

Abstract
Seven-transmembrane receptors (7TMRs; also known as G protein-coupled receptors) are the largest class of receptors in the human genome and are common targets for therapeutics. Originally identified as mediators of 7TMR desensitization, β-arrestins (arrestin 2 and arrestin 3) are now recognized as true adaptor proteins that transduce signals to multiple effector pathways. Signalling that is mediated by β-arrestins has distinct biochemical and functional consequences from those mediated by G proteins, and several biased ligands and receptors have been identified that preferentially signal through either G protein- or β-arrestin-mediated pathways. These ligands are not only useful tools for investigating the biochemistry of 7TMR signalling, they also have the potential to be developed into new classes of therapeutics.
Seven-transmembrane receptors (7TMRs), also called G protein-coupled receptors (GPCRs), are the most common class of receptors, with more than 800 members identified in the human genome1. They are also the most commonly targeted receptor class for medicinal therapeutics2. Drugs that activate 7TMRs are thought to modulate the proportion of receptors that are in an active signalling conformation relative to those in an inactive, non-signalling conformation. Based on the classical model for 7TMR activity, agonist binding to the 7TMR causes the receptor to adopt a conformation that results in the activation of associated heterotrimeric G proteins. This activation involves the exchange of bound GDP for GTP by the Gα subunit of the G protein, leading to dissociation of the heterotrimeric protein complex into Gα and Gβγ subunits. This dissociation then promotes the production of and consequent signalling by second messenger systems, such as those involving cyclic AMP, diacylglycerol and calcium3. Signalling by the activated conformation of the 7TMR is terminated by phosphorylation of the cytoplasmic loops and tail of the 7TMR, which is catalysed predominantly by GPCR kinases (GRKs). This results in the binding of arrestins (most commonly β-arrestin 1 and β-arrestin 2) and consequent desensitization followed by internalization into clathrin-coated pits4. Thus, in the classical model, heterotrimeric G proteins mediate signal transduction via the receptor, and β-arrestins mediate receptor desensitization and internalization (FIG. 1a).
Figure 1. G protein and β-arrestin-mediated signalling.

a | In the classical model for seven-transmembrane receptor (7TMR) activation, signalling is mediated by G proteins and desensitization is mediated by β-arrestins. b | In the current model for 7TMR activation, binding of a ligand results in activation of signalling by G proteins and β-arrestins, as well as desensitization and internalization by β-arrestins. c | In a system with biased agonism (β-arrestin-biased in this example), signalling only proceeds through one pathway. EGFR, epidermal growth factor receptor; GRK, G protein-coupled receptor kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase.
This classical model, however, is both over-simplified and incomplete. Over the past decade there has been a new appreciation regarding the capacity of β-arrestins to act not only as regulators of 7TMR desensitization, but also as multifunctional adaptor proteins that have the ability to signal through multiple mediators such as mitogen-activated protein kinases (MAPKs), SRC, nuclear factor-κB (NF-κB) and phosphoinositide 3-kinase (PI3K). This new perspective on 7TMR signalling represented a paradigm shift5 (FIG. 1b). In this model, both heterotrimeric G proteins and β-arrestins are capable of interacting with and recruiting intracellular signalling molecules, with desensitization of the ligand-bound receptor mediated by β-arrestins. It is still unclear whether the same receptor conformations that result in β-arrestin-mediated signal transduction also lead to receptor desensitization. Biochemical data suggest that the signalling mediated by β-arrestins has distinct functional and physiological consequences from that mediated by G proteins5. However, much of the work comparing β-arrestin- and G protein-mediated signalling has been performed in transiently transfected cell lines and not in primary cells or animal models, which limits our current understanding of the physiology of β-arrestin-mediated signalling.
It was originally thought that most ligands that bind to 7TMRs have balanced or unbiased activity for signalling through β-arrestins and G protein pathways; that is, they signal equally through both6. However, some receptor–ligand systems display bias towards one pathway over the other; that is, they preferentially signal through either the G protein- or β-arrestin-mediated pathway7. This behaviour is an example of biased agonism (FIG. 1c), which is also referred to as collateral efficacy, functional selectivity or stimulus trafficking8. Biased agonism has important implications for the design of therapeutics that target 7TMRs, as signalling through these parallel pathways is thought to have distinct functional consequences. For example, two drugs could both act as agonists of G protein-mediated functions, but may have differing effects on β-arrestin-mediated signalling. This could result in markedly different signalling profiles in vivo, examples of which will be discussed in this Review. Frequently, drug screening assays are designed to detect signalling downstream of G proteins, such as an increase in intracellular calcium levels typical of Gq-coupled receptors or increased cAMP levels typical of Gs-coupled receptors. Such assays, however, may not be sensitive to β-arrestin-mediated signalling, the target pathways of which have yet to be fully characterized. In this Review, we discuss recent advances in the characterization of β-arrestin-mediated signalling and biased agonism at 7TMRs, and address the implications of these for drug discovery and design involving this ubiquitous superfamily of receptors.
The range of β-arrestin-mediated signalling
There are four arrestin isoforms9. Arrestin 1 and arrestin 4 are expressed only in the retina where they regulate signalling of the photosensors rhodopsin and the colour opsins. By contrast, arrestin 2 and arrestin 3 (commonly referred to as β-arrestin 1 and β-arrestin 2, respectively) are expressed throughout the body. The amino-acid sequences of β-arrestin 1 and β-arrestin 2 are nearly 80% identical, with most of the differences in the carboxyl termini of the proteins.
The initial role for which arrestin activity was noted was in the desensitization of rhodopsin, a 7TMR that is the visual pigment responsible for monochromatic vision in the dark. Rhodopsin is activated into its signalling conformation by photon absorption, which allows it to bind the signalling protein transducin. This is followed by phosphorylation of the receptor10 by rhodopsin kinase11, a member of the GRK family of serine/threonine kinases12. This phosphorylation results in the binding of arrestin 1 (visual arrestin) to rhodopsin13, which sterically prevents further interaction of transducin with rhodopsin, thereby desensitizing the receptor.
A similar phosphorylation sequence of events was also observed in the β-adrenergic receptors14. In a reconstituted system with the β2-adrenergic receptor (also known as β2-adrenoceptor), it was subsequently shown that desensitization was dependent on GRKs and β-arrestins15. This mechanism was then demonstrated for several other 7TMRs, leading to the proposal of a classical paradigm for GPCR activity. In this scenario, signalling is performed by heterotrimeric G proteins followed by phosphorylation by GRKs, and desensitization is mediated by arrestins. However, it is now appreciated that β-arrestins act as multifunctional adaptor proteins that scaffold a diverse group of signalling proteins at the 7TMR and modulate the downstream activity of a number of signalling networks (reviewed in REF. 5). Selected examples are discussed below and highlighted in BOX 1.
Box 1. Selected examples of cellular functions that are mediated by β-arrestins.
Desensitization
Trafficking
Internalization: mediate7TMR internalization103, and act as clathrin adaptors104
Translocation: mediate translocation of Smoothened to the primary cilium44
Exocytosis: mediate chemokine (C-X-C motif) receptor 1-induced exocytosis in granulocytes105, and endothelin-1-stimulated translocation of glucose transporter 4 (GLUT4; also known as SLC2A4) to the plasma membrane106
Signalling
Kinase regulation: regulate mitogen-activated protein kinases (MAPKs)18–20, SRC16,17, phosphoinositide 3-kinase25 and AKT26,29
Transcriptional regulation: are binding partners for inhibitor of nuclear factor-κBα (IκBα)34,107; control histone deacetylation by p30038; and control β-catenin transcriptional regulation mediated by Wnts36
Chemotaxis: control protease-activated receptor 2-mediated chemotaxis23; angiotensin II type 1A receptor-mediated chemotaxis24; and CXCR4-mediated chemotaxis108.
Apoptotic/anti-apoptotic signalling: control substance P anti-apoptotic effects mediated by neurokinin 1 receptor17; retinal degeneration mediated by rhodopsin arrestin complexes109,110; inhibition of apoptotic signalling by N-formyl peptide receptor111; and anti-apoptotic signalling via BCL2-associated agonist of cell death (BAD) phosphorylation27
Epidermal growth factor receptor (EGFR) transactivation: mediate transactivation of EGFR by prostaglandin E4 receptor41, β1-adrenergic receptor42 and angiotensin II type 1A receptor43
Protein synthesis: regulate mRNA translation and protein synthesis by MAPK interacting serine/threonine kinase 1 (MNK1; also known as MKNK1)112
Regulation of MAPKs
The first example of β-arrestin-mediated signalling reported was β-arrestin 1 recruitment of activated SRC, a non-receptor tyrosine kinase, leading to the downstream activation of extracellular signal-regulated kinase (ERK)16,17. In the case of the neurokinin 1 (NK1) receptor, SRC recruitment by β-arrestin 1 is necessary for the prevention of apoptosis and the propagation of mitogenic signals17. For protease-activated receptor 2 (PAR2), β-arrestins recruit MAPK superfamily members upon agonist stimulation, resulting in the formation of a complex comprising activated receptor, β-arrestin 1, RAF1 and phosphorylated ERK18. Agonist binding to the angiotensin II type 1A (AT1A) receptor results in the formation of a β-arrestin 2, RAF1, MAPK/ERK kinase (MEK1) and ERK1/2 signalling complex19. Additionally, overexpression of β-arrestin 1 or β-arrestin 2 decreases phosphoinositide hydrolysis and increases ERK activation upon stimulation of the AT1A receptor with angiotensin II. However, this sequence of events is not accompanied by increased activity of the ELK1 transcription factor, which is typically associated with increased phosphorylated ERK generation mediated by G proteins20, which then leads to the transcription of immediate-early response genes.
Formation of these signalling complexes is likely to be of physiological relevance as ERK activation that is mediated by β-arrestins seems to have different consequences at the biochemical level from those activated by G proteins. β-arrestin-mediated phosphorylated ERK is retained in endocytic vesicles21 in a pathway that is spatially and temporally distinct from activation of phosphorylated ERK by G proteins. In HEK293 cells transiently transfected with the AT1A receptor, G protein activation of phosphorylated ERK is maximal at early time points (after ~2 minutes), whereas β-arrestin-mediated activity peaks later and is more protracted, accounting for 100% of phosphorylated ERK activity at 30 minutes22. β-arrestin-dependent ERK activation does not show the typical nuclear localization that is characteristic of G protein-activated ERK, and therefore does not result in activation of transcription factors such as ELK1 (REF. 20). Moreover, β-arrestin-dependent ERK activity seems to have predominant effects on chemotaxis and cytoskeletal rearrangements23,24.
Regulation of other kinase families
The β-arrestins can also regulate signalling through several other kinase families. For example, in mouse embryonic fibroblasts lacking both β-arrestin isoforms, the receptor tyrosine kinase insulin-like growth factor 1 receptor (IGF1R) is unable to stimulate PI3K activity, an effect that is rescued by expression of exogenous β-arrestin 1 (REF. 25).
AKT, a downstream target of PI3K, also has its activity upregulated and downregulated by β-arrestin-dependent mechanisms. In the case of protease-activated receptors, separate G protein and β-arrestin-1-dependent mechanisms for AKT activation have been identified, although the functional consequences of these differences are unclear26. β-arrestin 2 also regulates anti-apoptotic signalling through phosphorylation of BCL2-associated agonist of cell death (BAD) by AKT27. In diabetic mice, β-arrestin 2 is significantly downregulated, which results in decreased activity of SRC and AKT, leading to decreased insulin signalling28. However, for signalling mediated by the D2 dopamine receptor in the corpus striatum, a complex comprising β-arrestin 2, AKT and protein phosphatase 2A (PP2A; also known as PPP2R4) leads to dephosphorylation and inactivation of AKT and reciprocal activation of glycogen synthase kinase 3β (GSK3β), which is responsible for a subset of dopamine-dependent behaviours29.
β-arrestins probably regulate the activity of several other kinase families. For instance, a proteomic analysis of β-arrestins identified a wide range of kinase families that interact with either β-arrestin 1 or β-arrestin 2 (REF. 30).
Regulation of transcription
There are numerous examples of β-arrestins controlling transcription (reviewed in REF. 31). Transcription is modulated either indirectly through the regulation of signalling pathways that control transcription factors or directly through its activities in the nucleus. Both β-arrestin isoforms have amino-terminal nuclear localization signals, although it seems that β-arrestin 1 plays a more significant role in nuclear processes as it is present in both the nucleus and the cytosol. By contrast, β-arrestin 2 contains a nuclear export signal and is constitutively exported to the cytosol. However, β-arrestin 2 can be shuttled to the nucleus, as it can regulate transcription upon stimulation of the odorant receptor OR17-4 (also known as OR1D2) in spermatozoa32. β-arrestins are capable of binding and stabilizing IκB (an inhibitor of NF-κB nuclear translocation and transcriptional activation), and both β-arrestin isoforms can stabilize the IκB–NF-κB complex33,34. Overexpression of either β-arrestin results in inhibition of NF-κB-mediated transcription34, an effect that can be reversed by β-arrestin phosphorylation33.
β-arrestin 2 can also increase the activity of nuclear receptors, such as the retinoic acid receptors, by regulating their phosphorylation by ERK35. Through its regulation of Wnt signalling via the frizzled family of 7TMRs, β-arrestins regulate the activity of β-catenin, thereby modulating the activity of the TCF/LEF family of transcription factors36,37. A more direct role of transcription regulation has been demonstrated for β-arrestin 1, which binds to the promoter regions of several genes, including FOS and p27 (also known as PSMD9), upon stimulation of δ-opioid and κ-opioid receptors. This results in the recruitment of the histone acetyltransferase p300, through an interaction with the p300 binding partner cAMP responsive element binding protein (CREB), and leads to increased transcription of those genes38. Activated nuclear β-arrestin 1 can limit interferon-γ-stimulated transcription by recruiting T-cell tyrosine phosphatase, which leads to an increase in signal transducer and activator of transcription 1 (STAT1) tyrosine dephosphorylation39.
It is likely that further examples of β-arrestin scaffolding complexes that regulate transcription in the nucleus will be discovered, as it represents a common regulatory mechanism for a wide range of receptors and their downstream gene targets.
Other β-arrestin-regulated processes
The β-arrestins regulate a wide range of other cellular processes such as 7TMR transactivation of the epidermal growth factor receptor (EGFR), receptor trafficking (in addition to internalization), and the activity of small GTPases.
EGFR transactivation by 7TMRs is a well-characterized process that occurs via ligand-dependent and ligand-independent mechanisms40. The β-arrestin-mediated transactivation of EGFR occurs in a SRC-dependent manner upon agonist binding to the prostaglandin E4 (EP4) receptor41, which has been shown to lead to an increase in the metastatic progression of colorectal cancer. Similar β-arrestin-mediated activity occurs at the β1-adrenergic receptor42, in which activation of EGFR results in the activation of cardioprotective pathways as well as of the AT1A receptor43.
A key role for β-arrestins in the control of 7TMR trafficking has been demonstrated in the translocation of the Smoothened signalling complex to the primary cilium through the activity of kinesin motors44, a process that is distinct from receptor internalization or recycling.
Another pathway that is regulated by β-arrestins is the AT1A receptor-dependent activation of the small GTPase RhoA, which leads to the formation of stress fibres in a β-arrestin-2-dependent manner45.
Thus, the range of β-arrestin-mediated activity is wide, ranging from its classical function in desensitization, internalization and trafficking of receptors, to signalling via control of kinase signalling pathways, receptor transactivation and transcriptional regulation (BOX 1). β-arrestin-mediated signalling significantly expands the repertoire of 7TMR effectors from the well-characterized canonical pathways mediated by heterotrimeric G proteins to a wide array of other pathways mediated by β-arrestins that have yet to be fully explored.
The concept of biased agonism
Agonist binding to 7TMRs frequently results in the activation of multiple downstream effector pathways, a phenomenon that has been referred to as pluridimensional efficacy46. Initially, this behaviour was thought to be due to cell-specific effects and receptor heterogeneity47. However, it was then discovered that 7TMRs can couple to multiple G proteins and activate multiple signalling pathways47; for example, α2-adrenergic receptor subtypes can couple to both Gαs and Gαi (REF. 48). Moreover, signalling through these parallel pathways can differ depending on the ligand used to stimulate the receptor, thereby resulting in a biased response (reviewed in REF. 47).
An early example of biased agonism was demonstrated with two agonists of the muscarinic acetylcholine receptor: carbachol and pilocarpine. Binding of carbachol results in a balanced response that is mediated by both Gαs and Gαq. By contrast, other ligands such as pilocarpine do not lead to Gαs-mediated adenylyl cyclase stimulation, but do lead to phospholipase C (PLC) activity that is mediated by Gαq (REFs 49,50). Such ligands are said to exhibit Gαs bias over Gαq. These types of observations in a number of systems led to the proposal of a theoretical framework for this behaviour, termed agonist–receptor trafficking51 and which we refer to as biased agonism in this Review.
Biased agonism is a property of the ligand–receptor complex, and so a ligand or a receptor may be biased. A biased ligand (FIG. 2) favours one response over another (either G protein or β-arrestin) compared with the endogenous ligand, which is considered to be neutral. A biased receptor (FIG. 2) is only capable of signalling through a restricted subset of pathways that are typically available to that class of receptor. So far, most examples of β-arrestin-biased ligands have been generated by chemical modification of native ligands. These include SII angiotensin (Sar1, Ile4, Ile8)52,53, which signals through the AT1A receptor, and PTH-βarr ((D-Trp12,Tyr34)-PTH(7–34))54, which signals through the parathyroid hormone 1 (PTH1) receptor. Biased receptors that have been generated by mutating key residues involved in G protein coupling include the AT1A receptor mutant AT1AR(DRY/AAY), in which residues of the highly conserved DRY motif have been mutated to AAY52,55, and the β2-adrenergic receptor mutant β2AR(TYY), which contains mutations of three residues crucial for G protein coupling56. The existence of bias necessitates a more complex model of 7TMR activation to be developed (BOX 2).
Figure 2. Biased ligands and biased receptors.

In balanced signalling (a; green), binding of a ligand results in activation of signalling by G proteins and β-arrestins, as well as desensitization and internalization by β-arrestin alone. Two cases of biased agonism exist. In the case of a biased ligand (b), binding of a ligand (β-arrestin-biased, purple; G protein-biased, blue) to an unbiased receptor results in a biased response. In the case of a biased receptor (c), binding of an unbiased ligand to the biased receptor (β-arrestin-biased, purple; G protein-biased, blue) also results in a biased response. GRK, G protein-coupled receptor kinase.
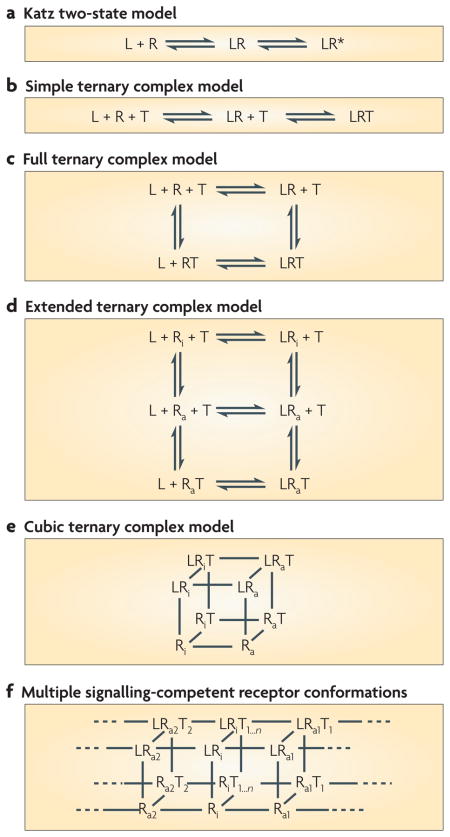
Box 2. Models of seven-transmembrane receptor signalling.
One of the oldest models for seven-transmembrane receptor (7TMR; R in the figure) activation is the Katz two-state model in which ligand (L) binding to the receptor results in formation of a ligand–receptor (LR) complex that generates an active receptor conformation (R*) that signals to downstream effectors (a). This model was subsequently modified to a simple ternary complex model, which included a ligand–receptor–G protein (or transducer; T) (LRT) complex (b). A more detailed ternary complex model113 accounted for the high and low affinity states of the receptor and receptor transducer complex that the ligand could bind to (c). The discovery of constitutively active 7TMR mutants resulted in the development of an extended ternary complex model114 (d), in which the receptor can adopt an active conformation (Ra) in the absence of ligand or transducer binding. A cubic model (e) of receptor activation includes distinct active and inactive receptor (Ri) conformations for transducer-bound receptor115, which increases the number of states compared with the extended ternary complex.
In all of these models, it is assumed that the receptor has a single signalling-competent conformation that results in activation of all signalling pathways. So, ligands can be classified as partial or full agonists, which increases the percentage of receptors in the active state; as neutral antagonists, which do not change the percentage of receptors in an active state compared with the inactive state; and as inverse agonists, which decrease the number of receptors in the active state relative to the inactive state.
To account for biased agonism, a model of 7TMR signalling must take into account the fact that receptors can signal through different pathways with different efficacies. The idea that this occurs through distinct receptor conformations is supported by studies demonstrating such states are associated with differential signalling capacity116,117. An additional state(s) for receptor activation is therefore required, with distinct conformations for signalling through G proteins or β-arrestins (Ra1, Ra2, and so on), as well as additional ternary complexes with the receptor bound to other signal transducers (T1, T2, and so on) such as β-arrestin (f). The concept of such additional states is supported by the demonstration of low and high ligand-affinity receptor states associated with β-arrestin binding similar to those observed for receptor complexes with heterotrimeric G proteins118.
A consequence of this model is the presence of biased agonists, which preferentially generate either a G protein or a β-arrestin ternary complex. Adding further to the complexity of multi-state models for receptor activation is the observation that some ligands bind at allosteric sites, which are structurally distinct from the orthosteric site, leading to effects on receptor activity. These allosteric modulators can significantly affect receptor activity and bias119.
Quantifying ligand bias
Quantifying ligand bias — that is, how much a certain ligand produces bias in a given cellular context — is an active area of research. Ligands that have full efficacy for G protein pathways and half such efficacy for β-arrestin pathways, as well as other ligands that have full efficacy for G proteins and no efficacy for β-arrestins, would all be considered to be G protein-biased ligands. How can we differentiate between these levels of bias? One of the simplest ways to quantify agonist bias is to plot β-arrestin activity against G protein activity57 (FIG. 3a). This activity could be related to efficacy, potency or another measure of quantifying the effective coupling of the ligand–receptor complex to a specific signalling pathway8,58. β-arrestin and G protein activity can also be represented as a matrix that incorporates data from multiple assays, or as ligand bias factors that compare β-arrestin activity against G protein activity in different assays (FIG. 3b). The quantification of the relative levels of bias is important in the identification of lead compounds and in the optimization of drug screening for biased agonists8.
Figure 3. Quantifying the pluridimensional efficacies of seven-transmembrane receptors.

a | One way to compare signalling between G protein- and β-arrestin-mediated pathways is to plot β-arrestin activity on the x axis and G protein activity on the y axis. Unbiased ligands would be expected to have equal levels of efficacy for β-arrestin- and G protein-mediated pathways, as shown by the green circles and the line. For biased ligands, there would be differing levels of β-arrestin- and G protein-mediated efficacies, as illustrated for β-arrestin-biased full agonists (grey), β-arrestin-biased partial agonists (yellow), G protein-biased partial agonists (blue) and G protein- biased full agonists (purple). b | Such data can also be represented in matrix form for m ligands (upper) or in terms of a bias factor for each ligand i (bottom).
There are still considerable gaps in our understanding of bias and in the different receptor conformations that are responsible for signalling to G proteins and β-arrestins. Some hypotheses explore regulated bias, in which G protein versus β-arrestin signalling can be controlled by GRKs or other cofactors. Although no clear examples of regulated bias have yet been identified, there is evidence that GRKs can be dynamically regulated, leading to effects on 7TMR signalling. For example, S-nitrosylation of GRK2 reduces its phosphorylation of β-adrenergic receptors and reduces the subsequent recruitment of β-arrestins to the receptor, resulting in the attenuation of receptor desensitization and internalization59. There is also evidence to suggest that the selective activity of GRKs mediates distinct responses by different ligands at the same receptor. The chemokine receptor CCR7 has two ligands, CCL19 and CCL21, which have different efficacies for calcium mobilization and receptor desensitization60. Binding of either CCL19 or CCL21 results in G protein coupling and β-arrestin recruitment in a GRK6-dependent manner. However, only CCL19 binding results in GRK3-dependent β-arrestin internalization and receptor desensitization61. These findings and others that follow will undoubtedly change our understanding of biased signalling by 7TMRs, and therefore will necessitate new models of their activity to be developed.
Biasing 7TMR physiology
The existence of 7TMR-mediated signal transduction that is G protein-independent requires a reassessment of the roles of specific biochemical inputs into all 7TMR-regulated physiological processes. Although the physiological consequences of β-arrestin-dependent signalling are only starting to be elucidated, several biased ligands and 7TMRs signal predominantly through either G protein- or β-arrestin-mediated pathways. These may serve to enhance our understanding of the different contributions of specific pathways to the regulation of physiological functions, and also as targeted agents for clinical therapies. In this section, we highlight the physiological consequences of this signalling in some of the better characterized systems.
β1- and β2-adrenergic receptors
Ligands for the Gαs-coupled β1- and β2-adrenergic receptors — which are involved in the pathogenesis of diseases such as heart failure and asthma — are essential therapeutic agents. Specifically, agonists of β-adrenergic receptors function as positive inotropes, and are frequently used for the acute treatment of systolic ventricular dysfunction in decompensated heart failure. By contrast, antagonists of these receptors, commonly referred to as beta-blockers, are used for the chronic treatment of heart failure, in which they act via poorly understood mechanisms to mediate cardioprotection. In addition, β2-adrenergic receptor agonists are used in the acute and chronic setting as bronchodilators in the treatment of obstructive lung diseases such as asthma and chronic obstructive pulmonary disease.
Recent studies have characterized the properties of medically relevant beta-blockers at the β1- and β2-adrenergic receptor level. In a study of 16 clinically relevant β2-adrenergic receptor antagonists62, the agents were roughly divided equally between weak partial agonists and inverse agonists with respect to Gαs activation; that is, no agent was a neutral antagonist. Of the inverse agonists, carvedilol was found to stimulate Gαs-independent, β-arrestin-2-dependent activation of ERK. In a similar study that specifically examined β1-adrenergic receptor-mediated EGFR transactivation — a process that was previously shown in the heart to be dependent upon β-arrestins42 — only alprenolol and carvedilol induced EGFR internalization63. Both drugs stimulated EGFR-dependent ERK activation, and these effects were dependent upon phosphorylation of the β1-adrenergic receptor and the presence of both β-arrestin 1 and β-arrestin 2. Thus, β-adrenergic receptor antagonists that are used in the clinical setting have divergent effects on Gαs- and β-arrestin-mediated signalling. However, the physiological ramifications of these findings, and the implications of β-arrestin-mediated signalling via the β-adrenergic receptors in general, are currently unknown.
AT1A receptor
One of the first examples of a β-arrestin-biased ligand was SII angiotensin, a synthetically modified form of angiotensin II that binds the AT1A receptor52,53. It is unable to activate Gαq signalling (as evidenced by the lack of phosphoinositide hydrolysis, calcium mobilization or diacylglycerol activity), but retains the ability to recruit β-arrestin 2, resulting in receptor internalization and activation of ERK in an entirely β-arrestin-2-dependent manner52. In a live ex vivo cardiomyocyte system, SII angiotensin exhibits positive inotropic and lusitropic properties64. Furthermore, while inhibition of the G protein-mediated pathway via protein kinase C (PKC) failed to affect the positive inotropic and lusitropic effects of SII angiotensin, deficiency of β-arrestin 2 or of the proximal kinase GRK6 abolished these effects. By contrast, the positive inotropic and lusitropic effects of angiotensin were markedly reduced by PKC inhibition, but were unaffected by deficiency of any of the β-arrestins or GRKs assessed64. Thus, angiotensin and SII angiotensin seem to be leading to the same positive effects in cardiomyocytes through pathways that are mediated by G proteins and β-arrestin 2, respectively. These data identify a role for G protein-independent, β-arrestin-dependent signalling (independent of effects on desensitization of G protein-mediated signalling), in the regulation of cardiovascular functions.
In a study designed to understand the role of the intracellular loops in signalling by the AT1A receptor to G proteins65, a mouse strain with cardiac-specific over-expression of a modified receptor with mutations in the second intracellular loop (called i2m) was shown to be uncoupled from Gαq. In comparison with mice over-expressing a wild-type AT1 receptor, overexpression of the i2m mutant resulted in marked ventricular dilation and eccentric hypertrophy, which was accompanied by diminished cardiomyocyte apoptosis. The effects on ventricular function were less clear, with only minor functional differences under resting conditions between the hearts from wild-type mice and i2m AT1 receptor-overexpressing mice. Although we can conclude that these differences were due to G protein-independent signalling, the authors did not test whether such signalling was β-arrestin-dependent. Interestingly, the hearts from the i2m mice revealed activation of SRC and cytoplasmic sequestration of activated ERK, which is consistent with selective β-arrestin-mediated signalling. From these studies it seems that AT1 receptor β-arrestin pathway-biased ligands may have beneficial effects on ventricular function while enhancing cardiomyocyte survival.
μ-opioid receptor
The μ-opioid receptor is the target for endogenous enkephalin peptides, as well as for exogenous opioid analgesics (that are agonists) such as morphine. In addition, antagonists of the μ-opioid receptor, such as naloxone and its derivatives, are used in the treatment of substance abuse. Enkephalins are balanced agonists for G protein- and β-arrestin-mediated activities, whereas morphine provokes considerably less receptor phosphorylation and internalization, which is consistent with bias towards G protein-mediated signalling66. However, in β-arrestin 2 knockout mice, morphine-induced analgesia is amplified and prolonged relative to wild-type mice, which is consistent with the presence of some morphine-induced β-arrestin-mediated desensitization67. Surprisingly, loss of β-arrestin 2 has no effect on tolerance induced by balanced agonists that strongly recruit β-arrestin 2, such as fentanyl and methadone68. Although, the role of β-arrestin 2 recruitment and signalling in the development of opioid tolerance is still unclear, β-arrestin 2 knockout mice are protected from the side effects of morphine, including respiratory depression and constipation, suggesting that β-arrestin-mediated pathways control these peripheral side effects69. Therefore, a purely G protein-biased agonist may be expected to have the antinociceptive effects of the opioid analgesics without some of their problematic side effects.
D2 dopamine receptor
D2 dopamine receptors are involved in mental illness, an effect that was originally thought to occur through Gαi/Gαo-mediated inhibition of adenylyl cyclase70. However, more recent behavioural and biochemical evidence has demonstrated that β-arrestin 2 plays a crucial role in signal transduction by D2 dopamine receptors through regulation of the AKT–GSK3 pathway71. Stimulation of D2 dopamine receptors results in the formation of a protein complex comprising β-arrestin 2, AKT and PP2A, which facilitates the dephosphorylation of AKT in response to dopamine29. This complex is a target of lithium — a drug used for the treatment of bipolar disorder and other psychiatric illnesses — the behavioural effect of which is lost in β-arrestin 2 knockout mice72. β-arrestin 2 knockout mice also display a number of defects in behaviours regulated by dopamine, including reduced apomorphine-induced climbing and reduced responsiveness to dopamine-dependent actions of amphetamine and morphine29,73. In addition, these knockout mice have a reduction in the typical novelty-induced locomotor hyperactivity phenotype of dopamine transporter knockout mice74.
5-HT2A and 5-HT2C receptors
The 5-hydroxytryptamine 2A (5-HT2A) and 5-HT2C members of the 5-HT (also known as serotonin) receptor family exhibit bias towards the activation of different G proteins (reviewed in REF. 75). Activation of these receptors by different agonists can lead to different levels of phospholipase A2 (PLA2)-mediated arachidonic acid release (mediated by Gαi) and PLC-mediated phosphoinositide hydrolysis (mediated by Gαq)76,77. β-arrestin 2 colocalizes with 5-HT2A receptors in rat prefrontal cortical neurons78, and β-arrestins are essential for mediating 5-HT2A receptor internalization by serotonin but not by the selective 5-HT2 receptor agonist, 2,5-dimethoxy-4-iodoamphetamine (DOI)79. Notably, generation of phosphorylated ERK by DOI is completely blocked by inhibition of PLC, whereas serotonin-mediated activation of phosphorylated ERK seems to be regulated by independent β-arrestin- and PLC-mediated pathways. In vivo, β-arrestin 2 knockout mice do not display a typical head-twitch response to high doses of serotonin, whereas treatment of these mice with DOI results in an equivalent head-twitch response to wild-type mice79. Therefore, it seems that DOI is a G protein-biased agonist for 5-HT2A receptors, although the full physiological consequences of this are unclear.
CXCR4
The chemokine (C-X-C motif) receptor CXCR4 is a Gαi-coupled 7TMR that binds the endogenous agonist ligand CXCL12 (also known as SDF-1a). A truncation mutation in the cytoplasmic tail of CXCR4 is found in WHIM (warts, hypogammaglobulinaemia, infections, myelokathexis) syndrome80, and leukocytes from patients with this syndrome display defective CXCR4 desensitization and enhanced chemotaxis. In WHIM syndrome, agonist-induced receptor internalization of CXCR4 — which is regulated by GRK3 and β-arrestin 2 (REF. 80) — is defective, although this internalization does not underlie the enhanced chemotaxis. Chemotaxis of CXCR4-bearing leukocytes from patients with WHIM syndrome requires β-arrestin 2 signalling, as additional mutation of residues that are necessary for β-arrestin 2 recruitment (without affecting G protein coupling) abolishes the augmented chemotactic responses of leukocytes derived from patients with WHIM syndrome. This suggests that the truncated receptor is capable of regulating β-arrestin-2-mediated signalling but not β-arrestin-2-mediated internalization or endocytosis.
PTH1 receptor
PTH is an 84 residue peptide and is the principal regulator of calcium and phosphate homeostasis. Binding of the N-terminal 34 residues of PTH, PTH(1–34), to the Gαs- and Gαq/11-coupled PTH1 receptor, results in full agonist activity. PTH-βarr, an inverse agonist of PTH1 receptor-mediated G protein-mediated signalling, results in β-arrestin-mediated ERK activation54. PTH-βarr also induces anabolic bone formation in mice in a similar manner to PTH(1–34), an effect that is attenuated in β-arrestin 2 knockout mice81. Thus, it seems that both G protein and β-arrestin-mediated pathways are involved in the anabolic response to PTH1 receptor activation. However, the biochemical differences between these two pathways have yet to be fully elucidated, and the full physiological impact of biased signalling at this receptor remains to be understood.
GPR109A
Niacin (also known as nicotinic acid or vitamin B3) — which stimulates the 7TMR GPR109A and acts through Gαi/Gαo proteins — lowers triglycerides and raises high-density lipoprotein, but its clinical use is limited by cutaneous flushing82. Niacin stimulation of GPR109A lowers cAMP levels and recruits β-arrestins to the receptor83. This leads to β-arrestin-mediated signalling to ERK/MAPKs and binding of β-arrestins to activated cytosolic PLA2. Although the interaction of β-arrestin 1 with PLA2 generates arachidonate, which ultimately leads to the flushing response, this signalling does not result in the lowering of serum free fatty-acid levels. As β-arrestin 1 mediates the cutaneous flushing and G proteins mediate the lowering of free fatty-acid levels in serum, it might be expected that a G protein-biased ligand would provide the beneficial effects of lowering lipid levels without the side effect of flushing. Indeed, a recently synthesized partial agonist of the receptor that retains its antilipolytic activity without vasodilatory side effects seems to work via a G protein-biased signalling mechanism84.
Discovery of biased agonists as drugs
G protein- and β-arrestin-biased agonism have important implications for the design of therapeutics that target 7TMRs, as signalling through these parallel pathways has distinct consequences at molecular and functional levels. As our understanding of these differences grows, there will be better appreciation of the importance and relevance of different aspects of biased signalling at 7TMRs. Although experiments have demonstrated significant functional differences between G protein- and β-arrestin-mediated signalling using biased agonists (for example, for AT1A receptor, PTH1 receptor and β2-adrenergic receptor), most have utilized genetically modified systems (for example, transgenic animals or small interfering RNA knockdown). Moreover, understanding of the physiology of β-arrestin-mediated signalling is, so far, largely limited to studies in cell culture or in small animals, with little or no data from large animal or human studies. Thus, the true therapeutic impact of biased agonists will not be known until they are tested in clinical trials. As described above, there are already several well-documented situations in which development and testing of such agents seems clearly justified. It seems likely that many more remain to be discovered.
The discovery of β-arrestin-mediated signalling necessitates a reassessment of the methods used for 7TMR drug discovery, otherwise potentially useful drugs that act primarily through non-canonical pathways, such as those mediated by β-arrestins, will continue to go unrecognized. Assays for drug discovery at 7TMRs have typically focused on identifying proximal responses that are typical of signalling by Gα and Gβγ subunits. These include the use of cell-based assays85 that measure reporter-gene expression or levels of second messengers that can be easily read with multi-well plate readers, for example, increases or decreases in second messengers such as cAMP or calcium86. As these assays of canonical G protein activity alone give an incomplete picture of a compound’s effects on cell signalling, several approaches directed towards addressing β-arrestin recruitment and signalling have been developed over the past few years87.
Redistribution assays
As β-arrestins are recruited to 7TMRs to facilitate their internalization, a number of assays (such as the Thermo Redistribution assay and the TransFluor assay88 from Molecular Devices) have been designed on the basis of visualizing agonist-stimulated changes in fluorescently-labelled 7TMR or β-arrestin distribution89 (TABLE 1).
Table 1.
Assays used in the discovery of β-arrestin-mediated signalling
| Assay type | Technology | strengths | Weaknesses |
|---|---|---|---|
| Redistribution |
|
|
|
| Proximity |
|
|
|
| Conformation |
|
|
|
| Signalling |
|
|
|
BRET, bioluminescence resonance energy transfer; FRET, fluorescence resonance energy transfer; HTS, high-throughput screening; MAPK, mitogen-activated protein kinase.
In β-arrestin redistribution assays, treatment with ligand results in one of two patterns for recruitment of fluorescently labelled β-arrestin (FIG. 4a). In the class A pattern, which is observed for receptors such as the β2-adrenergic receptor, β-arrestin is recruited to the plasma membrane and the 7TMR typically undergoes rapid recycling after internalization. In the class B pattern, which is observed for receptors such as the AT1A receptor, there is stronger and more prolonged binding of β-arrestin to the 7TMR such that following recruitment to clathrin-coated pits the receptor and β-arrestin remain bound together on the surface of endocytic vesicles and demonstrate a slow recycling pattern. The functional significance of this difference between class A and B patterns in β-arrestin recruitment is not completely understood. The prolonged association of β-arrestins with the receptor is thought to inhibit 7TMR resensitization, although other evidence argues for different mechanisms regulating receptor recycling (reviewed in REF. 90).
Figure 4. Assays for β-arrestin recruitment and activation.

a | In redistribution assays, changes in distribution of β-arrestins (green) or receptor (not shown) can be used as a response. Before stimulation, the receptor is usually localized to the membrane, whereas β-arrestins are found largely diffusely in the cytosol. In class A receptors, β-arrestins translocate to the membrane while the receptor is internalized into rapid recycling endosomes. In class B receptors, β-arrestins are internalized with the receptors into slow recycling endosomes. b | In proximity assays, the proximity between the β-arrestin and the receptor is monitored using a number of strategies. In resonance energy transfer (RET) assays, a donor probe and an acceptor probe can be attached to the receptor and β-arrestin, respectively. Upon recruitment of the β-arrestin, emission of the donor probe, by excitation with light in fluorescence RET (FRET) or with chemiluminescent substrate in bioluminescence RET (BRET), results in energy transfer to the acceptor probe. Proximity can also be assessed using reporter-based assays, such as the Tango assay. Upon recruitment of the β-arrestin, a protease — tobacco etch virus (TEV) in this example — covalently linked to the β-arrestin cleaves a site and releases a transcription factor that was attached to a modified receptor. This transcription factor then translocates to the nucleus resulting in reporter-gene expression. In assays based on enzyme complementation, the β-arrestin and the receptor are modified with fragments of an enzyme, which, upon recruitment of the β-arrestin, results in the formation of a functional enzyme. c | In conformation assays, changes in receptor or β-arrestin conformation are used to assess ligand activity and binding. In assays of receptor conformation, site-specific probes are incorporated in areas of the receptor that are thought to undergo significant structural change after ligand binding. Monitoring of FRET between these two fluorescent probes shows distinct changes associated with binding of the ligand. Similar strategies have been used to study the conformation of β-arrestin. Here, intramolecular BRET between luciferase (Luc) and yellow fluorescent protein (YFP) attached to the amino and carboxyl termini of the β-arrestin is used to monitor changes in β-arrestin conformation.
If the receptor is labelled in a redistribution assay, receptor internalization in response to agonist stimulation can be monitored (although no difference in β-arrestin recruitment pattern can be observed). In either labelling scheme, of β-arrestin or receptor, the formation of endocytic vesicles can be visualized by fluorescence microscopy or quantified by automated image acquisition and analysis.
Proximity assays
Proximity assays monitor changes in the proximity of β-arrestins to a 7TMR C terminus upon agonist binding (TABLE 1). Such assays are usually straightforward to scale up for high-throughput screening and, depending on the technology used and the system being studied, can be extremely sensitive to the β-arrestin–receptor interaction. Usually, proximity assays monitor the distance between the 7TMR and the β-arrestin by using reporter molecules attached to both the 7TMR and β-arrestin. Frequently, these reporters are spectroscopically active, allowing changes in intermolecular resonance energy transfer between fluorescent91 or bioluminescent92 probes — known as fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET), respectively — to be detected between suitably labelled 7TMRs and β-arrestins (FIG. 4b).
Other proximity assays are based on enzyme complementation or other strategies. For example, when β-galactosidase is fused to the C-terminal tail of the 7TMR and its complementarity deletion mutant is fused to β-arrestin93,94, β-arrestin recruitment results in complementation and hydrolysis of a chemiluminescent substrate (FIG. 4b). Alternatively, the C terminus of the 7TMR can be extended with a protease cleavage site and transcription factor with the corresponding protease linked to β-arrestin. β-arrestin recruitment results in cleavage of the C terminus of the 7TMR, release of the transcription factor and subsequent induction of reporter-gene expression95 (FIG. 4b). Proximity assays based on enzyme activity tend to be highly sensitive because of the significant signal amplification associated with enzyme activity or reporter-gene expression, thereby allowing the identification of weak partial agonists that are not identified by other assays.
Conformation assays
New methods for monitoring receptor and β-arrestin conformations associated with signalling are currently being developed87. Receptor conformation has been monitored through fluorescent labelling of loops or segments that are involved in receptor activation96. This has been performed for polymorphic variants of the β1-adrenergic receptor, which demonstrated discrete conformations depending on the type of ligand bound to the receptor97 (FIG. 4c). Therefore, monitoring β-arrestin conformation is likely to be a fruitful approach to screen both for 7TMR binding and for conformations that may be associated with specific signalling pathways, thus acting as a primary screen for ligand binding as well as a screen for downstream activity.
An intramolecular β-arrestin 2 BRET biosensor has been designed in which β-arrestin 2 is sandwiched between luciferase and yellow fluorescent protein (YFP). Upon 7TMR activation, β-arrestin 2 undergoes a conformational change that results in an alteration in distance or orientation between the luciferase and YFP, thereby changing the BRET signal98. This biosensor displays different BRET signatures associated with receptor binding to β-arrestin-biased agonists compared with balanced or unbiased agonists at the AT1A receptor, the β2-adrenergic receptor and the PTH1 receptor99 (FIG. 4c). These results suggest that distinct β-arrestin conformations are responsible for functionally selective responses. If these β-arrestin conformations could be correlated with distinct responses in a relevant system — for example, MAPK activation — such assays could yield insights into ligand binding and its downstream consequences, an area that is currently experiencing active investigation.
Signalling assays
As the full range of β-arrestin-mediated cell signalling has yet to be completely characterized, assays of cell signalling for drug screening are currently limited, but such assays may be fruitful in the future. Monitoring ERK1/2 phosphorylation, a well-characterized pathway regulated by β-arrestins, has been proposed as one method for assessing β-arrestin-mediated signalling7. Based on the presence of β-arrestin-mediated transcriptional activity, reporter assays could also be designed based on activation of selected signalling pathways. Other plausible strategies include assays of other characterized responses that are mediated by β-arrestins (such as chemotaxis24 or stress-fibre formation45), and performing high-throughput assays in the presence of inhibitors of G protein signalling (such as pertussis toxin for Gαi). Unfortunately, the applicability of such assays is limited by the current incomplete knowledge regarding β-arrestin-mediated signalling in many 7TMR systems.
Perspective on assays to measure β-arrestin activity
Currently, redistribution and proximity assays are the most well-developed and the easiest to apply to most 7TMR systems; however, they do have limitations (TABLE 1). One drawback of proximity assays is the requirement for labelling of the receptor with a probe, usually at the C terminus. Although such modifications usually do not affect ligand binding, theoretically they may have an effect on signalling and thus modify the recruitment of β-arrestins. This drawback of proximity assays is balanced by their easy scalability to high-throughput formats and their high sensitivity, which results from the amplification that is inherent to enzymatic or reporter systems. Redistribution assays may not require labelling of the receptor, but are not as straightforward to scale up in a high-throughput manner.
Conformational assays hold the promise of being surrogates for functional responses by the identification of active conformations of a receptor or a β-arrestin. Unfortunately, conformational assays lack sensitivity, thus limiting their utility for primary screening.
β-arrestin-mediated signalling assays need to be validated for the 7TMR and cell type being studied, but hold the promise of being as high throughput as the cell-based assays currently used for assaying G protein-mediated signalling. This field is still young and as we gain a better understanding of β-arrestin-mediated signalling pathways over the coming years, the role of these different assays in the drug development process will become clearer.
Concluding thoughts
Most classes of cell-surface receptors are appreciated to signal via various proximal effector molecules. Until recently, an exception to this rule was 7TMRs, which had been viewed as a simple on-off switch that used a single class of effector molecule: the heterotrimeric G proteins. However, converging lines of evidence demonstrate the existence of G protein-independent signal transduction and its unique biochemical and physiological effects.
Most prominent among these pathways is signal transduction mediated through the GRK–β-arrestin system, which not only desensitizes G protein-mediated signalling, but also initiates distinct signalling cascades. Recent studies highlight the complexity of signal transduction via the GRK–β-arrestin system, and the development of biased ligands suggests the ability to modulate 7TMR signal transduction efficaciously and specifically, selectively targeting a wide range of possible biochemical and physiological responses. Thus, a diverse array of ligands can potentially be developed for a given 7TMR, each of which may have unique signalling properties.
These biased ligands may be exploited for use as tools for understanding the basic biology of 7TMRs, and, potentially and most importantly, as fine-tuned therapeutics that maximize beneficial effects and minimize adverse effects100. For example, for conditions such as asthma or catecholamine-resistant shock, β-adrenergic receptor agonists that have the capacity to signal through G proteins without desensitization mediated by β-arrestins would be predicted to be more effective than current β-adrenergic receptor agonists that display significant tachyphylaxis. The potential therapeutic superiority of biased over unbiased ligands in these and other circumstances remains to be demonstrated in clinically relevant systems. As more work is done to better understand the full range of β-arrestin-mediated signalling from both biochemical and physiological perspectives, it seems likely that numerous other targets will emerge. A systems biology approach using mass spectrometry has yielded a number of possible roles for β-arrestins by identifying their binding partners30. Such experiments, combined with physiological studies of biased agonists in animal models, will yield much greater insights into these areas.
Acknowledgments
We thank E. Whalen, J. Violin, S. Ahn and A. Shukla for critical review of the manuscript. We thank D. Addison and E. Hall for secretarial assistance. This work was supported in part by National Institutes of Health (NIH) Grants HL16037 and HL70631 to R.J.L. R.J.L. is an Investigator with the Howard Hughes Medical Institute. S.R. is supported by NIH T32 training grant HL07101-34.
- G protein
A heterotrimeric protein that exchanges GDP for GTP in its α-subunit on agonist binding to a seven-transmembrane receptor, resulting in dissociation of the complex into Gα and Gβγ subunits. These subunits lead to the activation of second messenger systems through the regulation of enzymes such as adenylyl cyclase or phospholipase C
- Arrestins
Multifunctional adaptor proteins that are important in regulating desensitization and signalling by seven- transmembrane receptors and other transmembrane receptors
- β-catenin
A multifunctional adaptor protein. One of its roles includes the regulation of TCF/LeF transcription factors in response to signalling by Wnts through the frizzled seven-transmembrane receptors
- Transactivation
The activation of a transmembrane receptor that results from signalling caused by activation of another receptor. In the case of epidermal growth factor receptor (eGFR) transactivation, agonist binding at a number of seven-transmembrane receptors activates a pathway that releases a membrane- bound eGF ligand by proteolytic cleavage, which then activates the eGFR
- Biased agonist
A ligand that results in the activation of select, but not all, available signalling pathways that are known to be activated by the receptor
- Positive inotropes
An agent that increases the force of the heart’s contraction. Negative inotropes decrease the force of contraction
- Antagonist
A term that is commonly used to refer broadly to neutral antagonists, weak partial agonists and inverse agonists
- Partial agonist
A ligand that when bound to a receptor results in a submaximal response. Partial agonists can antagonize full agonists
- Inverse agonist
A ligand that decreases the signalling activity of the receptor on binding compared with the ligand-unbound state
- Neutral antagonist
A ligand that results in no change in activity of the receptor on binding compared with the ligand-unbound state
- Allosteric site
A binding site on a seven- transmembrane receptor that is different than the orthosteric site
- Orthosteric site
The binding site on a seven-transmembrane receptor to which the endogenous agonist binds
- Allosteric modulator
A ligand that binds to the allosteric site of the receptor and affects receptor responses to orthosteric ligands. Some allosteric modulators are capable of generating biased responses
- Lusitropic
Relates to the relaxation and filling of the heart. Positive lusitropic agents improve the heart’s relaxation and filling
- Full agonist
A ligand that completely activates the receptor on binding
Footnotes
Competing interests statement
The authors declare competing financial interests: see web version for details.
DATABASES
IUPHAR Databse of Receptors and Ion Channels: http://www.iuphar-db.org/index.jsp
5-HT2A | 5HT-2C | AT1A | β1-adrenergic | β2-adrenergic | CXCR4 | D2 | δ-opioid | EP4 | GPR109A | κ-opioid | NK1 | PAR2 | PTH1
UniProtKB: http://ca.expasy.org/sprot
β-arrestin 1 | β-arrestin 2
FURTHER INFORMATION
Laboaratory homepage of Robert J. Lefkowitz: http://www.lefkolab.org
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nature Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Ma P, Zemmel R. Value of novelty? Nature Rev Drug Discov. 2002;1:571–572. doi: 10.1038/nrd884. [DOI] [PubMed] [Google Scholar]
- 3.Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 5.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. β-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 6.Benovic JL, Staniszewski C, Mayor F, Jr, Caron MG, Lefkowitz RJ. β-adrenergic receptor kinase Activity of partial agonists for stimulation of adenylate cyclase correlates with ability to promote receptor phosphorylation. J Biol Chem. 1988;263:3893–3897. [PubMed] [Google Scholar]
- 7.Violin JD, Lefkowitz RJ. β-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Lefkowitz RJ, Whalen EJ. β-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Wilden U, Kuhn H. Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry. 1982;21:3014–3022. doi: 10.1021/bi00541a032. [DOI] [PubMed] [Google Scholar]
- 11.Shichi H, Somers RL. Light-dependent phosphorylation of rhodopsin. Purification and properties of rhodopsin kinase. J Biol Chem. 1978;253:7040–7046. [PubMed] [Google Scholar]
- 12.Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. β-adrenergic receptor kinase: primary structure delineates a multigene family. Science. 1989;246:235–240. doi: 10.1126/science.2552582. [DOI] [PubMed] [Google Scholar]
- 13.Wilden U, Hall SW, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stadel JM, et al. Catecholamine-induced desensitization of turkey erythrocyte adenylate cyclase is associated with phosphorylation of the β-adrenergic receptor. Proc Natl Acad Sci USA. 1983;80:3173–3177. doi: 10.1073/pnas.80.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benovic JL, et al. Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc Natl Acad Sci USA. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luttrell LM, et al. β-arrestin-dependent formation of β2 adrenergic receptor–Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 17.DeFea KA, et al. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin-dependent scaffolding complex. Proc Natl Acad Sci USA. 2000;97:11086–11091. doi: 10.1073/pnas.190276697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeFea KA, et al. β-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luttrell LM, et al. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. β-arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- 21.Tohgo A, et al. The stability of the G protein-coupled receptor–β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 22.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. This work demonstrates the different temporal and spatial patterns of β-arrestin- and G protein-mediated ERK. β-arrestin-mediated phosphorylated ERK peaks at late times in endosomes and G protein-mediated phosphorylated ERK peaks at early times with nuclear and cytoplasmic localization. [DOI] [PubMed] [Google Scholar]
- 23.Ge L, Ly Y, Hollenberg M, DeFea K. A β-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J Biol Chem. 2003;278:34418–34426. doi: 10.1074/jbc.M300573200. [DOI] [PubMed] [Google Scholar]
- 24.Hunton DL, et al. β-arrestin 2-dependent angiotensin II type 1A receptor-mediated pathway of chemotaxis. Mol Pharmacol. 2005;67:1229–1236. doi: 10.1124/mol.104.006270. [DOI] [PubMed] [Google Scholar]
- 25.Povsic TJ, Kohout TA, Lefkowitz RJ. β-arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis. J Biol Chem. 2003;278:51334–51339. doi: 10.1074/jbc.M309968200. [DOI] [PubMed] [Google Scholar]
- 26.Goel R, Phillips-Mason PJ, Raben DM, Baldassare JJ. α-Thrombin induces rapid and sustained Akt phosphorylation by β-arrestin1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. J Biol Chem. 2002;277:18640–18648. doi: 10.1074/jbc.M108995200. [DOI] [PubMed] [Google Scholar]
- 27.Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. β-arrestin-2 mediates anti-apoptotic signaling through regulation of BAD phosphorylation. J Biol Chem. 2009;284:8855–8865. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luan B, et al. Deficiency of a β-arrestin-2 signal complex contributes to insulin resistance. Nature. 2009;457:1146–1149. doi: 10.1038/nature07617. [DOI] [PubMed] [Google Scholar]
- 29.Beaulieu JM, et al. An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 30.Xiao K, et al. Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci USA. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma L, Pei G. β-arrestin signaling and regulation of transcription. J Cell Sci. 2007;120:213–218. doi: 10.1242/jcs.03338. [DOI] [PubMed] [Google Scholar]
- 32.Neuhaus EM, Mashukova A, Barbour J, Wolters D, Hatt H. Novel function of β-arrestin2 in the nucleus of mature spermatozoa. J Cell Sci. 2006;119:3047–3056. doi: 10.1242/jcs.03046. [DOI] [PubMed] [Google Scholar]
- 33.Luan B, Zhang Z, Wu Y, Kang J, Pei G. β-arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-κB activation. EMBO J. 2005;24:4237–4246. doi: 10.1038/sj.emboj.7600882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. β-arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc Natl Acad Sci USA. 2004;101:8603–8607. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piu F, Gauthier NK, Wang F. β-arrestin 2 modulates the activity of nuclear receptor RAR β2 through activation of ERK2 kinase. Oncogene. 2006;25:218–229. doi: 10.1038/sj.onc.1209024. [DOI] [PubMed] [Google Scholar]
- 36.Bryja V, Gradl D, Schambony A, Arenas E, Schulte G. β-arrestin is a necessary component of Wnt/β-catenin signaling in vitro and in vivo. Proc Natl Acad Sci USA. 2007;104:6690–6695. doi: 10.1073/pnas.0611356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosano L, et al. β-arrestin links endothelin A receptor to β-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc Natl Acad Sci USA. 2009;106:2806–2811. doi: 10.1073/pnas.0807158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang J, et al. A nuclear function of β-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–847. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 39.Mo W, et al. Nuclear β-arrestin1 functions as a scaffold for the dephosphorylation of STAT1 and moderates the antiviral activity of IFN-γ. Mol Cell. 2008;31:695–707. doi: 10.1016/j.molcel.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 40.Bhola NE, Grandis JR. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front Biosci. 2008;13:1857–1865. doi: 10.2741/2805. [DOI] [PubMed] [Google Scholar]
- 41.Buchanan FG, et al. Role of β-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA. 2006;103:1492–1497. doi: 10.1073/pnas.0510562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noma T, et al. β-arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardio-protection. J Clin Invest. 2007;117:2445–2458. doi: 10.1172/JCI31901. This paper suggests an important role for β-arrestin-mediated signalling in the heart. The β-arrestin-mediated pathway downstream of the β1-adrenergic receptor protects against cardiomyopathy induced by catecholamine infusion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. Independent β-arrestin2 and Gq/protein kinase Cζ pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J Biol Chem. 2009;284:11953–11962. doi: 10.1074/jbc.M808176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovacs JJ, et al. β-arrestin-mediated localization of smoothened to the primary cilium. Science. 2008;320:1777–1781. doi: 10.1126/science.1157983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barnes WG, et al. β-arrestin 1 and Gαq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem. 2005;280:8041–8050. doi: 10.1074/jbc.M412924200. [DOI] [PubMed] [Google Scholar]
- 46.Galandrin S, Bouvier M. Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol. 2006;70:1575–1584. doi: 10.1124/mol.106.026716. In this work, the authors used a set of ligands (agonists, antagonists and inverse agonists) for the adrenergic receptors to identify ligand-specific differences in the balance between cAMP and ERK signalling. [DOI] [PubMed] [Google Scholar]
- 47.Roth BL. In: Functional Selectivity of G Protein-Coupled Receptor Ligands. Neve K, editor. Springer; New York: 2009. pp. 3–7. [Google Scholar]
- 48.Eason MG, Kurose H, Holt BD, Raymond JR, Liggett SB. Simultaneous coupling of α2-adrenergic receptors to two G-proteins with opposing effects. Subtype-selective coupling of α2C10, α2C4, and α2C2 adrenergic receptors to Gi and Gs. J Biol Chem. 1992;267:15795–15801. [PubMed] [Google Scholar]
- 49.Fisher A, et al. Selective signaling via unique M1 muscarinic agonists. Ann NY Acad Sci. 1993;695:300–303. doi: 10.1111/j.1749-6632.1993.tb23070.x. [DOI] [PubMed] [Google Scholar]
- 50.Gurwitz D, et al. Discrete activation of transduction pathways associated with acetylcholine m1 receptor by several muscarinic ligands. Eur J Pharmacol. 1994;267:21–31. doi: 10.1016/0922-4106(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 51.Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. One of the first papers to approach the question of biased agonism from a theoretical perspective. [DOI] [PubMed] [Google Scholar]
- 52.Wei H, et al. Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holloway AC, et al. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- 54.Gesty-Palmer D, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 55.Gaborik Z, et al. The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology. 2003;144:2220–2228. doi: 10.1210/en.2002-0135. [DOI] [PubMed] [Google Scholar]
- 56.Shenoy SK, et al. β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 57.Drake MT, et al. β-arrestin-biased agonism at the β2-adrenergic receptor. J Biol Chem. 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- 58.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Whalen EJ, et al. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 60.Kohout TA, et al. Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 61.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci USA. 2009;106:9649–9654. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wisler JW, et al. A unique mechanism of beta-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim IM, et al. Beta-blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA. 2008;105:14555–14560. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajagopal K, et al. β-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhai P, et al. Cardiac-specific overexpression of AT1 receptor mutant lacking Gαq/Gαi coupling causes hypertrophy and bradycardia in transgenic mice. J Clin Invest. 2005;115:3045–3056. doi: 10.1172/JCI25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Groer CE, et al. An opioid agonist that does not induce μ-opioid receptor–arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bohn LM, et al. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. One of the first examples of a physiological response that was regulated by β-arrestins in an animal model. [DOI] [PubMed] [Google Scholar]
- 68.Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- 69.Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 70.Enjalbert A, Bockaert J. Pharmacological characterization of the D2 dopamine receptor negatively coupled with adenylate cyclase in rat anterior pituitary. Mol Pharmacol. 1983;23:576–584. [PubMed] [Google Scholar]
- 71.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 72.Beaulieu JM, et al. A β-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- 73.Bohn LM, et al. Enhanced rewarding properties of morphine, but not cocaine, in β(arrestin)-2 knock-out mice. J Neurosci. 2003;23:10265–10273. doi: 10.1523/JNEUROSCI.23-32-10265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 75.Berg KA, Clarke WP. In: Functional Selectivity of G Protein-Coupled Receptor Ligands. Neve K, editor. Springer; New York: 2009. pp. 155–176. [Google Scholar]
- 76.Berg KA, et al. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- 77.Moya PR, et al. Functional selectivity of hallucinogenic phenethylamine and phenylisopropylamine derivatives at human 5-hydroxytryptamine (5-HT)2A and 5-HT2C receptors. J Pharmacol Exp Ther. 2007;321:1054–1061. doi: 10.1124/jpet.106.117507. [DOI] [PubMed] [Google Scholar]
- 78.Gelber EI, et al. Structure and function of the third intracellular loop of the 5-hydroxytryptamine2A receptor: the third intracellular loop is α-helical and binds purified arrestins. J Neurochem. 1999;72:2206–2214. doi: 10.1046/j.1471-4159.1999.0722206.x. [DOI] [PubMed] [Google Scholar]
- 79.Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on β-arrestin-2 interactions in vivo. Proc Natl Acad Sci USA. 2008;105:1079–1084. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balabanian K, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105:2449–2457. doi: 10.1182/blood-2004-06-2289. [DOI] [PubMed] [Google Scholar]
- 81.Gesty-Palmer D, et al. A β-arrestin biased agonist of a parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1ra1. doi: 10.1126/scitranslmed.3000071. One of the first studies to demonstrate the effects of a biased agonist in an animal model. Compared with the unbiased agonist PTH(1–34), which stimulates both bone formation and resorption, the biased agonist PTH-βarr stimulates bone formation only. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bodor ET, Offermanns S. Nicotinic acid: an old drug with a promising future. Br J Pharmacol. 2008;153 (Suppl 1):68–75. doi: 10.1038/sj.bjp.0707528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Walters RW, et al. β-arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119:1312–1321. doi: 10.1172/JCI36806. An example in which development of a biased agonist could lead to a therapeutic with fewer side effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Semple G, et al. 3-(1H-tetrazol-5-yl)-1,4,5,6- tetrahydro-cyclopentapyrazole (MK-0354): a partial agonist of the nicotinic acid receptor, G-protein coupled receptor 109a, with antilipolytic but no vasodilatory activity in mice. J Med Chem. 2008;51:5101–5108. doi: 10.1021/jm800258p. [DOI] [PubMed] [Google Scholar]
- 85.Eglen RM, Bosse R, Reisine T. Emerging concepts of guanine nucleotide-binding protein-coupled receptor (GPCR) function and implications for high throughput screening. Assay Drug Dev Technol. 2007;5:425–451. doi: 10.1089/adt.2007.062. [DOI] [PubMed] [Google Scholar]
- 86.Siehler S. Cell-based assays in GPCR drug discovery. Biotechnol J. 2008;3:471–483. doi: 10.1002/biot.200800001. [DOI] [PubMed] [Google Scholar]
- 87.Verkaar F, et al. G protein-independent cell-based assays for drug discovery on seven-transmembrane receptors. Biotechnol Annu Rev. 2008;14:253–274. doi: 10.1016/S1387-2656(08)00010-0. A review on current technologies used for high-throughput screening of 7TMRs with a focus on assays of β-arrestin activity. [DOI] [PubMed] [Google Scholar]
- 88.Hudson CC, Oakley RH, Sjaastad MD, Loomis CR. High-content screening of known G protein-coupled receptors by arrestin translocation. Methods Enzymol. 2006;414:63–78. doi: 10.1016/S0076-6879(06)14005-7. [DOI] [PubMed] [Google Scholar]
- 89.Henriksen U, Fog J, Loechel F, Praestegaard M. Profiling of multiple signal pathway activities by multiplexing antibody and GFP-based translocation assays. Comb Chem High Throughput Screen. 2008;11:537–544. doi: 10.2174/138620708785204081. [DOI] [PubMed] [Google Scholar]
- 90.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 91.Krasel C, Bunemann M, Lorenz K, Lohse MJ. β-arrestin binding to the β2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J Biol Chem. 2005;280:9528–9535. doi: 10.1074/jbc.M413078200. [DOI] [PubMed] [Google Scholar]
- 92.Bertrand L, et al. The BRET2/arrestin assay in stable recombinant cells: a platform to screen for compounds that interact with G protein-coupled receptors (GPCRs) J Recept Signal Transduct Res. 2002;22:533–541. doi: 10.1081/rrs-120014619. [DOI] [PubMed] [Google Scholar]
- 93.Olson KR, Eglen RM. Beta galactosidase complementation: a cell-based luminescent assay platform for drug discovery. Assay Drug Dev Technol. 2007;5:137–144. doi: 10.1089/adt.2006.052. [DOI] [PubMed] [Google Scholar]
- 94.Luker KE, Gupta M, Luker GD. Imaging CXCR4 signaling with firefly luciferase complementation. Anal Chem. 2008;80:5565–5573. doi: 10.1021/ac8005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barnea G, et al. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci USA. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vilardaga JP, Bunemann M, Krasel C, Castro M, Lohse MJ. Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nature Biotech. 2003;21:807–812. doi: 10.1038/nbt838. [DOI] [PubMed] [Google Scholar]
- 97.Rochais F, et al. Real-time optical recording of β1-adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol. J Clin Invest. 2007;117:229–235. doi: 10.1172/JCI30012. A study of the β1-adrenergic receptor that monitored distinct structural responses of polymorphic receptor variants in response to treatment with a clinically used beta blocker. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Charest PG, Terrillon S, Bouvier M. Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep. 2005;6:334–340. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shukla AK, et al. Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci USA. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. β-arrestin: a protein that regulates β-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 102.Perry SJ, et al. Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 103.Ferguson SS, et al. Role of β-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 104.Goodman OB, Jr, et al. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. One of the first papers to describe a role for β-arrestins as adapter proteins, in this case for clathrin, leading to receptor endocytosis. [DOI] [PubMed] [Google Scholar]
- 105.Barlic J, et al. Regulation of tyrosine kinase activation and granule release through β-arrestin by CXCRI. Nature Immunol. 2000;1:227–233. doi: 10.1038/79767. [DOI] [PubMed] [Google Scholar]
- 106.Imamura T, et al. β-arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J Biol Chem. 2001;276:43663–43667. doi: 10.1074/jbc.M105364200. [DOI] [PubMed] [Google Scholar]
- 107.Gao H, et al. Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol Cell. 2004;14:303–317. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 108.Lagane B, et al. CXCR4 dimerization and β-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112:34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 109.Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin–arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 110.Kiselev A, et al. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 111.Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins block G protein-coupled receptor-mediated apoptosis. J Biol Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- 112.DeWire SM, et al. β-arrestin-mediated signaling regulates protein synthesis. J Biol Chem. 2008;283:10611–10620. doi: 10.1074/jbc.M710515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 114.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- 115.Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. III resurrecting efficacy. J Theor Biol. 1996;181:381–397. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- 116.Audet N, et al. Bioluminescence resonance energy transfer assays reveal ligand-specific conformational changes within preformed signaling complexes containing δ-opioid receptors and heterotrimeric G proteins. J Biol Chem. 2008;283:15078–15088. doi: 10.1074/jbc.M707941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vilardaga JP, Steinmeyer R, Harms GS, Lohse MJ. Molecular basis of inverse agonism in a G protein-coupled receptor. Nature Chem Biol. 2005;1:25–28. doi: 10.1038/nchembio705. [DOI] [PubMed] [Google Scholar]
- 118.Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist–receptor–arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- 119.Schwartz TW, Holst B. Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends Pharmacol Sci. 2007;28:366–373. doi: 10.1016/j.tips.2007.06.008. [DOI] [PubMed] [Google Scholar]