of a migratory factor or chemokine following infection with CVB3 or after CNS damage. Identifying such a migratory factory would be imperative for new treatments involving stem cell therapies. Controversy exists in the field of “transdifferentiation,” whereby some researchers have shown that BMDCs can differentiate into neurons, cardiomyoctes, and other cell lineages (Brazelton et al., 2000; Mezey et al., 2000). In contrast, others have interpreted these results differently (Castro et al., 2002). They suggest that BMDCs fuse to sick or dying neurons and repair functionality without “transdifferentiation.” Intriguing results from Nataf et al. (2006) describe a population of myeloid progenitor cells in close association with fibroblastic-like cells of unknown function expressing nestin within choroid plexus stroma cultures. Furthermore, choroid plexus ependymal cells were recently shown to harbor neural progenitor cells (Itokazu et al., 2006). We hypothesize that these neonatal nestin+ myeloid cells responding to CVB3 infection may represent a unique primitive progenitor cell which might assist with CNS repair in response inflammation, damage, or infection with particular viruses. Neonatal myeloid progenitor cells may exhibit unique characteristics and function compared with their fully differentiated counterparts within the adult host. For example, neonatal monocytes show modified expression levels of genes involved in the functional immune response when compared with adult monocytes (Strunk et al., 2004; Lawrence et al., 2007).
To clarify the peripheral origin of these myeloid cells which appear to be highly susceptible to CVB3 infection, we intrahepatically inoculated bone marrow-derived cells or newborn liver-derived cells from actin promoter-GFP transgenic mice into 1- or 3-d-old C57BL/6 recipient animals. Recipient animals were subsequently intracranially inoculated with a recombinant CVB3 expressing dsRED protein (dsRED-CVB3). We demonstrated the migration of myeloid-like cells expressing GFP with similar morphological characteristics and anatomical location within the CNS, as shown for mice infected with eGFP-CVB3. In contrast, no GFP+ donor cells were observed in mock-infected neonatal mice. The migration of adoptively transferred GFP-expressing cells which traffic into the CNS following dsRED-CVB3 inoculation suggests a peripheral origin for myeloid-like cells seen within the choroid plexus following infection.
We performed an Illumina BeadArray Whole Mouse Genome analysis of the neonatal brain following infection with two contrasting RNA viruses in hopes of identifying novel chemokines and cytokines induced specifically by CVB3 infection which might be responsible for nestin+ myeloid cell recruitment into the CNS. As determined by our cDNA microarray analysis, CVB3 replication in the neonatal CNS appeared to induce a specific combination of chemokines which may lead to myeloid cell recruitment. This recruitment may be amplified in the neonatal host, where development is ongoing and cellular plasticity may be more evident.
Microarray analysis indicated that several chemokines were induced in eGFP-CVB3-infected samples at 12 h postinfection. In particular, the β-chemokines CCL12, CCL7, CCL9, and CCL4 were highly induced in response to CVB3 infection. These chemokines were not upregulated following infection with an unrelated RNA virus (LCMV), nor have their upregulation been described in the literature for other neurotropic microbes, including HIV and T. brucei, which primarily induce CCL2 (Courtioux et al., 2006; Campbell and Spector, 2008). Also, CCL12 protein was expressed at high levels within the choroid plexus and the ependymal cell layer following CVB3 infection, and intracranial inoculation of CCL12 into neonatal mice increased the number of myeloid cells within the choroid plexus independent of CVB3 infection. These results suggest that the CCL12 chemokine may play an essential role in the recruitment of nestin+ myeloid cells. In contrast, CXCL12 expression levels previously shown to mediate hematopoietic stem cell chemotaxis (Nervi et al., 2006) were relatively unchanged (data not shown). Although relatively low levels of chemokines were induced following wtCVB3 infection, we hypothesize that differential viral kinetics of the slower replicating recombinant CVB3 (eGFP-CVB3) versus wtCVB3 may be partly responsible for these differences. Nonetheless, myeloid cell recruitment was observed for both recombinant and wtCVB3s. Future studies will address the role of CCL12 in myeloid cell recruitment through the choroid plexus, and the ability of these recruited myeloid cells to assist in the establishment of CVB3 persistence in the CNS.
Footnotes
This work was supported by National Institutes of Health Awards R01 NS054108 (to R.F.), R01 AI-42314 (to J.L.W.), RO1 NS051247 (to K.S.D.), and an Achievement Rewards for College Scientists Foundation Scholarship (to J.M.T.-G.). No conflicts of interest exist between the subject matter and the authors included in the manuscript. Microarray analysis was performed at the Biogem Core Facility of the University of California San Diego—Director Gary Hardiman. We thank Dr. Ricardo Zayas and Roma Munday (Biology Department, San Diego State University) for assistance with using a motorized Zeiss Axio Observer fluorescent microscope with ApoTome.
References
- Alvarez-Buylla A, Seri B, Doetsch F. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull. 2002;57:751–758. doi: 10.1016/s0361-9230(01)00770-5. [DOI] [PubMed] [Google Scholar]
- Berger JR, Chumley W, Pittman T, Given C, Nuovo G. Persistent coxsackie B encephalitis: report of a case and review of the literature. J Neurovirol. 2006;12:511–516. doi: 10.1080/13550280601090546. [DOI] [PubMed] [Google Scholar]
- Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000;290:1775–1779. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Spector SA. CCL2 increases X4-tropic HIV-1 entry into resting CD4+ T cells. J Biol Chem. 2008;283:30745–30753. doi: 10.1074/jbc.M804112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capela A, Temple S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron. 2002;35:865–875. doi: 10.1016/s0896-6273(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Castro RF, Jackson KA, Goodell MA, Robertson CS, Liu H, Shine HD. Failure of bone marrow cells to transdifferentiate into neural cells in vivo. Science. 2002;297:1299. doi: 10.1126/science.297.5585.1299. [DOI] [PubMed] [Google Scholar]
- Chen N, Hudson JE, Walczak P, Misiuta I, Garbuzova-Davis S, Jiang L, Sanchez-Ramos J, Sanberg PR, Zigova T, Willing AE. Human umbilical cord blood progenitors: the potential of these hematopoietic cells to become neural. Stem Cells. 2005;23:1560–1570. doi: 10.1634/stemcells.2004-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtioux B, Boda C, Vatunga G, Pervieux L, Josenando T, M'Eyi PM, Bouteille B, Jauberteau-Marchan MO, Bisser S. A link between chemokine levels and disease severity in human African trypanosomiasis. Int J Parasitol. 2006;36:1057–1065. doi: 10.1016/j.ijpara.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Dar A, Kollet O, Lapidot T. Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp Hematol. 2006;34:967–975. doi: 10.1016/j.exphem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Dickson PW, Aldred AR, Marley PD, Bannister D, Schreiber G. Rat choroid plexus specializes in the synthesis and the secretion of transthyretin (prealbumin). Regulation of transthyretin synthesis in choroid plexus is independent from that in liver. J Biol Chem. 1986;261:3475–3478. [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Esfandiarei M, Luo H, Yanagawa B, Suarez A, Dabiri D, Zhang J, McManus BM. Protein kinase B/Akt regulates coxsackievirus B3 replication through a mechanism which is not caspase dependent. J Virol. 2004;78:4289–4298. doi: 10.1128/JVI.78.8.4289-4298.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood–brain barrier: a potential mechanism of HIV-CNS invasion and neuroAIDS. J Neurosci. 2006;26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A, Frisén J. Amphiregulin is a mitogen for adult neural stem cells. J Neurosci Res. 2002;69:757–762. doi: 10.1002/jnr.10410. [DOI] [PubMed] [Google Scholar]
- Feuer R, Whitton JL. Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr Top Microbiol Immunol. 2008;323:149–173. doi: 10.1007/978-3-540-75546-3_7. [DOI] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol. 2003;163:1379–1393. doi: 10.1016/S0002-9440(10)63496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Hassett DE, Whitton JL. Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med Microbiol Immunol. 2004;193:83–90. doi: 10.1007/s00430-003-0192-z. [DOI] [PubMed] [Google Scholar]
- Feuer R, Pagarigan RR, Harkins S, Liu F, Hunziker IP, Whitton JL. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J Neurosci. 2005;25:2434–2444. doi: 10.1523/JNEUROSCI.4517-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Ruller CM, An N, Tabor-Godwin JM, Rhoades RE, Maciejewski S, Pagarigan RR, Cornell CT, Crocker SJ, Kiosses WB, Pham-Mitchell N, Campbell IL, Whitton JL. Viral persistence and chronic immunopathology in the adult central nervous system following coxsackievirus infection during the neonatal period. J Virol. 2009;83:9356–9369. doi: 10.1128/JVI.02382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Chang L, Goldman RD. The dynamic and motile properties of intermediate filaments. Annu Rev Cell Dev Biol. 2003;19:445–467. doi: 10.1146/annurev.cellbio.19.111401.092306. [DOI] [PubMed] [Google Scholar]
- Ho MK, Springer TA. Tissue distribution, structural characterization, and biosynthesis of Mac-3, a macrophage surface glycoprotein exhibiting molecular weight heterogeneity. J Biol Chem. 1983;258:636–642. [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- Itokazu Y, Kitada M, Dezawa M, Mizoguchi A, Matsumoto N, Shimizu A, Ide C. Choroid plexus ependymal cells host neural progenitor cells in the rat. Glia. 2006;53:32–42. doi: 10.1002/glia.20255. [DOI] [PubMed] [Google Scholar]
- Kleeberger W, Bova GS, Nielsen ME, Herawi M, Chuang AY, Epstein JI, Berman DM. Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res. 2007;67:9199–9206. doi: 10.1158/0008-5472.CAN-07-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama H, Inoue T, Watanabe R, Yasuoka H, Kawakami Y, Ogawa S, Ikeda Y, Mikoshiba K, Kuwana M. Cardiomyogenic potential of mesenchymal progenitors derived from human circulating CD14+ monocytes. Stem Cells Dev. 2005;14:676–686. doi: 10.1089/scd.2005.14.676. [DOI] [PubMed] [Google Scholar]
- Kodama H, Inoue T, Watanabe R, Yasutomi D, Kawakami Y, Ogawa S, Mikoshiba K, Ikeda Y, Kuwana M. Neurogenic potential of progenitors derived from human circulating CD14+ monocytes. Immunol Cell Biol. 2006;84:209–217. doi: 10.1111/j.1440-1711.2006.01424.x. [DOI] [PubMed] [Google Scholar]
- Kramer M, Schulte BM, Toonen LW, Barral PM, Fisher PB, Lanke KH, Galama JM, van Kuppeveld FJ, Adema GJ. Phagocytosis of picornavirus-infected cells induces an RNA-dependent antiviral state in human dendritic cells. J Virol. 2008;82:2930–2937. doi: 10.1128/JVI.02376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Ben-Arie N. A novel role for the choroid plexus in BMP-mediated inhibition of differentiation of cerebellar neural progenitors. Mech Dev. 2006;123:67–75. doi: 10.1016/j.mod.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Kuwana M, Okazaki Y, Kodama H, Izumi K, Yasuoka H, Ogawa Y, Kawakami Y, Ikeda Y. Human circulating CD14+ monocytes as a source of progenitors that exhibit mesenchymal cell differentiation. J Leukoc Biol. 2003;74:833–845. doi: 10.1189/jlb.0403170. [DOI] [PubMed] [Google Scholar]
- Lawrence S, Tang Y, Frank MB, Dozmorov I, Jiang K, Chen Y, Cadwell C, Turner S, Centola M, Jarvis JN. A dynamic model of gene expression in monocytes reveals differences in immediate/early response genes between adult and neonatal cells. J Inflamm (Lond) 2007;4:4. doi: 10.1186/1476-9255-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- Massengale M, Wagers AJ, Vogel H, Weissman IL. Hematopoietic cells maintain hematopoietic fates upon entering the brain. J Exp Med. 2005;201:1579–1589. doi: 10.1084/jem.20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier F, Kitasako JT, Hatton GI. Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. J Comp Neurol. 2002;451:170–188. doi: 10.1002/cne.10342. [DOI] [PubMed] [Google Scholar]
- Mezey E, Chandross KJ, Harta G, Maki RA, McKercher SR. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science. 2000;290:1779–1782. doi: 10.1126/science.290.5497.1779. [DOI] [PubMed] [Google Scholar]
- Nagai M, Yaoita E, Yoshida Y, Kuwano R, Nameta M, Ohshiro K, Isome M, Fujinaka H, Suzuki S, Suzuki J, Suzuki H, Yamamoto T. Coxsackievirus and adenovirus receptor, a tight junction membrane protein, is expressed in glomerular podocytes in the kidney. Lab Invest. 2003;83:901–911. doi: 10.1097/01.lab.0000073307.82991.cc. [DOI] [PubMed] [Google Scholar]
- Nataf S, Strazielle N, Hatterer E, Mouchiroud G, Belin MF, Ghersi-Egea JF. Rat choroid plexuses contain myeloid progenitors capable of differentiation toward macrophage or dendritic cell phenotypes. Glia. 2006;54:160–171. doi: 10.1002/glia.20373. [DOI] [PubMed] [Google Scholar]
- Nervi B, Link DC, DiPersio JF. Cytokines and hematopoietic stem cell mobilization. J Cell Biochem. 2006;99:690–705. doi: 10.1002/jcb.21043. [DOI] [PubMed] [Google Scholar]
- Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, Mocarski ES. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood. 2006;107:30–38. doi: 10.1182/blood-2005-05-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Refojo D, Echenique C, Müller MB, Reul JM, Deussing JM, Wurst W, Sillaber I, Paez-Pereda M, Holsboer F, Arzt E. Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proc Natl Acad Sci U S A. 2005;102:6183–6188. doi: 10.1073/pnas.0502070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero JR. Pediatric group B coxsackievirus infections. Curr Top Microbiol Immunol. 2008;323:223–239. doi: 10.1007/978-3-540-75546-3_10. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Sawyer MH, Leake JA. Acute disseminated encephalomyelitis associated with enteroviral infection. Pediatr Infect Dis J. 2004;23:1174–1175. [PubMed] [Google Scholar]
- Shingo T, Gregg C, Enwere E, Fujikawa H, Hassam R, Geary C, Cross JC, Weiss S. Pregnancy-stimulated neurogenesis in the adult female forebrain mediated by prolactin. Science. 2003;299:117–120. doi: 10.1126/science.1076647. [DOI] [PubMed] [Google Scholar]
- Strunk T, Temming P, Gembruch U, Reiss I, Bucsky P, Schultz C. Differential maturation of the innate immune response in human fetuses. Pediatr Res. 2004;56:219–226. doi: 10.1203/01.PDR.0000132664.66975.79. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verboon-Maciolek MA, Groenendaal F, Cowan F, Govaert P, van Loon AM, de Vries LS. White matter damage in neonatal enterovirus meningoencephalitis. Neurology. 2006;66:1267–1269. doi: 10.1212/01.wnl.0000208429.69676.23. [DOI] [PubMed] [Google Scholar]
- Vitry S, Bertrand JY, Cumano A, Dubois-Dalcq M. Primordial hematopoietic stem cells generate microglia but not myelin-forming cells in a neural environment. J Neurosci. 2003;23:10724–10731. doi: 10.1523/JNEUROSCI.23-33-10724.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol. 2005;3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]