

Abstract
Glial fibrillary acidic protein (GFAP) is the principle intermediate filament (IF) protein in astrocytes. Mutations in the GFAP gene lead to Alexander disease (AxD), a rare, fatal neurological disorder characterized by the presence of abnormal astrocytes that contain GFAP protein aggregates, termed Rosenthal fibers (RFs), and the loss of myelin. All GFAP mutations cause the same histopathological defect, i.e. RFs, though little is known how the mutations affect protein accumulation as well as astrocyte function. In this study, we found that GFAP accumulation induces macroautophagy, a key clearance mechanism for prevention of aggregated proteins. This autophagic response is negatively regulated by mammalian target of rapamycin (mTOR). The activation of p38 MAPK by GFAP accumulation is in part responsible for the down-regulation of phosphorylated-mTOR and the subsequent activation of autophagy. Our study suggests that AxD mutant GFAP accumulation stimulates autophagy, in a manner regulated by p38 MAPK and mTOR signaling pathways. Autophagy, in turn, serves as a mechanism to reduce GFAP levels.
INTRODUCTION
Alexander’s disease (AxD) is a rare and fatal leukodystrophy that is characterized by widespread accumulation of Rosenthal fibers (RFs), protein aggregates within the cytoplasm and cytoplasmic processes of astrocytes (1,2). Genetically, AxD is determined by sporadic gain of function mutations in the gene encoding GFAP (3,4). Patients affected by AxD are usually heterozygous for GFAP mutations (5,6), and these mutations behave in a genetically dominant manner. In both transgenic astrocytes (Messing et al., 1998) and cell cultures (7–9), overexpressed wild type (wt) or AxD R239C mutant (mt) GFAP accumulates and spontaneously forms cytoplasmic inclusions. These inclusions share some characteristics with the RFs in AxD brain, containing stress protein kinases, small heat shock proteins and IF-associated proteins (9,11) (Tian, 2006). The R239C mutation alters the normal solubility and organization of GFAP networks (7), but little is known about the underlying mechanism and why the wild type proteins also accumulate and form aggregates.
Autophagy is a membrane-trafficking mechanism that delivers cytoplasmic constituents into the lysosome for degradation (10,11). Macroautophagy (referred to as autophagy hereafter) is initiated by the formation of autophagosomes, which sequester cytoplasmic contents including ribosomes, soluble proteins and organelles. Autophagosomes are transported to and fuse with lysosomes, thus maturing into degradative autolysosomes, where their contents are degraded. A growing body of evidence has substantiated the hypothesis that autophagy is a protein quality control mechanism through which mammalian cells may capture and degrade mutant proteins, including those prone to aggregate in neurodegenerative diseases, such as Alzheimer Disease, polyglutamine disease (CAG repeat), and Parkinson disease (10).
In this study, we asked if GFAP accumulation activated the autophagy–lysosome pathway and if there was evidence that this pathway was activated in the pathology of AxD. Our results indicate an induction of autophagy in response to abnormal GFAP accumulation in astrocytic cells, in AxD brain, and in mt GFAP knock-in mouse brains. The induction of autophagy was regulated by the coordination of the p38 MAPK stress cascade and mTOR signaling pathways. Finally, by modulating autophagic activity through pharmacological and genetic means, we provide direct evidence for the autophagic clearance of accumulated GFAP protein.
RESULTS
Signs of autophagy in mt GFAP expressing U251 cells, in AxD brains and R236H/+ mutant GFAP mouse brains
As we described previously (9), in U251 astrocytoma cells overexpressing GFAP, the wt GFAP formed mostly a filamentous network while the mt GFAP formed dot-like aggregates superimposed on the filamentous network (Fig. 1A and B, left panel). To determine whether those protein aggregates formed by mt GFAP are associated with autophagy, we used LC3 as a specific marker for autophagosomes. LC3 is the mammalian autophagic protein that localizes to the autophagosome membrane as well as to the cytosol (Kabeya et al., 2000). Overexpression of GFP tagged LC3 (GFP-LC3) is a well-accepted, straightforward and specific assessment of autophagosome formation without perturbing autophagosome number or function (12). Here, we introduced an RFP-tagged LC3 construct into wt or mt GFAP expressing U251 cells. Immunofluorescence revealed that the RFP-LC3 (red) overlapped with many of the GFAP-positive, punctate aggregates (green) in the mt and occasionally in the wt expressing cells (Fig. 1A).
Figure 1.
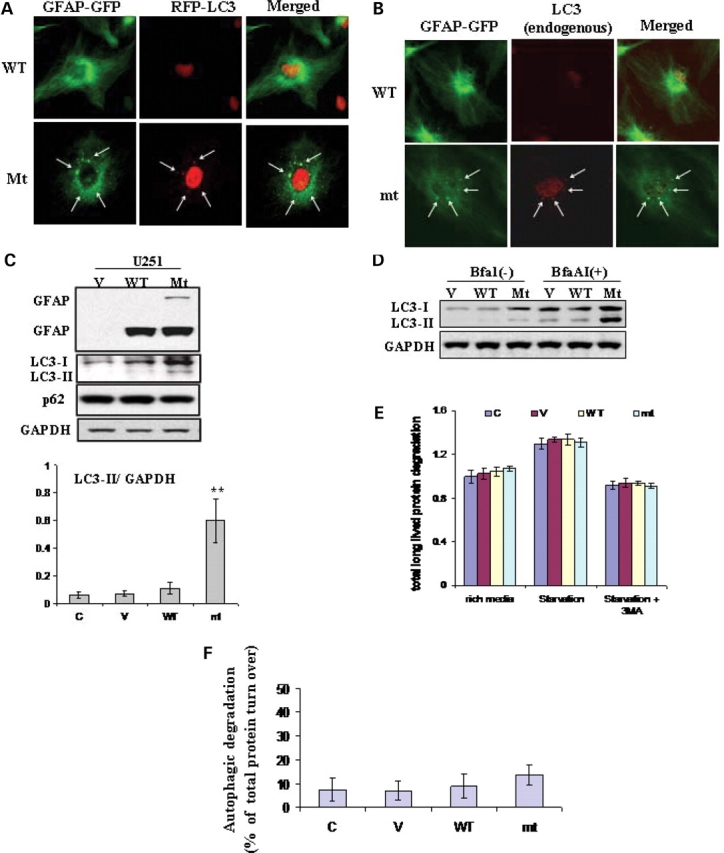
Evidence for autophagy in U251 astrocytoma cells stably expressing R239C AxD mutant GFAP. (A) Co-localization of RFP-LC3 with cytoplasmic GFAP aggregates, indicated by arrows. U251 cells stably expressing EGFP-C1 vector, wt or mt GFAPs were transiently transfected with RFP-LC3. Forty-eight hours after transfection, cells were treated with cytoskeletal buffer and fixed with 4% PFA and then examined for GFAP (green) and LC3 (red) fluorescence. (B) Overlap of endogenous LC3 and GFAP aggregates in GFAP expressing U251 cells. U251 cells were treated with cytoskeletal buffer and fixed with 4% PFA, then were immunostained for LC3. Merged images on right show LC3 in red, GFAP-GFP in green and the overlap in yellow, aggregates were indicated by arrows. (C) Immunoblotting analysis of LC3 in U251 cells stably expressing GFAPs (upper panel). Each gel labeled with V, vector-control; WT, WT GFAP; Mt, mt GFAP. Lower panel: quantitative immunoblot analysis of LC3 activation, represented by LC3-II to GAPDH ratio, under normal growth conditions in U251 astrocytoma cells stably transfected with wild-type and mutant GFAP. ** P < 0.001. (D) Increased autophagic flux in mt GFAP expressing U251cells, represented by the differences between LC3-II level between BfaA1 treated and non-treated cells. (E) Long-lived bulk protein degradation. Compared to U251 cells under normal growth conditions, no significant differences were observed in cells overexpressing wt and mt GFAPs, and cells upon amino acid- and serum-deprivation in the presence or absence of 3-MA macroautophagy inhibitor. n = 6 wells, mean ± SEM two-tailed ANOVA. (F) The proportion of total protein turnover due to autophagy in different cell lines.
We also asked if the GFAP aggregates were associated with endogenous LC3. U251 cells highly expressing wt or mt GFP-GFAPs were treated with a PHEM cytoskeleton buffer so that most of the free, soluble cytoplasmic protein was removed, leaving behind the nuclei and GFAP aggregates in the cytoplasm (9). By monitoring GFP fluorescence, we found that the GFAP filamentous network in cells with wt GFAP and the aggregates in cells with mt GFAP were well preserved in the cytosol (Fig. 1B). Punctate structures positive for both GFAP and LC3 were found in the mt GFAP expressers, suggesting an overlap of this autophagic marker with GFAP in some of the inclusions.
Endogenous LC3 is processed posttranslationally into LC3-I, an 18 kDa cytosolic form. LC3-I is then converted to LC3-II, a 16 kDa membrane-bound form that associates with autophagosome membranes. The level of endogenous LC3 was thus examined by western blotting (Fig. 1C, upper). Both LC3-I and LC3-II increased dramatically in U251 astrocytoma cells stably expressing mt GFAP, but slightly in wt GFAP expressers. As the LC3-II levels correlate with autophagosome number per cell (Kabeya et al., 2000), we also quantified the autophagosome number by normalizing the level of LC3-II versus the loading control, GAPDH. The LC3-II level was increased markedly in mt GFAP expressers, compared to those in cells expressing the wt GFAP or an EGFP vector control (Fig. 1, lower), suggesting that mt GFAP accumulation increased the number of autophagosomes.
It is likely that autophagosomes accumulated in response to an inhibition of autophagic degradation, for example, blockage of autophagosome-lysosome fusion. To further address this possibility, we determined total autophagic flux, which is more accurately represented by differences in the amount of LC3-II between cells in the presence and absence of lysosomal protease inhibitors (13). Cells were treated with Bafilomycin AI, an inhibitor for autophagosome–lysosome fusion. Proteins were extracted from both treated and untreated cells and subjected to LC3-II immunoblotting analysis. As shown in Fig 1D, the BfaA1 treatment increased the amount of autophagosomes in all three cell lines, as indicated by an increased LC3-II level, compared to untreated controls. Particularly, in mt GFAP expressors where the increased LC3-II preexisted, BfaA1 caused a further accumulation of LC3-II, indicating an enhancement of the autophagic flux (A1) in mutant GFAP expressors.
We then wanted to determine if the increased formation of autophagosomes was accompanied by an increased level of total protein degradation. First, we examined endogenous p62/SQSTM1, an autophagy–lysosome substrate. The p62 protein is associated with protein aggregates in a number of aggregation diseases, and there is a general correlation between the inhibition of the autophagy–lysosomal protein degradation and increased levels of p62 (14,15). GFAP overexpression did not alter the levels of endogenous p62 (Fig. 1C). We then measured the degradation rate of long-lived proteins in mt GFAP expressing cells, and compared it with those in wt GFAP expressers, EGFP vector controls and non-transfected U251 cells. No significant differences were detected in the overall, long-lived (24 h) protein turnover rates among different cell lines (Fig. 1E). Further stimulating autophagy, under the condition of amino acid- and serum-deprivation, produced a similar increase in overall protein degradation in all cells lines (Fig. 1E). To estimate how much of the proteolysis is due to induced and basal level of autophagy, we measured bulk protein turnover in the same groups of cells incubated with 3-Methyladenine (3-MA), an autophagy inhibitor (16). Co-administration of 3-MA inhibited both basal (under normal growth conditions) and induced (under starvation) autophagic protein degradation, and brought down the levels of overall protein degradation to the same levels in all four cell lines (Fig. 1E). The capacity of autophagic protein degradation is estimated as the difference between the amino acid-starved cells without and with 3-MA (Fig. 1F). We did not find a significantly increased autophagic turnover, represented as a proportion of total bulk protein turnover. By this assay, however, autophagy represented only a small proportion of total protein degradation (7–9% in mock, vector and wt controls, and 13% in mt cells), and therefore bulk turnover appears to be a relatively insensitive assay for autophagy. Furthermore, other consequences of mt GFAP expression, such as proteasome inhibition (9), may up- or down-regulate other proteolytic pathways, making the results of this assay more difficult to interpret (see Discussion). These observations also may imply that mt GFAP-stimulated autophagosome formation is used against highly selective targets for removal and degradation of GFAP aggregates, rather than for a global increase in autophagy degradation.
We also looked for evidence of autophagy in AxD human brain tissues. Hematoxylin and eosin (H&E) staining revealed eosinophilic structures (RFs, indicated by arrows) stained bright red with eosin in AxD brain (Fig. 2A-II). RFs are intracellular, non-membrane-bound inclusions found only in astrocytes located in white matter, periventricular areas, deep gray and stem nuclei, subpial zones, and particularly densely around blood vessels, and are absent in healthy control brains (Fig. 2A-I). GFAP immunostaining identified morphologically normal astrocytes in control brains (Fig. 2A-III), but many abnormal astrocytes containing RFs in AxD (Fig. 2A-IV). Immunostaining showed that LC3 was located surrounding the RFs (Fig. 2A-VI), indicating a close proximity of LC3 to RFs inside the affected astrocytes. This was consistent with our ultrastructural examination, which showed organelles consistent with autophagosomes and autolysosomes near RFs in astrocytes (see below). In addition, western-blot analysis showed increased LC3-II levels in brain tissues from three AxD patients (with R239C, R416W and R239H mutations), when compared with that in tissue of a non-neurological disease control subject (Fig. 2B) and another control patient with Werdnig-Hoffman disease (non-AxD, non-leukodystrophy neurological disease without RFs, data not shown).
Figure 2.
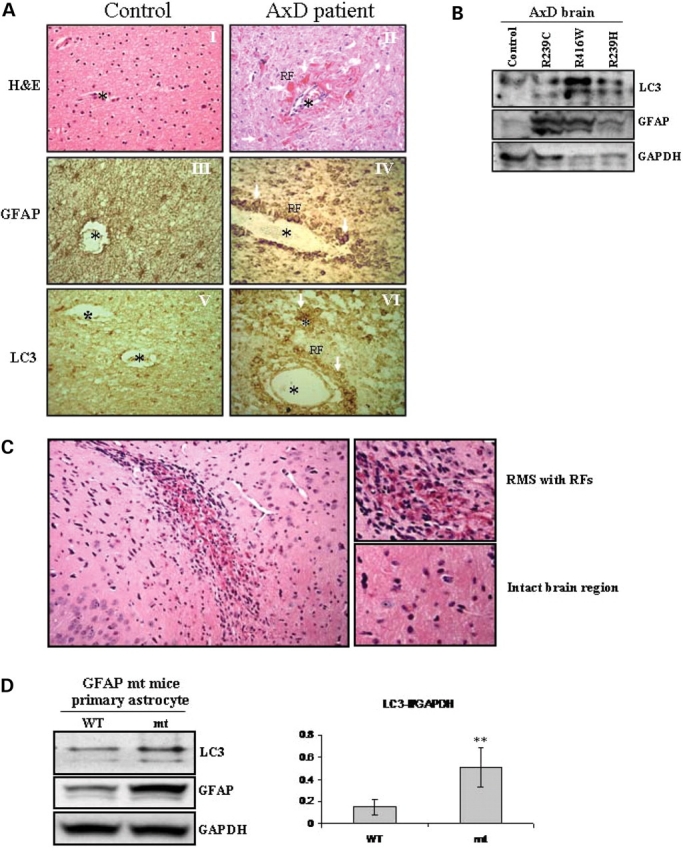
Signs of autophagy in brain tissue of AxD patients and R236H/+ mt GFAP knock-in mice. (A) Immunohistochemistry of brain tissue of an AxD case (R239H) and a control subject, using antibodies against GFAP and LC3. Photograph from a patient showed extensive brightly eosinophilic staining red Rosenthal fibers (arrows) in a perivascular distribution (H&E, 40×). Compared to healthy control’s white matter, GFAP immunostaining disclosed many RFs in the AxD white matter, especially in perivascular areas. Correlated with the GFAP staining, the signal for LC3 staining was increased in areas enriched in RFs. *, blood vessel, ▴, RFs. (B) Immunoblotting analysis of LC3 in AxD patients with different GFAP mutations. (C) H&E staining showed RFs in the rostral migratory stream of a GFAP knock-in mouse. Note that RFs are never seen in wild-type mice (17). (D) Immunoblotting analysis of LC3 in astrocytes isolated from the whole brains of wild-type or R239H/+ GFAP mutant mice. Note that only a small fraction of astrocytes in GFAP mt mouse brain contained RFs. In spite of this fact, we still detected a slight increase of protein level of LC3-II. The protein level of LC3-I was also increased in astrocytes from GFAP mutant mice. →, GFAP aggregates.
Mice heterozygous for the R236H GFAP mutation (R236H/+), which corresponds to the R239H mutation in human AxD, develop RFs in a fraction of astrocytes in hippocampus, corpus callosum, rostral migratory stream (RMS), olfactory bulbs, subpial and periventricular regions (17). Though these mutant mice possess a normal lifespan and do not show spontaneous neurological deficits, they are more susceptible to kainate-induced seizures than are normal littermates. In these mice the RMS, olfactory bulb, hippocampus and white matter contained large numbers of RFs, whereas other areas appeared normal (Fig. 2C). We isolated astrocytes from whole forebrains of neonatal R236H/+ mice, and detected an increase in LC3-II in the R236H/+ mutant astrocytes, compared to those from GFAP+/+ wt mice (Fig. 2D). Therefore, in human tissues and in a mouse heterozygous for an AxD mutation, we found evidence for autophagy.
Formation of autophagic vacuoles in AxD brain tissue, mouse mutants and cell cultures overexpressing mt GFAP
We next examined white matter from an AxD patient, mouse mutants and cells overexpressing mt GFAP by electron microscopy to determine if organelles consistent with autophagosomes were indeed present. In AxD brain, astrocyte processes contained RFs, displacing cytoplasmic organelles (Fig. 3A-I). Note that RFs are dense structures associated with skeins of intermediate filaments, but are not membrane bound. Close to RFs were many vacuoles with a range of morphologies similar to autophagic vacuoles (AVs). AV morphologies included (18) typical autophagosomes, the double membrane organelles containing electron dense material, with an average size of 0.5 µm in diameter (Fig. 3A-II, IV, V) (14), autolysosomes, the fusion product of autophagosomes and lysosomes characterized by single membrane vesicles with amorphous dense material in the process of degradation, and with an average size greater than 0.5 µm in diameter (Fig. 3A-VI, VII). We also examined the CNS of R236H/− mutant mice and found structures consistent with autophagosomes in close proximity to RFs inside astrocytes (Fig. 3B). These observations were consistent with our results from immunohistochemistry (see above), where the LC3 immunostaining appeared at the edge of RFs when examined at the light microscope level. In U251 cell stably expressing high GFAP levels, the wt GFAP mostly formed 10 nm filaments that resembled normal GFAP intermediate filaments (Fig. 3C-I, II), while mt GFAP tended to form membrane-free dense structures (Fig. 3C-VI, VII,), which resembled the GFAP inclusions observed previously in some primary astrocytes that overexpressed wt GFAP (8) as well as RFs in AxD brain. Both autophagosomes and autolysosomes (Fig. 3C, V–X) were frequently seen in mt GFAP expressing cells (Fig. 3C-VI), but occasionally in wt expressers (Fig. 3C-III).
Figure 3.
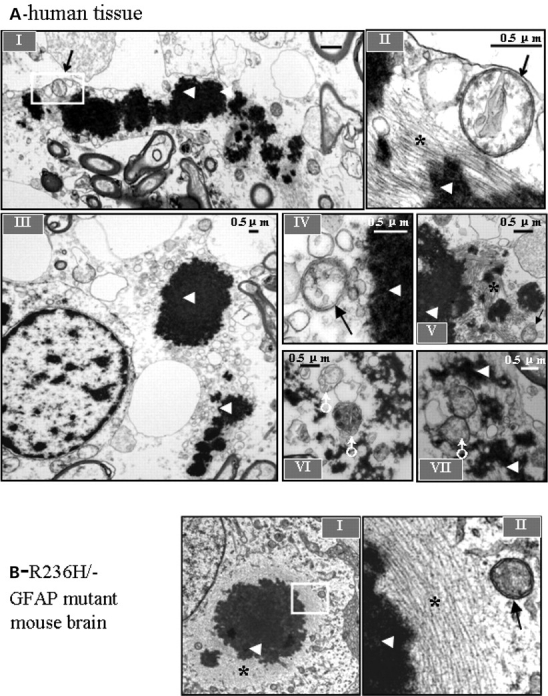
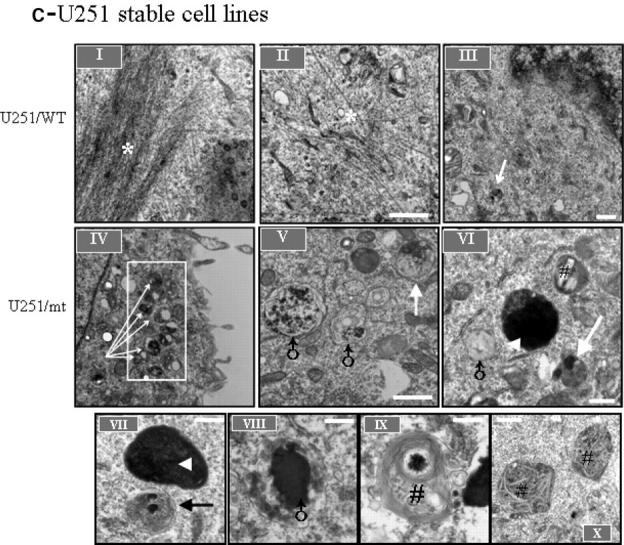
Transmission electron microscopy of AxD white matter, R239H/- GFAP mutant mouse brain and GFAP aggregate-containing U251 cells. (A) Typical autophagic vacuoles (AVs) in RF-containing astrocytes in L359V AxD brain. A-I, abnormal astrocyte process containing RFs. A-II, a typical autophagosome, with double-limiting membranes, adjacent to the skein of GFAP intermediate filaments, as shown in boxed area in A-I. A-III, another instance of affected astrocytes with RFs and multiple vesicular vacuoles. A-IV, A-V: autophagosomes close to RFs. A-VI, A-VII, vacuoles with single-limiting membranes. (B) A representative autophagosome in a RF-containing astrocyte in a R236H/- GFAP mutant mouse brain (B-II), as shown in boxed area in B-I. Note vesicular structures are located in the vicinity of RFs and the skeins of GFAP filaments (B-I). (C) GFAP filaments and AVs in GFAP aggregate-containing U251 cells. Note that AVs were frequently seen in mt cells (C-IV), and occasionally in wt GFAP expressing cells (C-III). C-V, VI,VII,VIII, XI and X, representative AVs in mt GFAP expressing U251 cells. * filaments; →, autophagosomes; ▴, Rosenthal fibers or GFAP aggregates; ♂, vacuoles with single-limiting membranes, reflecting autolysosomes; #, multilamellar bodies. Scale bar: 0.5 µm, Magnification: 25 000×.
Autophagy regulates GFAP accumulation
We next addressed whether autophagy, which is induced by the accumulation of GFAP, might in turn regulate GFAP accumulation. First, we examined the effects of 3-MA, an inhibitor of autophagy (19), on GFAP accumulation in U251 astrocytoma cells stably expressing GFAP. While the drug showed no significant effect on cell viability (Fig. 4B), 3-MA increased the GFAP protein level (Fig. 4A). When exposed to 3-MA, U251 cells stably expressing wt GFAP showed denser GFAP filamentous bundles and cells expressing mt GFAP showed more punctate aggregates per cell (Fig. 4C).
Figure 4.
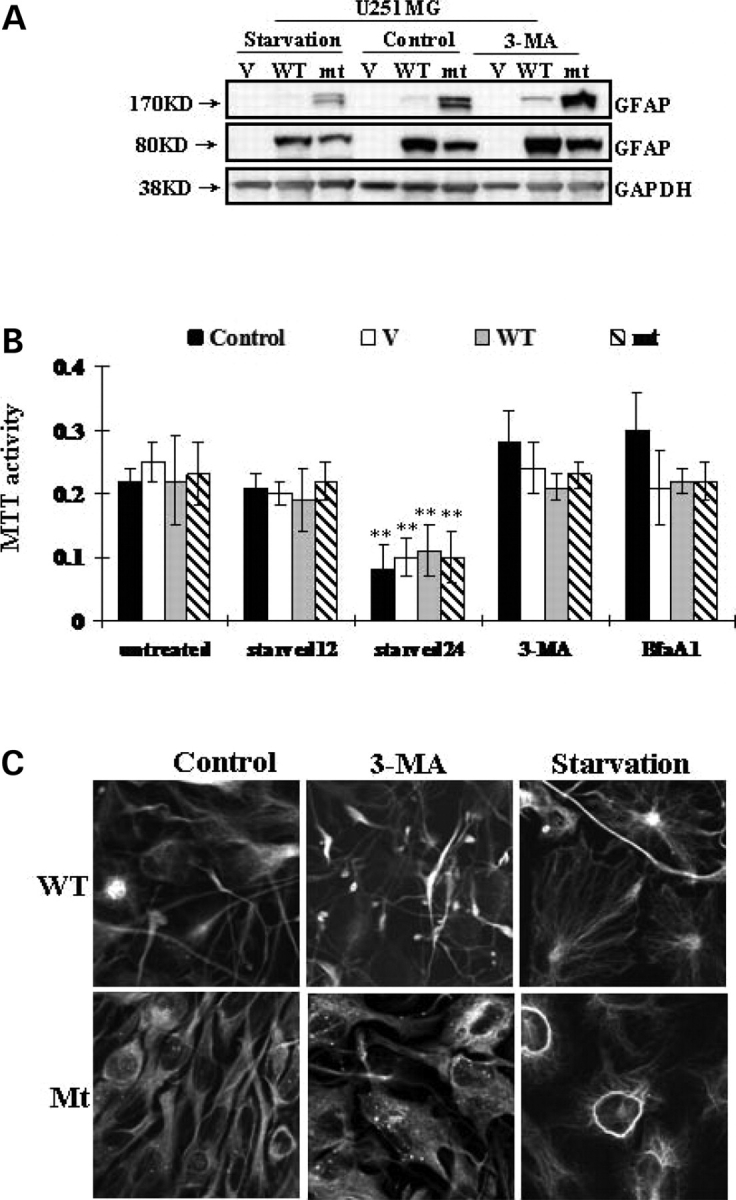
Effects of 3-MA and starvation on GFAP accumulation. (A) U251 cells stably expressing vector control (V), wt GFAP(WT) and mt GFAP(mt) were incubated with medium with or without 3-MA or starved for 12 h. Total cell lysates were subjected to SDS–PAGE and western-blotting analysis with antibodies against GFAP and GAPDH. (B) U251 cells were subject to 3-MA and BfaA1 for 24 h, to starvation for 12 (starved12) or 24 (starved24) hours. Cell viability was represented by methylthiazoletetrazolium (MTT) activity. Each result represents a mean ± SD of MTT activity from three independent experiments carried out in triplicate. * By student's t-test, and compared with untreated controls, P < 0.05. Control: untransfected U251 cells. (C) Morphological changes of GFAP expressing U251 cells. Cells were incubated with 3-MA or starved, then fixed and processed for GFP fluorescence. More GFAP filamentous bundles occurred in wt and dot aggregates in mt after 3-MA treatment. Starved cells are more spread out, with fewer GFAP aggregates.
We also tested whether starvation, a conventional physiological inducer of autophagy, could alter the protein levels of accumulated GFAP in U251 cells. No apparent cell death occurred 12 h after starvation. When starved for 24 h, however, cell death did occur (Fig. 4B). The level of GFAP was decreased compared to that in control samples (Fig. 4A). Starved U251 cells stably expressing GFAP showed a smaller percentage of cells with inclusions (data not shown), and most of the cells did not contain GFAP aggregates but showed filamentous bundles instead (Figs. 4C).
Finally, we studied GFAP accumulation in Atg5-deficient mouse embryonic fibroblasts (Atg5−/− MEFs), which lack autophagosome formation and autophagic activity (20). GFAP-GFP constructs were utilized for morphological study. In wild-type MEF cells, wt GFAP overexpressers showed a pattern of filamentous bundles and very few larger aggregates (∼1 µm). Wt GFAP aggregates were usually present in low numbers, most often only one per cell. In contrast, the mt GFAP formed more small aggregates (<1 µm), which were scattered around the cytoplasm and superimposed on a filamentous network, and some larger inclusions (1∼5 µm) localized close to nuclei (Fig 5A and B). In the Atg5−/− MEFs, both wt and mt GFAP formed many more and larger cytoplasmic inclusions close to the nuclei (Fig. 5A and B). We further examined the protein levels of wt or mt GFAP in transfected cells. Wild-type or Atg5−/− cells were transiently transfected with pcDNA3/GFAP constructs. An EGFP-C1 vector plasmid was cotransfected as a control for transfection efficiency. We found that the Atg5−/− cells accumulated far more GFAP protein than their wild-type MEF counterparts, for both wt and mt GFAPs (Fig. 5C). In both wt and Atg5−/− MEFs, mt GFAP accumulated more than wt GFAP, including the formation of a higher MW band of GFAP protein (Fig. 5C, upper band), as we have shown previously (9). This result suggested that the Atg5 deficiency slowed the cells’ ability to degrade GFAP. In contrast to wild-type cells, Atg5−/− cells showed no conversion of LC3-1 to LC3-II during GFAP accumulation, although there was an increase in the amount of LC3 protein (Fig. 5D). GFAP overexpression significantly increased LC3-II levels in the autophagy competent Atg5+/+ cells, but not in the autophagy-deficient Atg5−/− cells.
Figure 5.

Autophagy deficiency leads to more GFAP aggregation. (A) Representative GFAP filaments or inclusions in wild-type and Atg5−/− MEFs. →, filamentous pattern+small dot aggregates, filamentous pattern, ▸ filamentous pattern+large inclusions. WT: wt GFAP; mt: mt GFAP. (B) Percentage of cells with different phenotypes in wild-type and Atg5−/− MEFs. Results are average ± SD from three independent experiments, with at least 400 transfected cells counted in each. Compared with wt and by student's t-test, P < 0.05. (C and D) Immunoblot analysis of GFAP (C) and LC3 (D). In wt or mt GFAP expressing Atg5−/− MEFs. Wild type and Atg5−/− MEFs were transfected with pcDNA3/GFAP constructs. An EGFP-C1 vector was cotransfected to normalize the transfection efficiency, as indicated by GFP level. Forty-eight hours later, total cell lysates were prepared and subjected to western-blotting analysis for GFAP (C) and LC3 (D).
GFAP accumulation induces autophagy via mTOR signaling
To understand how GFAP accumulation induces autophagy, the classic mTOR signaling pathway was studied. Western blotting analysis of whole cell lysates showed a significant decrease of phospho-Akt (ser 473), phospho-mTOR (ser 2448) and phospho-pS6K in mt GFAP expressing U251 cells, but not in wt expressers (Fig. 6A and B). In contrast, the total mTOR levels and total S6K levels were not changed as a result of GFAP expression. mTOR was not sequestered into the GFAP inclusions, as assayed by immunohistochemistry, and an immunoprecipitation assay using either anti-GFAP or anti-mTOR antibodies revealed no interaction between GFAP and mTOR (not shown). These results suggest that GFAP accumulation causes a down-regulation of mTOR function, and this reduced mTOR function might exert a role, at least in part, on the regulation of autophagy. However, the lower activity of p-mTOR in the mt GFAP over-expressing cells was not due to its sequestration into aggregates, which was reported to contribute to the reduced soluble p-mTOR in a Huntington disease cell model system (21).
Figure 6.

Implication of mTOR in autophagic mt GFAP degradation. (A) Reduced phosphorylation of mTOR, Akt and p70S6K in mt GFAP overexpressing U251 cells. Protein extracts from vector control(V), wt (WT) and mt (mt) GFAP expressing U251 cells were subjected to western-blotting analysis with antibodies against phosphor-mTOR (p-mTOR), total mTOR (T-mTOR), phospho-p70S6K (p-S6K), total p70S6K (T-S6K), phosphor-Akt (p-Akt) and total Akt (T-Akt). (B) Quantitative analysis of p-mTOR, p-Akt and p-S6K in U251 cells expressing EGFP-C1 vector (V), wt (WT) and mt GFAPs (mt). In comparing cells transfected with vector only as control (100%), protein levels of p-mTOR, p-Akt and p-S6K were reduced markedly in mt GFAP expressing cells. (C) Inhibiting mTOR stabilized GFAP protein. U251 cells stably expressing GFAPs were exposed to fresh medium with Rapamycin (Upper), with those starved cells as positive control. (D) Effects of mTOR SiRNA on m-TOR level and GFAP protein level. EGFP-C1 vector, wt and mt GFAP expressing U251 cells were transfected with control SiRNA or mTOR SiRNA. Proteins were extracted at 48 h post-transfection and subjected to immunoblotting analysis. (E) Quantitation of levels of T-mTOR and p-mTOR in vector control cells transfected with control SiRNA and mTOR SiRNA. Control SiRNA has no effects on the level of p-mTOR, while mTOR SiRNA decreased the p-mTOR. (F) Effects of mTOR SiRNA on GFAP levels. Protein levels of wt or mt GFAP in U251 cells transfected with control SiRNA and mTOR SiRNA were quantified and plotted. (G) Effects of Av-Akt on GFAP accumulation. U251 cells stably expressing GFAP (wt or mt) were transfected with PCMV-AvAkt constructs. Forty-eight hours later, protein was harvested and subjected to immunoblotting analysis (upper). The optical density of p-mTOR protein bands was quantified and plotted to analyze the effects of Akt constructs on mTOR phosphorylation (lower). Av-Akt, constitutively active form of Akt; Myr AvAkt: a myristylated constitutively active form of Akt; E40K AvAKt, constitutively active Akt E40K mutant.
The role of mTOR in the regulation of autophagy-mediated clearance of accumulated proteins remains controversial. For the autophagic response to mt Huntingtin, some studies suggest an mTOR-dependent process (21), while others indicate an mTOR-independent induction (22). Therefore, the link between mTOR and the regulation of GFAP protein levels was further investigated. Pharmacologically inhibiting mTOR with rapamycin, an inhibitor of mTOR, decreased GFAP levels (Fig. 6C), a similar effect to that observed during amino acid starvation (Fig. 4A). To test the direct involvement of mTOR in this effect, we also employed RNA interference to knock-down mTOR levels in GFAP-expressing cells (Fig. 6D). The efficiency of mTOR SiRNA was examined in cells transfected with the vector plasmid. The control SiRNA showed no effects on mTOR level, while the mTOR SiRNA exhibited a significant inhibitory effect on both total mTOR and p-mTOR levels (Fig. 6D and E). SiRNA also reduced p-mTOR levels in both wt and mt GFAP expressing cells, and led to decreased GFAP levels (Fig. 6D and F).
Effects of AKT on mTOR signaling and GFAP level
Since mTOR is one of downstream substrates of the class I phosphatidylinositol 3-kinase (PI3K)/ protein kinase B (Akt) signaling pathway (23,24), we employed constructs expressing constitutively active forms of Akt (Myr and E40K) as potential mTOR activators. As shown by western-blot analysis, the expression of the active form of Akt resulted in increased phospho-mTOR, as well as in increased GFAP levels (Fig. 6G). These results are consistent with a role of Akt in activating mTOR, and thus decreasing autophagy, which in turn increases GFAP protein levels. The transfection efficiency was determined by blotting all samples with an anti-HA antibody, since the Akt constructs contained a small HA tag. Exogenous Akt protein levels were similar in all cells (Fig. 6G, upper).
P38 MAPK acts as a negative upstream signal regulating mTOR-mediated autophagy during GFAP accumulation
Our previous study demonstrated that the mixed-lineage protein kinase 3 (MLK3) was activated in Alexander disease (9). MLK3 is a member of the mitogen-activated protein (MAP) kinase group that has been implicated in multiple signaling cascades, including the c-Jun NH(2)-terminal kinase (JNK) and p38 MAP kinase pathways (25,26). Recent evidence supports a link between p38/MAPK and autophagy, though the underlying mechanism remains unknown (27,28). Based on these findings, we looked for a possible interaction between the cascade response of the MAPK stress signaling and the mTOR-mediated autophagy in response to mt GFAP accumulation.
First, we tested the effects of several pharmacological kinase inhibitors or activators on GFAP accumulation. GFAP-expressing cells were treated with a specific p38 inhibitor (SB203580), a specific JNK inhibitor (SP600125) and a potential p38/JNK activator, anisomysin. Cells were also co-treated with SB203580 and SP600125 simultaneously, so that the possible role of activation of the p38 pathway alone could be evaluated. As shown in Figure 7A and B, inhibiting p38 with SB203050 stimulated mt GFAP accumulation and the percentage of cells with GFAP inclusions. In contrast, stimulating p38/JNK with anisomysin while simultaneously inhibiting JNK with SP600125 showed the opposite effects. Inhibiting JNK alone with SP600125 had no significant effects on GFAP accumulation (not shown on western). These data suggested that activating p38/MAPK decreased GFAP levels, while inhibiting p38 increased GFAP levels.
Figure 7.

p38 negatively regulates mTOR-dependent autophagic GFAP degradation. (A and B) Effects of p38/MAPK on GFAP accumulation and aggregation. U251 cells stably expressing wt or mt GFAP were exposed to fresh medium or medium with SB203580 (5 uM), SP600125 (10 uM) and anisomysin (200 nm) overnight. (A) Percentage of cells with aggregates upon exposure to different chemicals. Results are average ± SD from three independent experiments, with at least 400 transfected cells counted in each. A larger percentage of mutant GFAP expressing cells, compared to wt expressing cells, contained aggregates under all treatment conditions. Different treatments were then compared within the mt GFAP expressing group, using DMSO as control. ** Compared to DMSO control, by student's t-test, P < 0.001. (B) Western-blotting analysis of GFAP protein levels. Note that activating p38 decreased GFAP levels while inhibiting p38 increased GFAP levels. (C) Immunoblots of phospho-p38 (p-p38) in U251 astrocytoma stably expressing GFAP (left) and in white matter from AxD patients (right panel). (D) Western-blotting analysis of the effects of AvMKK3 and dnMKK3 on the mTOR mediated autophagy induced by mt GFAP accumulation. Activation of p38 by AvMKK3 down-regulated, while inhibition of p38 via dnMKK3 up-regulated, levels of p-mTOR. The LC3-II level showed the opposite tendency relative to that of p-mTOR. (E) Quantitation of levels of p-mTOR, p-P70S6K and LC3-II/GAPDH in U251, U251/wt and U251/mt cells expressing AvMKK3, dn MKK3 and a pRSV vector, respectively. Compared to pRSV vector control cells, *P < 0.05, **P < 0.001, by two-tailed ANOVA.
Next, we asked if p38/MAPK is activated by mt GFAP accumulation. By western blotting, we detected large increases in p-p38 in wt and mt GFAP overexpressers, when compared to cells transfected with vector alone, though total p38 levels remained constant (Fig. 7C, left). An elevation of p-p38 was also detected in AxD white matter from the three patients (Fig. 7C, right).
Our pilot study using different pharmacological inhibitors (above) suggested a plausible role of p38/MAPK in regulating mTOR-dependent autophagic GFAP degradation. We then directly examined the ability of p38 kinase to stimulate the formation of autophagosomes. p38/MAPK was activated by introducing two constitutively active MKK3 mutants (AvMKK3 and AvMKK6), which directly phosphorylate and activate p38 (29). The effects of dominant negative MKK 3/6 constructs (dnMKK3 and dnMKK6), which are kinase-dead mutants and function by blocking endogenous p38 activation, were also tested. U251 cells stably expressing the EGFP vector, wt GFAP or R239C mt GFAP were transiently transfected with a vector control for MKK3 constructs (pRSVi—vector control), AvMKK3 and dnMKK3. All cell lines were analyzed to compare the relative protein levels of p-p38, p-mTOR, total mTOR, p-S6K and LC3 (Fig. 7D and E). In cells transfected with pRSVi only, overexpression of wt and mt GFAP led to an increase in the level of p-p38, relative to non-GFAP expressing cells. The Av-MKKs produced a substantial increase of p-p38 in all cell lines (GFP, wt, and mt), relative to their pRSV-VEC controls. In contrast, the level of p-p38 was drastically lower in dn-MKK3 transfected cells than those in control cells. Effects of AvMKK6 or dn MKK6 were the same as those of their MKK3 equivalents (data not shown).
High levels of p-p38, produced by the Av-MKK3 constructs, correlated with low levels of p-mTOR, p-p70S6K, and high levels of LC3-II/GAPDH. In contrast, low levels of p-p38, produced by the dnMKK3 construct, correlated with high levels of p-mTOR and low levels of LC3-II/GAPDH (Fig. 7E). These effects were obvious not only in U251 astrocytes overexpressing AxD R239C mt GFAP, but also in wt GFAP expressing cells with far fewer protein aggregates as well as the EGFP-vector expressing cells, which had very low levels of endogenous GFAP. Thus, the effects of activating or inactivating p38 on p-mTOR and LC3-II levels were not a consequence of the accumulation of mutant GFAP. p-p38 might act as a negative regulator for the mTOR-mediated autophagy in U251 astrocytes, irrespective of the abnormal accumulation of mt GFAP. Interestingly, wt GFAP expressing cells also exhibited an enhanced autophagic activity, though slightly, relative to EGFP-vector cells, as indicated by the level of LC3-II/GAPDH (Fig. 7D and E), suggesting that GFAP accumulation itself, regardless of the presence of a mutation, might stimulate autophagy via an mTOR-independent pathway.
P38 regulates autophagy in astrocytes in an mTOR-dependent manner
Our findings noted above showed that p38 blockage could upregulate mTOR signaling, and thus inhibit mutant GFAP-induced autophagy. To ask whether the effect of activating the p38/MAPK cascade on autophagic degradation of mt GFAP in astrocytes is dependent on mTOR signaling, we employed mTOR SiRNA and p38 siRNAs. The expression of mTOR siRNA had no effects on levels of p-p38, suggesting that mTOR is located downstream of p38 (Fig. 8). The expression of mTOR siRNA increased LC3 levels in all three lines, as expected. Expressing the p38 siRNA by itself resulted in increases in p-mTOR levels in all three lines and blocked the conversion of LC3-I to LC3-II. When we co-expressed both p38 siRNA and mTOR SiRNA, the inhibitory effect of the p38siRNA on autophagy was blocked, suggesting that p-mTOR is required for the p38-mediated regulation of autophagy.
Figure 8.

p38 regulates autophagy in astrocytes in an mTOR dependent manner. U251 cells stably expressing EGFP-C1 vector control (V), wt GFAP (wt) and mt GFAP (mt) were transiently transfected with mTOR siRNA, p38 SiRNA or both. Forty-eight hours later, protein was extracted and subjected to immunoblotting analysis. The expression of mTOR siRNA had no effects on levels of p-p38, but increased LC3 levels in all three lines. Expressing the p38 siRNA increased p-mTOR levels in all three lines and blocked the conversion of LC3-I to LC3-II. Co-expressing both p38 siRNA and mTOR SiRNA blocked the inhibitory effect of the p38 siRNA on autophagy.
DISCUSSION
Aberrant accumulation of GFAP induces autophagy
The autophagy pathway has been implicated in neurodegenerative disorders as an important pathway for the clearance of abnormally accumulated intracellular proteins, such as huntingtin and mutant α-synuclein (11,19,30). In this study, we demonstrated the induction of autophagy in response to abnormal GFAP accumulation in astrocytic cells, in AxD brain and in mt GFAP knock-in mouse brains. This observation adds AxD to a growing list of neurological diseases characterized by intracellular cytoplasmic protein inclusions and activated autophagic responses.
The induction of autophagy was supported by the following observations. First, AVs, either immature (presumably autophagosomes) or mature (autolysosomes), were identified in RF-containing astrocytes in AxD and in the R239H/+ GFAP knock-in mutant mouse brain, and also in mt GFAP expressing U251 astrocytoma cells. These structures were uncommon in wt GFAP-overexpressing U251 cells. Second, there was a strikingly increase in LC3-II in mt GFAP expressers, AxD brains and astrocytes from the knock-in mice. This increase was not seen in white matter of control individuals and astrocytes from wt mice. The further accumulation of LC3-II in the presence of lysosomal protease inhibitors indicated an enhancement of the autophagic flux (A1) in the context of mt GFAP accumulation. Third, we observed increased levels of both wt and R239C GFAP in the Atg−/− cells, and decreased GFAP levels in cells in which autophagy had been stimulated, either via starvation or by inhibiting mTOR, suggesting that GFAP degradation is in part processed via autophagy.
We also found that there was no change in p62/SQSTM1 levels. Inhibition of autophagy correlates well with increased p62 protein levels, and steady-state levels of this protein reflect the autophagic status. However, p62 is also degraded via the proteasome and therefore the p62 level may increase when the proteasome is inhibited. In our previous study, we showed that proteasome activity is inhibited by mt GFAP accumulation (9). Therefore, in our system, analysis of p62 alone can only be utilized to assist in assessing the impairment of autophagy, but not to monitor changes in autophagic flux. Based on the fact that there is a significant decrease in proteasome activity in mt GFAP-expressing cells, we can further suggest that the unchanged p62 level under the circumstance of proteasome inhibition could be balanced by increased autophagic degradation. This assumption is coincident with our autophagy flux data.
The bulk protein turnover was not affected by wt or mt GFAP expression. It might appear paradoxical that the increased autophagosome production is not correlated with an elevated degradation rate of total long-lived protein. However, the autophagic protein degradation was responsible for only a modest proportion of bulk protein degradation in U251 astrocytes. This low proportion might make it more difficult to detect a significant increase in protein turnover due to autophagy in the cells expressing mt GFAP. Another plausible explanation is that a rate-limiting step of the autophagic pathway that is downstream of the biogenesis of autophagosomes is not changed in the presence of GFAP overexpression. Therefore, the unchanged lysosomal degradation would result in a decrease in degradation efficiency per AV. The less efficient degradation would be compensated, however, by the increased numbers of autophagosomes (Talloczy et al., unpublished observations). The bulk protein turnover assay is further complicated by the possibility that other proteolytic pathways in these cells could be activated. For example, the expression of mt GFAP inhibits proteasome activity (9), which in turn may stimulate autophagy (and perhaps other degradative pathways as well) to keep protein turnover at a constant rate. It is also possible that the bulk turnover of protein does not reflect the specific kinetics of GFAP turnover. GFAP is a long-lived protein. Turnover kinetics in cultures of rat astrocytes suggest two pools, one (45% of the total) with a t1/2 of 18 h, the other (55% of the total) 6 days (Chiu and Goldman, 1984). In vivo turnover in mouse spinal cord appears to be even longer, since 40% of the radioactive amino acid incorporation into GFAP remains 9 weeks after labeling (DeArmond et al., 1986). Further studies are warranted to assess GFAP turnover rates.
The accumulation of mt GFAP elicited a significantly stronger autophagic response than did the accumulation of wt GFAP. We do not yet know why this is the case. The dependence of proteins on autophagic degradation correlates with their propensity to aggregate (29). Consistently, we observed an increased propensity for R239C mt GFAP to form inclusions (9). Recent studies also suggest that autophagosome formation is strongly activated by proteasome impairment (11,31). We have found that GFAP accumulation inhibits proteasome activity and that mt GFAP produces a greater inhibition than does wt GFAP (9). It may be, therefore, that the mt GFAP, in inhibiting the proteasome system, induces a more dramatic autophagic response than wt GFAP.
Signaling pathways underlying the GFAP-induced autophagy: the role of mTOR
Several signaling pathways, including the TOR signaling pathway, ATG1 complex and the Vps34/class III PI3K complex, are involved in the induction of autophagy (10,32). The sequestration of mTOR into Htt inclusions is responsible for the decreased soluble mTOR levels in some HD models, thus impairing mTOR function and activating autophagy (21). Inhibiting mTOR induces autophagy (24) and subsequently reduces the toxicity of polyglutamine expansions in fly or mouse models of HD (21). Our observations are consistent with a role for mTOR in regulating autophagy and the autophagy-mediated clearance of accumulated GFAP. Thus, we detected reduced phosphorylated mTOR during mt GFAP accumulation, although we did not observe mTOR sequestration with the GFAP inclusions. Inhibiting mTOR, using either rapamycin or mTOR SiRNA, decreased the levels of p-mTOR, and increased the number of autophagosomes, thus reducing GFAP levels. In contrast, activating mTOR increased the number of cells bearing GFAP inclusions, as well as the GFAP protein levels, indicating a decrease in autophagic clearance of GFAP.
Signaling pathways underlying the GFAP-induced autophagy: the role of P38 MAPK
It appears that the loss of p-mTOR protein is a consequence of the GFAP aggregation, and multiple upstream signal inputs to mTOR could lead to such an event. First, we identified the classical upstream kinase, p Akt, as an essential player in downregulating p-mTOR. Accumulation of mt GFAP led to a significant decrease in p-Akt level. Expression of constitutively active Akt constructs increased p-mTOR levels and increased GFAP protein level, presumably by inhibiting the autophagic response.
We also provided evidence about the existence of cross-talk between the p38/MAPK cascade and mTOR signaling in the setting of GFAP accumulation. The possible role of p38 MAPK in the regulation of autophagy has been inferred in a recent study, where activation of p38 MAPK by hypoosmolarity led to an inhibition of autophagy (28). However, a subsequent study presented an opposite finding that, in yeast, loss of Hog1, the p38 homologue, led to inhibition of autophagy upon hypo- and hyper-osmotic stress (27). More recently, a novel cell type-specific role of p38 in the control of autophagy and cell death in colorectal cancer cells was reported (33). However, the molecular mechanisms that link Hog1/p38 MAPK to autophagy are not yet known.
Our studies demonstrated that activation of p38 upregulates autophagic protein degradation in the context of AxD mutant GFAP accumulation in astrocytes. This result appeared to contradict Comes’ (33) findings that pharmacologically inhibiting p38 by SB203580 treatment induced autophagy and subsequent cell death in colorectal cancer cells. However, in their studies, SB203580 treatment failed to cause autophagic cell death in C3H10T1/2 and NIH3T3 mouse fibroblasts, C2C12 mouse myoblasts and human RD rhabdomyosarcoma cells. It appears that p38 blockade induces growth inhibition, autophagy and cell death in colorectal cancer cells in a cell type-specific manner. In our astrocyte cell models, GFAP accumulation activates stress response kinases, MLK3 and its downstream kinases, SAPK/JNK (9) and p38 (present study). Pharmacological modulation of the JNK and p38 pathways suggested that p38 activation, but not JNK activation, will trigger autophagy and a decline inGFAP. The effects of p38 in regulating autophagy are mTOR-dependent: the inhibition of p38 not only increased levels of p-mTOR, but also reduced the autophagic response, as shown by a decreased level of LC3-II. In contrast, activating p38 produced an opposite effect, decreasing p-mTOR and increasing LC3-II levels. Thus, p38 activation produced by GFAP accumulation can negatively regulate mTOR activity, thereby stimulating the autophagic degradation of GFAP. The molecular mechanism by which p38 influences mTOR phosphorylation and autophagy is unknown.
We detected a simultaneous decrease in p-Akt and an increase in p-P38 in U251 cells overexpressing AxD mt GFAP. Inhibiting p38 resulted in increased levels of p-mTOR. This observation seems to contradict a previous finding that PI3K-mediated activation of p38 kinase could increase Akt Ser473 phosphorylation (34), which in turn tends to upregulate p-mTOR. These effects may be cell-context dependent. Thus, the p38 kinase cascade regulates PI3K-mediated Akt activation in human neutrophils (34), but has no such effect on HEK293 cells (18). We find in U251 cells expressing mt GFAP, that the levels of p-PI3K, p-PDK and PTEN (components of the PI3K-Akt signaling cascade) are unchanged, compared with those in cells expressing wt GFAP or the GFP vector (unpublished experiments). We propose that, in our cell models, p38 activation is initiated by MLK2/3 phosphorylation, a communication between p38 and mTOR pathways, either through a direct interaction between p38 and mTOR molecules, or via a cross-talk between downstream substrates of the p38 and mTOR molecule is likely. A number of factors such as a better understanding of the downstream effectors of p38 cascade, including heat shock proteins, p53, NFkB, and the presence of mTOR regulation via pathways other than Akt and p38 signaling have to be considered in the future. In support of this proposal, induction of p53 has been reported to inhibit mTOR signaling via an AMPK dependent mechanism (35). We have observed in an AxD brain that p53 is upregulated, and we have also seen the AMPK activated in mt GFAP expressing cells (unpublished observations).
A proposed model
We propose a model in which autophagy is induced by GFAP accumulation, via the coordinate regulation between p38/MAPK and mTOR signaling pathways, and in which this induction in turn serves as a mechanism to bring down GFAP levels (Fig. 9). In this model, MLK/MAPK stress signaling, especially the P38/MAPK signaling, which is induced by GFAP accumulation, down-regulates mTOR function and subsequently activates the autophagy pathway. However, we do not exclude other possible mechanisms that cells might employ to induce autophagy to compensate for GFAP accumulation. One could be that wt or mt GFAP accumulation impairs proteasome function (9), and proteasome inhibition can, in turn, activate autophagy (11). Other possibilities include the class III PI3K—Beclin 1/Atg6 complex, which has been shown to regulate autophagy in Huntington disease (Shibata et al., 2006), and molecular chaperones, such as heat shock proteins hsp27, which can suppress protein misfolding and aggregation (Meriin and Sherman, 2005) and interact with p38/MAPK-MK2-Akt complex (34). All these systems are presumably coordinated in the degradation of aggregated proteins (31). In our cell cultures as well as AxD brains, we have detected elevated MAPK stress signaling, elevated small heat shock proteins, impaired proteasome activity and increased autophagic responses. Further studies will be aimed at understanding how these molecular pathways can work coordinately to bring down GFAP levels.
Figure 9.

A proposed model of activation of autophagy by GFAP accumulation.
MATERIALS AND METHODS
Reagents
Cell-culture medium DMEM and molecular biological reagents were obtained from Invitrogen and Qiagen. Primary antibodies included anti-GFAP polyclonal antibody, anti-cathepsin D (pAb, DAKO), anti-FLAG (Sigma), anti p-mTOR, mTOR, p-Akt; Akt, p-p38, p38 antibody (pAb, Cell Signaling Technology), anti-LC3 antibody (pAb, Santa Cruz, and provided by Dr Noboru Mizushima), anti-GAPDH monoclonal antibody (mAb, Encor) and Guinea pig anti-p62/ SQSTM1 pAb (American Research Products). The fluorescence-conjugated and HRP conjugated secondary antibodies were from Chemicon and Amersham Bioscience. 3-MA, BfaA1, EBSS, leupeptin, pepstatin, SB203580, SP100062, anisomycin, anti-FLAG M2 agarose affinity gel and protein G Sepharose were from Sigma-Aldrich.
Brain tissue
Tissues stored at −80°C from cerebral hemispheric white matter of control subjects or AxD cases with different mutations were obtained postmortem. For immunohistochemistry, the tissue was fixed in formalin and then embedded in paraffin. All AxD cases were diagnosed based on histopathological examination and confirmed by the molecular genetic analysis for GFAP mutation. Immunostaining and Western-blot analyses were carried out with fixed and frozen CNS tissues from Alexander patients with R239C, R239H and R416W mutations. All were children who manifested the disease as infants and expired within the first decade (10 years, 29 months and 7 years). All showed similar and typical, pathology for Alexander disease, with severe leukodystrophy and numerous RFs. Electron microscopy was carried out with CNS tissue from a patient with juvenile Alexander disease (carrying an L359V mutation), who died at 21 years of age. All post-mortem intervals were less than 10 h. Controls included frozen central nervous system tissue from two children, one with no neurological disease, 20 months of age, 18 h postmortem interval and the other with Werdnig-Hoffman disease (non-AxD, non-leukodystrophy neurological disease without RFs), 22 months, 6 h postmortem interval. Brain tissues from R236H mutant GFAP knock-in mice were obtained as described (17).
Cell cultures and transfection
cDNA clones encoding human wt GFAP or R239C mt GFAP were inserted into the EGFP-C1 expression vector, resulting in a fusion of GFP to the N-terminal of GFAP. Different AKT constructs, including E40K active, dominant-negative(d/n), and myristylated forms, were kindly provided by Dr Thomas Franke (New York University, New York, NY, USA). Different MKK3/6 constructs, including active forms of MKK3 and MKK6, dominant-negative forms of MKK3 and MKK6, were kindly provided by Dr Roger J Davis (University of Massachusetts Medical School, Worcester, MA, USA). mTOR SiRNA kit was purchased from Cell Signaling Technology.
Three cell lines were used in this study: mouse embryonic fibroblasts (MEFs, from wt and Atg5−/− mice, provided by Dr Noboru Mizushima, Tokyo Metropolitan Institute of Medical Science, Japan), and two GFAP positive cell lines [U251 human astrocytoma cells and primary astrocytes from R236H/+ heterozygous mutant GFAP mice (17)]. Among them, MEF cells were transiently transfected with the GFAP constructs using Lipofectamine-plus. Forty-eight hours after transfection, cells were harvested or fixed for either western blotting or immunostaining. Inhibitors or activators were applied to cultures at 24 h post-transfection and incubated for a further 24 h.
To generate permanent cell lines, U251 astrocytoma cells were transfected with the GFAP-GFP plasmid using calcium phosphate. Clonal cell lines were selected and maintained in DMEM supplement with 10% FCS, antibiotics, 1 mg/ml G418.
Immunoprecipitation and western blotting
Co-immunoprecipitation and western immunoblotting were performed as previously described (9). Quantitative immunoblot analysis was performed to assay LC3 activation, represented by LC3-II to GAPDH ratio, mTOR activation, represented by p-mTOR/total mTOR, and p70S6K activation, represented by p- p70S6K to total p70S6K ratio. Each result represents a mean ± SD of three independent experiments.
Analysis of cell survival
The cells were plated into 96-well plates and cell viability was measured by MTT assay according to standard protocol. Data were presented as the percent of living cells. All experiments were performed in triplicate.
Autophagic protein degradation assay
Autophagic proteolytic activity assay was modified from published protocol (16). U251 astrocytoma cells were seeded at 2 × 104 cells/well in 12-well plates, 6 wells for each condition. Cells were radiolabeled in presence of [3H]-leucine (1 µCi/ml) for 48 h. Cells were washed and then subjected to amino acid and serum starvation or incubated in growth media in presence of excess cold leucine (2.8 mm). At 0, 1, 12 and 24 h, aliquots of media were measured for acid-soluble radioactivity. The autophagy inhibitor, 3-MA (10 mm), was used to determine autophagic protein degradation levels. At the end of the incubation, the trichloroacetic acid-precipitable radioactivity of the cell monolayers was measured and the percentage of protein degradation at each time point was calculated (36). Mean percentage of total radiolabeled protein degradation and standard error of the mean was calculated from six wells, and subjected to statistical analysis (two-tailed ANOVA).
Immunocytochemistry and immunohistochemistry
Immunostaining of cultured cells was performed as described before (7). A cytoskeleton buffer (PHEM buffer: 100 mm Pipes, 2 mm EGTA, 1 mm MgCl2, 0.5% Triton-× 100, PH6.8) was utilized to characterize the cytoskeleton. Cells were visualized using a Zeiss LSM510 confocal microscope.
Immunohistochemistry was performed according to general protocols. Ten-micrometer thick consecutive sections were prepared. Sections were blocked in TBS-T/5% BSA for 1 h and subsequently incubated with antibodies against LC3 (1:50), GFAP (1:100) and p-mTOR (1:100) overnight at 4°C. Negative control sections were held in PBS during the primary incubation. Sections were then incubated with an HRP-labeled secondary antibody for 1 h at room temperature, and developed with 3,3′-diaminobenzidine (DAB).
Transmission electron microscopy
Cultured cells were fixed in 2.5% glutaraldehyde for 1 h, postfixed in 1% osmium tetroxide for 1 h at 4°C and processed for embedding in the culture dish. Cells were then gently scraped and embedded in blocks of eponaraldite. Thin sections were stained with 4% aqueous uranyl acetate and lead citrate and examined on a Philips CM12 electron microscope, glutaraldehyde fixed, osmium tetroxide post fixed brain tissues from an AxD patient with L359C mutation was examined under EM.
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed using the two-tailed ANOVA or Student’s t-test. P < 0.05 was considered statistically significant.
FUNDING
This work was supported by National Institutes of Health Grant PO1NS42803 and P30-HD03352.
ACKNOWLEDGEMENTS
We are grateful to Dr N. Mizushima (Tokyo Metropolitan Institute of Medical Science, Japan) for Atg5 deficient and wild-type MEFs and LC3 antibodies, to Dr Roger J Davis (University of Massachusetts Medical School, Worcester, MA, USA) for MKK3/6 constructs and to Dr Thomas Franke (New York University) for AKT constructs. We also thank Kristy Brown, Department of Pathology, Columbia University, for help with our electron microscopy study.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Goldman J.E., Corbin E. Isolation of a major protein component of Rosenthal fibers. Am. J. Pathol. 1988;130:569–578. [PMC free article] [PubMed] [Google Scholar]
- 2.Head M.W., Corbin E., Goldman J.E. Overexpression and abnormal modification of the stress proteins alpha B-crystallin and HSP27 in Alexander disease. Am. J. Pathol. 1993;143:1743–1753. [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner M., Johnson A.B., Boespflug-Tanguy O., Rodriguez D., Goldman J.E., Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 4.Johnson A.B. Alexander disease: a leukodystrophy caused by a mutation in GFAP. Neurochem. Res. 2004;29:961–964. doi: 10.1023/b:nere.0000021240.30518.2c. [DOI] [PubMed] [Google Scholar]
- 5.Li R., Messing A., Goldman J.E., Brenner M. GFAP mutations in Alexander disease. Int. J. Dev. Neurosci. 2002;20:259–268. doi: 10.1016/s0736-5748(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 6.Li R., Johnson A.B., Salomons G., Goldman J.E., Naidu S., Quinlan R., Cree B., Ruyle S.Z., Banwell B., D’Hooghe M., et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann. Neurol. 2005;57:310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- 7.Messing A., Head M.W., Galles K., Galbreath E.J., Goldman J.E., Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- 8.Hsiao V.C., Tian R., Long H., Der Perng M., Brenner M., Quinlan R.A., Goldman J.E. Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J. Cell Sci. 2005;118:2057–2065. doi: 10.1242/jcs.02339. [DOI] [PubMed] [Google Scholar]
- 9.Koyama Y., Goldman J.E. Formation of GFAP cytoplasmic inclusions in astrocytes and their disaggregation by alphaB-crystallin. Am. J. Pathol. 1999;154:1563–1572. doi: 10.1016/s0002-9440(10)65409-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang G., Xu Z., Goldman J.E. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J. Biol. Chem. 2006;281:38634–38643. doi: 10.1074/jbc.M604942200. [DOI] [PubMed] [Google Scholar]
- 11.Tian R., Gregor M., Wiche G., Goldman J.E. Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am. J. Pathol. 2006;168:888–897. doi: 10.2353/ajpath.2006.051028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marino G., Lopez-Otin C. Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol. Life Sci. 2004;61:1439–1454. doi: 10.1007/s00018-004-4012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rideout H.J., Lang-Rollin I., Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int. J. Biochem. Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N., Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 17.Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q.J., Ding Y., Kohtz D.S., Mizushima N., Cristea I.M., Rout M.P., Chait B.T., Zhong Y., Heintz N., Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talloczy Z., Jiang W., Virgin H.W.t., Leib D.A., Scheuner D., Kaufman R.J., Eskelinen E.L., Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl Acad. Sci. USA. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagemann T.L., Connor J.X., Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J. Neurosci. 2006;26:11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravikumar B., Duden R., Rubinsztein D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ravikumar B., Vacher C., Berger Z., Davies J.E., Luo S., Oroz L.G., Scaravilli F., Easton D.F., Duden R., O’Kane C.J., Rubinsztein D.C. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto A., Cremona M.L., Rothman J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corradetti M.N., Inoki K., Guan K.L. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J. Biol. Chem. 2005;280:9769–9772. doi: 10.1074/jbc.C400557200. [DOI] [PubMed] [Google Scholar]
- 26.Sarbassov dos D., Ali S.M., Sabatini D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 27.Brancho D., Ventura J.J., Jaeschke A., Doran B., Flavell R.A., Davis R.J. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol. Cell Biol. 2005;25:3670–3681. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brancho D., Tanaka N., Jaeschke A., Ventura J.J., Kelkar N., Tanaka Y., Kyuuma M., Takeshita T., Flavell R.A., Davis R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prick T., Thumm M., Kohrer K., Haussinger D., Vom Dahl S. In yeast, loss of Hog1 leads to osmosensitivity of autophagy. Biochem. J. 2006;394:153–161. doi: 10.1042/BJ20051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.vom Dahl S., Dombrowski F., Schmitt M., Schliess F., Pfeifer U., Haussinger D. Cell hydration controls autophagosome formation in rat liver in a microtubule-dependent way downstream from p38MAPK activation. Biochem. J. 2001;354:31–36. doi: 10.1042/0264-6021:3540031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raingeaud J., Whitmarsh A.J., Barrett T., Derijard B., Davis R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webb J.L., Ravikumar B., Atkins J., Skepper J.N., Rubinsztein D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 33.Qin Z.H., Gu Z.L. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol. Sin. 2004;25:1243–1249. [PubMed] [Google Scholar]
- 34.Qin Z.H., Wang Y., Kegel K.B., Kazantsev A., Apostol B.L., Thompson L.M., Yoder J., Aronin N., DiFiglia M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003;12:3231–3244. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- 35.Chiu F.C., Goldman J.E. Synthesis and turnover of cytoskeletal proteins in cultured astrocytes. J. Neurochem. 1984;42:166–174. doi: 10.1111/j.1471-4159.1984.tb09713.x. [DOI] [PubMed] [Google Scholar]
- 36.DeArmond S.J., Lee Y.L., Kretzschmar H.A, Eng LF. Turnover of glial filaments in mouse spinal cord. J. Neurochem. 1986;47:1749–1753. doi: 10.1111/j.1471-4159.1986.tb13084.x. [DOI] [PubMed] [Google Scholar]
- 37.Rubinsztein D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 38.Iwata A., Riley B.E., Johnston J.A., Kopito R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 39.Kamada Y., Funakoshi T., Shintani T., Nagano K., Ohsumi M., Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comes N., Matrone A., Lastella P., Nico B., Susca F.C., Bagnulo R., Ingravallo G., Modica S., Lo Sasso G., Moschetta A., Guanti G., Simone C. A novel cell-type specific role of p38a in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007;14:693–702. doi: 10.1038/sj.cdd.4402076. [DOI] [PubMed] [Google Scholar]
- 41.Rane M.J., Coxon P.Y., Powell D.W., Webster R., Klein J.B., Pierce W., Ping P., McLeish K.R. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J. Biol. Chem. 2001;276:3517–3523. doi: 10.1074/jbc.M005953200. [DOI] [PubMed] [Google Scholar]
- 42.Alessi D.R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Z., Zhang H., Levine A.J., Jin S. The coordinate regulation of the p53 and p-mTOR pathways in cells. Proc. Natl Acad. Sci., USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shibata M., Lu T., Furuya T., Degterev A., Mizushima N., Yoshimori T., MacDonald M., Yankner B., Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 45.Meriin A.B., Sherman M.Y. Role of molecular chaperones in neurodegenerative disorders. Int. J. Hyperthermia. 2005;21:403–419. doi: 10.1080/02656730500041871. [DOI] [PubMed] [Google Scholar]
- 46.Gronostajski R.M., Goldberg A.L., Pardee A.B. The role of increased proteolysis in the atrophy and arrest of proliferation in serum-deprived fibroblasts. J. Cell Physiol. 1984;121:189–198. doi: 10.1002/jcp.1041210124. [DOI] [PubMed] [Google Scholar]