

Abstract
The hereditary spastic paraplegias (SPG1-33) comprise a cluster of inherited neurological disorders characterized principally by lower extremity spasticity and weakness due to a length-dependent, retrograde axonopathy of corticospinal motor neurons. Mutations in the gene encoding the large oligomeric GTPase atlastin-1 are responsible for SPG3A, a common autosomal dominant hereditary spastic paraplegia. Here we describe a family of human GTPases, atlastin-2 and -3 that are closely related to atlastin-1. Interestingly, while atlastin-1 is predominantly localized to vesicular tubular complexes and cis-Golgi cisternae, mostly in brain, atlastin-2 and -3 are localized to the endoplasmic reticulum (ER) and are most enriched in other tissues. Knockdown of atlastin-2 and -3 levels in HeLa cells using siRNA (small interfering RNA) causes disruption of Golgi morphology, and these Golgi structures remain sensitive to brefeldin A treatment. Interestingly, expression of SPG3A mutant or dominant-negative atlastin proteins lacking GTPase activity causes prominent inhibition of ER reticularization, suggesting a role for atlastin GTPases in the formation of three-way junctions in the ER. However, secretory pathway trafficking as assessed using vesicular stomatitis virus G protein fused to green fluorescent protein (VSVG-GFP) as a reporter was essentially normal in both knockdown and dominant-negative overexpression conditions for all atlastins. Thus, the atlastin family of GTPases functions prominently in both ER and Golgi morphogenesis, but they do not appear to be required generally for anterograde ER-to-Golgi trafficking. Abnormal morphogenesis of the ER and Golgi resulting from mutations in atlastin-1 may ultimately underlie SPG3A by interfering with proper membrane distribution or polarity of the long corticospinal motor neurons.
INTRODUCTION
The hereditary spastic paraplegias (HSPs) are a clinically and genetically diverse cluster of inherited neurological disorders in which the primary manifestations are progressive spasticity and weakness of the lower limbs due to a length-dependent retrograde axonopathy of the corticospinal upper motor neurons (1–5). The autosomal dominant HSP SPG3A is the second most common HSP overall, and most common young-onset form (6–11). It is caused by well over 30 different mutations, predominantly missense mutations, in the atlastin-1 gene. Based on its similarity to proteins in the dynamin superfamily of large GTPases (4,6,12–14) and on recent studies in heterologous cell culture systems (15), atlastin-1 has been implicated in intracellular membrane trafficking, particular at the ER(endoplasmic reticulum)-to-Golgi interface (12,15). However, atlastin-1 is predominantly localized to the central nervous system, and its expression levels are much lower in peripheral tissues (6,12), indicating that it may have specific functions in the central nervous system.
To gain further insight into the cellular functions of atlastin-1, we have investigated two closely related human proteins, atlastin-2 and atlastin-3 (12,14). Although these proteins are highly similar structurally to atlastin-1, their distributions among tissues vary substantially. We have found that knockdown of atlastin-2 and -3 in cells causes abnormalities in Golgi morphology, most commonly fragmentation. Interestingly, expression of SPG3A mutant and dominant-negative forms of atlastin-1, -2 or -3 that lack GTPase activity resulted in prominent changes in ER morphology, with loss of typical reticularization. However, both dominant-negative and atlastin siRNA knockdown studies revealed essentially normal ER-to-Golgi trafficking. Thus, the atlastins constitute a family of large GTPases that function in morphogenesis of the ER and Golgi apparatus. Since a number of SPG3A mutations in atlastin-1 impair its GTPase activity (13), abnormal morphogenesis of the ER and Golgi may underlie this form of HSP.
RESULTS
Altastin family of GTPases
We investigated several large GTPases highly homologous to the SPG3A protein atlastin-1. In humans there are three atlastin family members, which we have named atlastin-1, -2 and -3 (12,14). Indeed, a ClustalW phylogenetic tree of these proteins indicates that this division is conserved in a variety of rodents and higher mammals (Fig. 1A). However, some species such as Drosophila melanogaster (D. melanogaster) (16), Caenorhabditis elegans (C. elegans), and the sea urchin Strongylocentrotus purpuratus (S. purpuratus) express only one atlastin, indicating that the three atlastins in higher species may have at least partially overlapping functions. Indeed, in humans and rodents, atlastin-1 is expressed at very low levels outside of the central nervous system (6,12).
Figure 1.

(A) Atlastin protein phylogeny. Species names and GenBank accession numbers are to the right. In higher species, there are three atlastin family members: atlastin-1 (green), atlastin-2 (yellow), and atlastin-3 (lavender), while a number of lower species have only one atlastin family member (top). The tree was constructed using ClustalW (v. 1.4) and MacVector 7.2. (B) Anti-peptide antibodies were raised against divergent regions of human atlastin-1, -2 and -3 as well as to a region common to all three (AT1-3) and used for immunoblotting extracts from cells overexpressing the indicated atlastins. Though atlastin proteins are expressed endogenously in COS7 cells, the endogenous proteins are not seen in immunoblots at these short exposure times; however, they are seen at longer exposures (unpublished data). Anti-atlastin-2ab (αAT2ab) is directed against a sequence present in both known C-terminal splice variants of atlastin-2: -2a and -2b. (C) Extracts were prepared from untransfected (Untrans) COS7 cells and cells transfected with AT1, AT2b, or AT3 (Trans) and then immunoblotted with the corresponding antibodies; sizes of the endogenous proteins were assessed by immunoblotting untransfected human 293 cell extracts. For atlastin-3, two different translation start sites were examined, and the shorter form (Trans-S) closely matched the size of the endogenous protein in 293 cells (arrowhead). An asterisk indicates a cross-reactive band. (D and E) Extracts from the indicated adult human tissues (D) and immortalized and primary human cell lines (E) were probed with the indicated anti-atlastin antibodies. Calnexin levels were monitored in (E) as a control for protein loading (Sk muscle, skeletal muscle; Sm muscle, smooth muscle).
To study the biochemical properties, distributions, and functions of the atlastin family of GTPases, we generated anti-peptide antibodies specific for atlastin-2 and -3; we had previously generated specific antibodies to atlastin-1 (12,13). As shown in Figure 1B, these antibodies were subtype-specific in their detection of recombinant proteins overexpressed in COS7 cells. Atlastin-2 exists in two splice forms with different C-termini (isoforms 2a and 2b; Supplementary Material, Fig. S1), and one antibody against atlastin-2 was specific for the 2b isoform (Fig. 1B). Since the atlastin-2b antibody had much higher affinity than the anti-atlastin-2ab antibody, we used this antibody preparation for subsequent experiments investigating atlastin-2. An antibody generated against a peptide sequence identical in all three atlastins detected all subtypes equally, confirming that expression levels of the recombinant proteins were similar (Fig. 1B). Antibodies against atlastin-1 and atlastin-2b detected endogenous proteins of identical size to the recombinant proteins overexpressed in 293 cells (Fig. 1C). However, two different start sites for translation initiation have been suggested for human atlastin-3 (Supplementary Material, Fig. S1). Overexpressing short and long forms using these different start sites and comparing their migrations on SDS–PAGE to that of the endogenous atlastin-3 protein in 293 cell extracts indicated that the shorter form resulting from use of the more 3′ initiator ATG codon corresponds most closely in size to the endogenous protein. Thus, numbering for the human atlastin-3 amino acid sequence commences at this latter initiator methionine (Supplementary Material, Fig. S1).
We examined the distributions of atlastin proteins in human tissues and cell lines. In human tissues, we found that while atlastin-1 is largely localized to brain, atlastin-2 and -3 were expressed at higher levels in peripheral tissues, and much less so in brain. Similarly, in multiple cell lines tested the atlastin proteins had mostly complementary distributions, with some overlap (Fig. 1D and E).
Homo-oligomerization of atlastins
Previously, we suggested that the atlastin-1 protein is an integral membrane protein with both N- and C-termini facing the cytoplasm using protease protection assays. Also, using Triton X-114 phase partitioning and membrane association assays, we found that the C-terminal hydrophobic domains are necessary and sufficient for atlastin-1 membrane localization (12). We similarly found that both atlastin-2 and -3 are integral membrane proteins (Fig. 2A and B) with N- and C-termini facing the cytoplasm, using the same approaches (Fig. 2C and D).
Figure 2.

Membrane topology of atlastin proteins. (A) Triton X-114 phase partitioning. Homogenates from COS7 cells transfected with atlastin-1, -2, or -3 were partitioned with Triton X-114 and aliquots of the starting material (Homog) as well as aqueous and detergent (Deterg) phases were immunoblotted with the indicated antibodies. The cytoplasmic protein PLCγ was monitored as a control for phase partitioning. (B) Hydrophobic domains of the atlastins are required for membrane association. Post-nuclear supernatants from COS7 cells expressing either Myc-tagged full-length (FL) or the indicated Myc-tagged deletion constructs for atlastin-1, -2 and -3 were fractionated into membrane (Memb) and soluble (Sol) fractions and immunoblotted with anti-Myc antibodies. Deletion of both predicted transmembrane domains (ΔH-H) markedly reduced membrane association, while C-terminal fragments containing these domains (H-H) were sufficient for membrane association. ΔH-H: atlastin-1 (1–447); atlastin-2a (1–474); atlastin-3 (1–417). H-H: atlastin-1 (448–558); atlastin-2a (475–579); atlastin-3 (418–515). Amino acid residues are numbered as in Supplementary Material, Fig. S1. (C) Protease protection assays. Microsomal (P3) fractions from cells transfected with N-terminally Myc-tagged atlastin-1, -2, or -3 were treated with proteinase K (Prot K) as indicated and then immunoblotted for Myc (αN-term), the corresponding C-terminal atlastin antibodies (αC-term), calnexin, and Grp78. A small portion of the transmembrane calnexin protein is oriented to the cytoplasm and thus it is partially degraded, while Grp78 is wholly within the ER lumen and protected from proteolysis. (D) Schematic structure of the proposed membrane topology for the atlastin proteins (Cyto, cytoplasm).
Atlastin-1 exists as an oligomer, most likely a tetramer based on chemical cross-linking and gel-exclusion fast protein liquid chromatography (FPLC) studies (12). We found similar chemical cross-linking patterns for atlastin-2 and -3 and similar elution patterns on gel-exclusion FPLC (Fig. 3A and B). Importantly, co-immunoprecipitation studies of atlastin-1, -2 and -3, all of which are present in human 293 cells (Fig. 1E), demonstrated that anti-atlastin-1 antibodies only co-immunoprecipitate atlastin-1, whereas atlastin-2 antibodies only co-precipitate atlastin-2 (Fig. 3C). Thus, endogenously these proteins occur as homomeric complexes. On the other hand, upon overexpression these proteins form heterologous complexes, and in yeast two-hybrid assays, each atlastin showed the capacity to interact with the others as well (Supplementary Material, Fig. S2).
Figure 3.
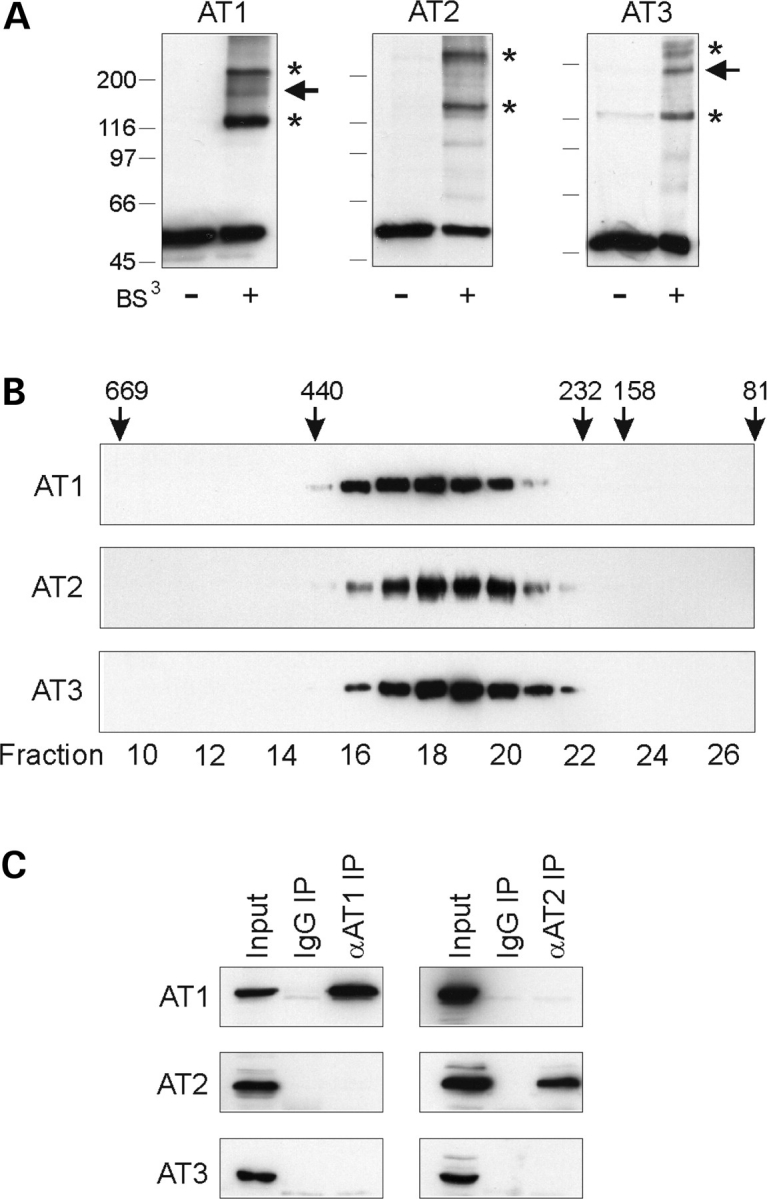
Atlastins form homomeric protein complexes in cells. (A) Chemical cross-linking. Extracts from COS7 cells expressing Myc-tagged atlastin-1 (AT1), -2b (AT2), and -3 (AT3) were cross-linked with 0.25 mm bis(sulfosuccinimidyl)suberate (BS3) and immunoblotted with anti-Myc antibodies. Asterisks denote probable dimeric and tetrameric forms, while an arrowhead identifies a putative trimer. Migrations of molecular mass standards (in kDa) are indicated in the left. (B) Extracts from COS7 cells expressing atlastin-1, -2 and -3 were subjected to gel-exclusion FPLC. Aliquots of each collected fraction were immunoblotted with anti-Myc antibodies. Elution peaks for marker proteins (in kDa) are shown across the top. Fraction numbers are indicated along the bottom. (C) Co-immunoprecipitation of endogenous atlastin proteins. Triton X-100 solubilized 293 cell extracts were immunoprecipitated (IP) with antibodies raised in guinea pigs or goats against atlastin-1 or -2b, respectively, or else control IgG, and then immunoblotted for atlastin-1, -2b, or -3 using antibodies raised in rabbits. Input represents 20% of the starting material.
Atlastin-2 and -3 localize to the ER along microtubules
To determine whether the fact that atlastins form homomers in vivo reflects distinct subcellular localizations, we examined the distributions of atlastin-2 and -3 in HeLa cells. In contrast to the atlastin-1, atlastin-2 and -3 exhibited very little co-localization with the cis/medial-Golgi marker GM130 (Fig. 4A). However, in HeLa cells transfected with the temperature-sensitive ts045 vesicular stomatitis virus G protein fused to green fluorescent protein (VSVG-GFP) construct and held at 40°C to label the ER, both atlastin-2 and atlastin-3 showed prominent localization to the ER (Fig. 4B); similar results were seen using anti-calnexin antibodies to label the ER (unpublished data). Even so, the atlastin-2 staining pattern consistently appeared more reticular, whereas atlastin-3 staining was more punctate.
Figure 4.
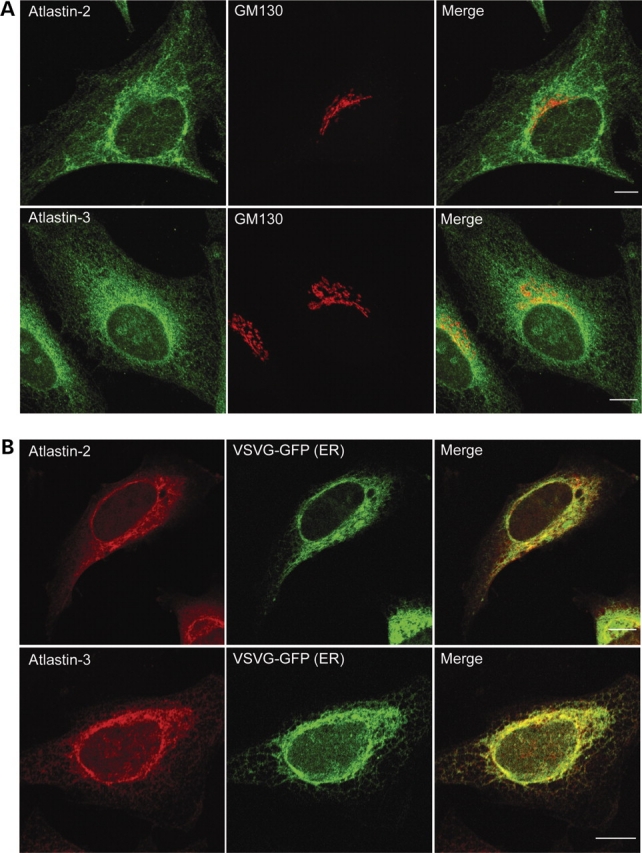
Atlastin-2 and -3 localize to the ER. (A) HeLa cells were co-immunostained for endogenous atlastin-2 or -3 (green) and the cis/medial-Golgi marker GM130 (red). Merged images are to the right. (B) HeLa cells were transfected with VSVG-GFP (green) and fixed during the 40°C incubation to reveal the ER. Cells were then immunostained for endogenous proteins using antibodies specific for atlastin-2 and -3 (red). Merged images are at the right. Bars, 10 µm.
We examined the localization of atlastin-2 and -3 by immunogold electron microscopy and found that gold particles not only decorated ER tubules and vesicular tubular complexes (VTCs) in the vicinity of the Golgi apparatus, but also localized to presumptive ER membranes extending along the length of microtubules (Fig. 5). Although both atlastins were associated to the same membrane compartments ultrastructurally, overall differences were observed in the density of gold particles. For instance, the atlastin-2 staining pattern revealed a particularly high proportion of gold particles associated with ER membranes along microtubules as compared with atlastin-3 staining (Fig. 5). Neither atlastin-2 nor atlastin-3 was visualized on the nuclear envelope (unpublished data).
Figure 5.
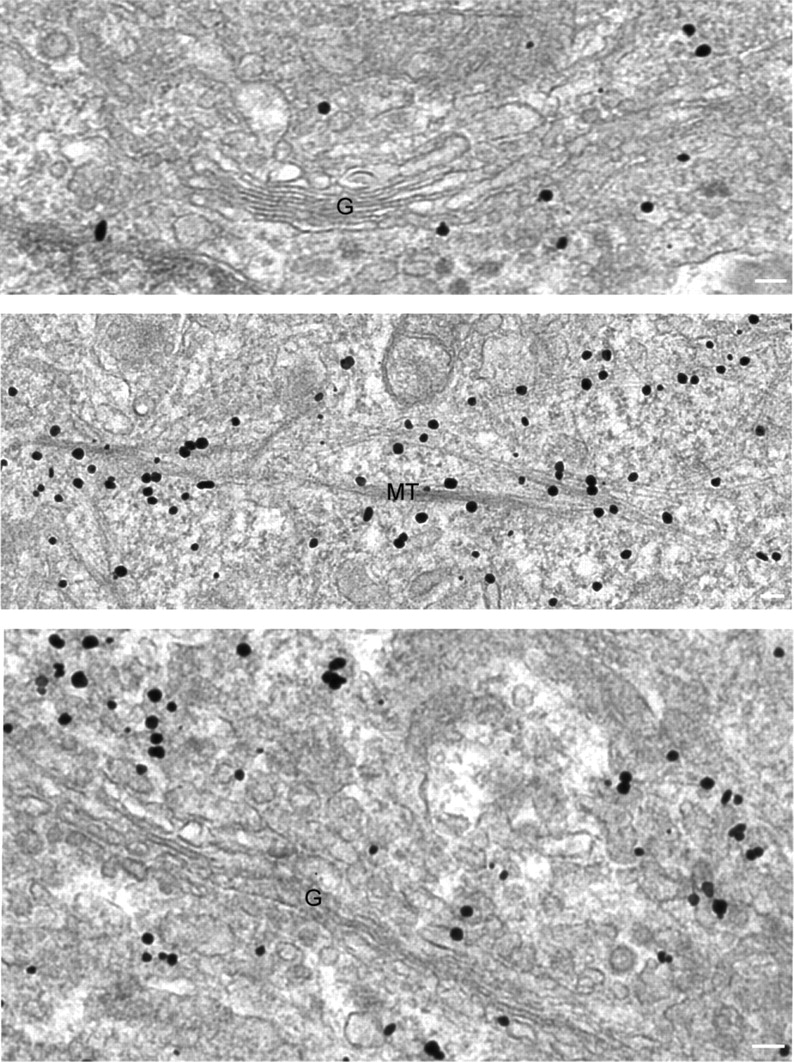
Atlastin-2 and -3 localize to ER membranes and along microtubules by immunogold electron microscopy. Electron microscopic analysis of HeLa cells immunostained for endogenous atlastin-2 (top and middle panels) or atlastin-3 (bottom panel) reveals gold particles not only at ER and VTCs/cis-Golgi (top and bottom panels), but also decorating membranes along microtubules (middle panel) (G, Golgi apparatus; MT, microtubules). Bars, 100 nm.
Knockdown of atlastin-2 and -3 disrupts Golgi morphology
To assess the functional roles of the atlastins in intracellular membrane trafficking and morphology, we examined the effects of depleting atlastin-2 or -3 in HeLa cells using siRNAs; HeLa cells express atlastin-2 and -3 abundantly, but have extremely low levels of atlastin-1 (Fig. 1E). We were able to achieve significant knockdown of atlastin-2 and -3 at 72 h after transfection and found that upon the specific knockdown of either of these isoforms, there was no up-regulation of the other atlastin within these cells (Fig. 6A). We also achieved significant double knockdown of both atlastin-2 and -3 at 72 h after transfection (Fig. 6B). We then examined the cells for morphological changes, and we found a substantial change in Golgi morphology upon depletion of atlastin-2, -3, or both (Fig. 6C and D, and unpublished data). On the other hand, ER morphology was normal except in a small subset of double knockdown cells where the ER clearly was more tubular, with loss of typical reticularization (unpublished data).
Figure 6.
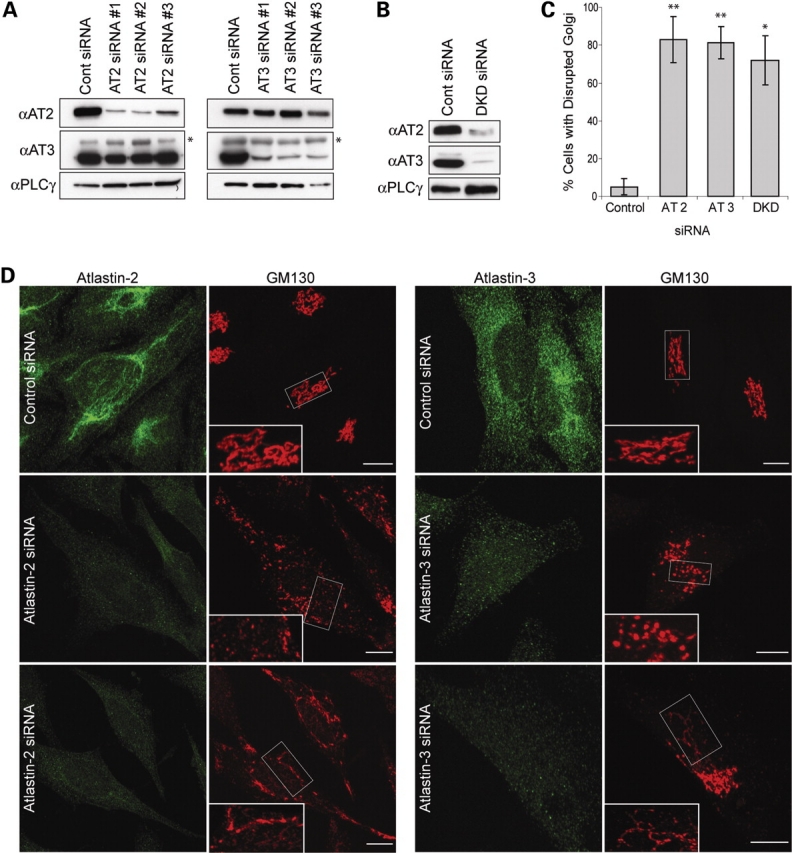
siRNA-mediated knockdown of atlastin-2 and -3 disrupts the Golgi apparatus. (A) Lysates from HeLa cells transfected with three different siRNAs specific for atlastin-2 (AT2) or else control siRNA were immunoblotted for endogenous atlastin-2 and -3 at 72 h post-transfection (left panels). Equal protein loading was monitored by immunoblotting for PLCγ. There is no upregulation of atlastin-3 protein expression upon knockdown of atlastin-2. Lysates from HeLa cells transfected with three different siRNAs specific for atlastin-3 (AT3) or else control siRNA were immunoblotted for atlastin-2 and -3, with PLCγ levels monitored as a loading control (right panels). Atlastin-2 expression is not upregulated upon knockdown of atlastin-3. An asterisk in atlastin-3 immunoblots identifies a cross-reactive band. (B) Lysates from HeLa cells transfected with both AT2 siRNA #1 and AT3 siRNA #3 or else control siRNA were immunoblotted for atlastin-2 and -3, with PLCγ levels monitored as a control for protein loading. (C) Graphical representation of percentage of cells with disrupted Golgi morphology in atlastin-2, -3, or double knockdown (DKD) conditions as compared with control siRNA-transfected cells (n = 3; 100 cells per condition). *P < 0.01; **P < 0.001. (D) HeLa cells transfected with control siRNA or atlastin-2 siRNA #1 were co-immunostained for atlastin-2 and GM130 (left panels) 72 h after transfection. HeLa cells were also transfected with control siRNA or atlastin-3 siRNA #3 and co-immunostained for atlastin-3 and GM130 (right panels). Boxed areas are enlarged in the insets to show fragmentation or tubular Golgi extending beyond the perinuclear region. Bars, 10 µm.
Two main Golgi phenotypes as assessed by GM130 staining were detected with both the atlastin-2 and atlastin-3 specific siRNAs. One was a widespread distribution of fragmented GM130-positive structures (Fig. 6D, middle panels), whereas the other was characterized by a more elongated, tubular pattern (Fig. 6D, lower panels). The proportion of cells showing each pattern differed among the knockdown groups, with the atlastin-2 siRNA group producing a higher percentage of cells with the tubular phenotype and the atlastin-3 siRNA and the double knockdown groups having a majority of cells with a more dispersed, ‘mini-stack’ distribution pattern. Mini-stacks are small, organized stacked structures with Golgi proteins adjacent to ER exit sites. After microtubule disassembly using drugs such as nocodazole, electron microscopy studies have shown that VTCs form initially, but over time, a stacked morphology more typical of the Golgi complex appears (17). The Golgi apparatus was considered ‘disrupted’ in those cells exhibiting either a fragmented or tubular Golgi morphology. Importantly, disruption of Golgi morphology was present in ∼80% of atlastin-2 and -3 siRNA cells, significantly higher than the percentage in control siRNA cells (P < 0.001; Fig. 6C).
Abnormal Golgi structures in atlastin-2 or -3 depleted cells are brefeldin A sensitive
To determine whether the GM130-positive structures are within the ER or else membrane compartments formed after exiting the ER, we used brefeldin A (BFA), a drug commonly used to block transport of secretory cargoes from the ER to the Golgi apparatus by inhibiting the Arf1 GTPase. HeLa cells transfected with control, atlastin-2, or atlastin-3 siRNAs were re-transfected with an YFP-Golgi construct and then treated with BFA a day later. Live images of the cells were acquired every 30 s during the BFA treatment to assess any redistribution of cargo protein from the abnormal membrane compartments present in the atlastin-2 and -3 siRNA cells (Fig. 7). Surprisingly, both the punctate and the tubular structures identified in the atlastin-2 and -3 siRNA groups were BFA-sensitive, confirming that upon loss of either atlastin, proteins are still able to exit the ER. Redistribution of YFP-Golgi can be followed over 25 min following BFA treatment in control siRNA cells (Supplementary Material, Video 1), and a similar redistribution can be clearly seen for the same duration following BFA treatment in atlastin-2 and atlastin-3 siRNA cells (Supplementary Material, Videos 2 and 3).
Figure 7.

Tubular Golgi structures in atlastin siRNA cells are sensitive to brefeldin A (BFA). HeLa cells transfected with either control or atlastin-2 siRNAs were re-transfected with an YFP-Golgi expression construct and then treated with BFA 24 h later. Live cell images were acquired during the BFA treatment to determine whether the tubular structures seen in atlastin-2 siRNA cells were sensitive to BFA. As shown, the distribution of YFP-Golgi changes when comparing cells before BFA treatment (upper panels) to after treatment (lower panels). Similar results were also seen in cells depleted of atlastin-3 using siRNA (unpublished data). See also Supplementary Material, Videos 1–3. Bars, 10 µm.
VSVG trafficking is not impaired in cells lacking atlastin-2 and -3
Because depletion of atlastin-2 or -3 altered the Golgi morphology, we next assessed whether knockdown of atlastin-2, -3, or both impaired protein trafficking through the Golgi apparatus, using ts045 VSVG-GFP as a reporter. Two days after transfection of HeLa cells with control, atlastin-2, atlastin-3, or both atlasin-2 and -3 siRNAs, all groups were transfected with VSVG-GFP and placed overnight at a temperature (40°C) that is non-permissive for VSVG exiting the ER. Cells were then transferred to a permissive temperature (32°C) and examined at various time points. In control cells, VSVG was observed at the plasma membrane as early as 90 min after the temperature change, and the majority of VSVG was present at the plasma membrane by 180 min, consistent with previous reports (18). VSVG trafficked normally to the plasma membrane in all atlastin knockdown groups examined (Fig. 8, and unpublished data), suggesting that atlastins are not required for general secretory pathway trafficking from the ER to the plasma membrane.
Figure 8.
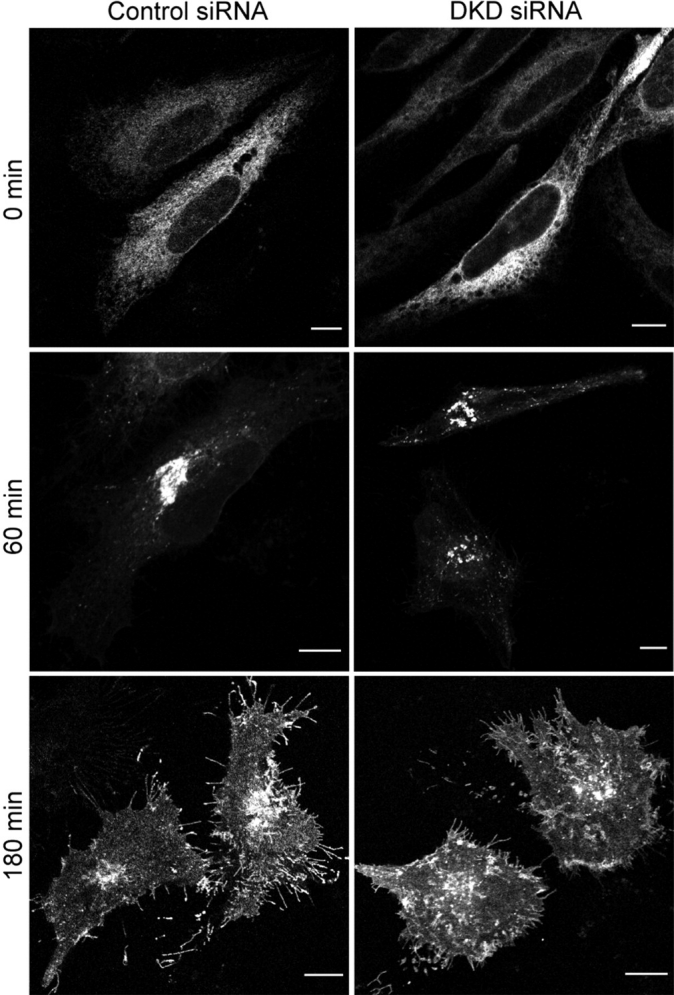
VSVG trafficking is not impaired in cells lacking both atlastin-2 and -3. HeLa cells were transfected with control siRNA (left panels), or with both atlastin-2 and -3 siRNAs (DKD (double knockdown); right panels), and both cell groups were then re-transfected with ts045 VSVG-GFP. The VSVG can be seen at perinuclear pre-Golgi and/or Golgi structures in both cell groups 60 min after moving to a permissive temperature. VSVG trafficked to the plasma membrane by 180 min in control cells as well as atlastin-2 and -3 DKD cells. Bars, 10 µm.
Dominant-negative atlastin proteins disrupt ER morphology
We found that upon expression of Myc-atlastin-1, Myc-atlastin-2, or Myc-atlastin-3 a normal, reticular ER morphology was identified (Fig. 9, top panels). However, when an SPG3A mutant form of atlastin-1 containing a mutation in a critical residue in the RD loop of the GTP-binding domain (R217Q) as well as the corresponding mutant forms of atlastin-2 and -3 (Supplementary Material, Fig. S1) were expressed in HeLa cells, the appearance of the ER changed to a more tubular, less reticular morphology (Fig. 9, middle panels). We also created dominant-negative forms of these proteins with point mutations in a key lysine residue required for GTP binding in other dynamin-related GTPases (12,19), which resulted in a very similar, and even slightly more pronounced, ER phenotype (Fig. 9, bottom panels).
Figure 9.

Dominant-negative atlastin proteins prominently disrupt ER morphology. HeLa cells expressing Myc-tagged atlastin-1, -2 or -3 (upper panels) exhibit a typical reticular ER morphology, as revealed by co-labeling with VSVG-GFP at 40°C. However, expression of Myc-tagged SPG3A atlastin-1 mutant R217Q and the equivalent mutations atlastin-2R244Q or atlastin-3R187Q result in a more tubular, elongated ER morphology with much less branching (middle panels). This profound effect on ER morphology was also seen to the same, if not greater, extent upon expression of Myc-tagged dominant-negative mutations atlastin-1K80A, atlastin-2K107A, or atlastin-3K47A (bottom panels). Bars, 10 µm.
Dominant-negative atlastin proteins disrupt Golgi morphology but not protein trafficking
We found that overexpression of either wild-type or SPG3A mutant forms (20) of all three atlastin proteins can result in a fragmented Golgi phenotype; however, these results were variable. Fortunately, we were able to identify a very consistent trend using the dominant-negative mutants. Overexpression of any of the dominant negative constructs resulted in significant Golgi fragmentation (Fig. 10A) in ∼60–65% of transfected cells (Fig. 10B). Importantly, we did not detect any impairment of VSVG trafficking to the Golgi mini-stacks and cell surface upon overexpression of any of the wild-type or mutant atlastin proteins (Fig. 10C, and unpublished data).
Figure 10.

Dominant-negative atlastin proteins disrupt Golgi morphology but do not alter anterograde secretory trafficking. (A) Expression of Myc-tagged dominant-negative (DN) atlastin-1, -2 or -3 (left panels) in HeLa cells results in Golgi fragmentation in a majority of transfected cells (right panels), as revealed by co-immunostaining for Myc-epitope and GM130. An arrow identifies a morphologically normal Golgi apparatus in an untransfected cell. (B) Graphical representation of percentage of cells with disrupted Golgi morphology in DN atlastin-1, -2, or -3 expressing cells versus control cells (n = 3; 100 cells per condition). *P < 0.01; **P < 0.001. (C) HeLa cells either singly transfected with ts045 VSVG-GFP or else co-transfected with VSVG-GFP and Myc-tagged atlastin-3 DN exhibit VSVG-GFP within ER before being moved to the permissive temperature (left panels). 180 min after being moved to the permissive temperature, cells from both groups are able to traffic VSVG to the plasma membrane despite the abnormal ER morphology (right panels). Similar results were seen when using atlastin-1 and -2 DN constructs (unpublished data). Bars, 10 µm.
Interaction of atlastin proteins with spastin
Atlastin-1 has been shown to interact with microtubule-severing AAA ATPase spastin that is mutated in the most common form of autosomal dominant hereditary spastic paraplegia, SPG4 (21,22). Since atlastin-2 and -3 localize prominently to ER membranes along microtubules (Fig. 5), we investigated whether atlastin-2 and -3 interact with spastin as well. Using yeast two-hybrid tests, we found a robust interaction of atlastin-1 with spastin, as published previously (21,22). However, neither atlastin-2 nor -3 interacted with spastin (Fig. 11A). The C-terminal 150 residues of atlastin-1 were sufficient for the interaction, but we were unable to narrow down the interaction domain further using the yeast two-hybrid system (Fig. 11B). Lauring and co-workers (22) were able to identify the C-terminal cytoplasmic domain of atlastin-1 as the region mediating the spastin interaction using fusion protein pull down assays, and this region is divergent among atlastin-1, -2 and -3 (Supplementary Material, Fig. S1).
Figure 11.

Interactions of atlastin family members with the SPG4 protein spastin. (A and B) Yeast two-hybrid tests showing interactions of the spastin bait construct with full-length atlastins (A) and the indicated atlastin-1 deletion constructs, with residues 1-558 representing the full-length atlastin-1 protein (B), as revealed by growth selection on -His/Leu/Trp plates. The -Leu/Trp panel demonstrates similar transformation efficiencies for each reaction. Sequential 10-fold dilutions of yeast are shown for all panels. Strength of interaction was also assessed semi-quantitatively by determining the time for colonies to turn blue in X-gal filter lift assays: +++, <30 min; ++, 30–60 min, +, 60–120 min; and −, no significant activity.
DISCUSSION
We have investigated the functions of the atlastin family of GTPases, emphasizing studies of atlastin-2 and -3. We had previously reported that the SPG3A protein atlastin-1 co-localizes prominently with p115, a protein present at VTCs and cis-Golgi (12,13,23), mostly in brain, and also that atlastin-1 is present in growth cones and required for axon formation and elongation during development (13). Interestingly, atlastin-2 and -3 are present widely in peripheral tissues and localize predominantly to ER membranes. Our findings suggest that, while all atlastins are present at the ER, VTCs, and cis-Golgi cisternae to some degree, there are differences in distributions of the majority of each particular isoform that may correlate with some functional specificity of each atlastin along the ER-to-Golgi axis. Indeed, although both atlastin-2 and -3 localize to ER, we noted a difference in distribution between these two in that the atlastin-2 distribution on ER appeared more closely aligned with microtubules. Also, atlastin-1 localizes predominantly with VTCs/cis-Golgi as well as within growth cones (12,13), where dynamic ER has the ability to rapidly move into or away from the distal parts (24,25). Clearly, however, there may be functional overlap among the highly homologous atlastins. Consistent with this notion, species such as Drosophila and C. elegans have only one atlastin protein.
Because our experiments were carried out in HeLa cells, and levels of atlastin-1 are extremely low in these cells, we focused on knocking down atlastin-2 and -3 to assess for any intracellular changes. With the specific depletion of either atlastin-2 or -3 we saw two different types of changes in Golgi morphology, but no consistent ER phenotype. In cells where atlastin-2 and -3 double knockdowns were performed, we saw a similar degree of Golgi disruption, but also saw a more tubular ER morphology in a small subset of cells (unpublished data). In complementary experiments, we examined the structural and functional changes that occurred upon overexpression of either wild-type, SPG3A-type mutant (20), or dominant-negative forms of all three atlastins. In all cases, overexpression of the mutant, but not wild-type, atlastin proteins resulted in a change in ER morphology to a more tubular phenotype with very few interconnections by three-way junctions, which are points of homotypic fusion events in smooth ER that result in a polygon-like structure (26–28). The fact that all six atlastin mutants defective in GTPase activity produced the same morphological change, while the wild-type atlastins did not, may reflect the fact that each overexpressed mutant atlastin protein can oligomerize with all three endogenous atlastins (Supplementary Material, Fig. S2), thus impairing GTPase activity of heteromeric complexes comprising all of the atlastins. In assessing Golgi morphology, we found that overexpression of both the wild-type and SPG3A-type constructs caused fragmentation of the Golgi into what appeared to be ‘mini-stacks,’ but this phenotype varied among the mutant forms. These differences may reflect functional specificity of the different protein or variations in expression levels within the cells. Expression of dominant-negative atlastin proteins produced a more consistent effect on Golgi morphology. However, on examining effects on protein trafficking in these overexpression paradigms, we found that there did not seem to be inhibition of VSVG trafficking to the plasma membrane, even in cells with significantly altered ER morphology.
One explanation for the differences between dominant-negative studies and siRNA depletion studies, particularly with respect to changes in ER morphology, is that the loss of a single isoform may have more modest effects because the other isoforms can compensate. Furthermore, even in the atlastin-2 and -3 double knockdown cells the degree of knockdown of each isoform may not be sufficient in a majority of the cells to elicit a phenotype, and there is also a low level of atlastin-1 in HeLa cells. On the other hand, the changes in Golgi morphology upon knockdown of a single atlastin may reflect more specialized functions of each isoform at the level of VTCs or Golgi apparatus. This notion is consistent with the fact that although both Golgi phenotypes were present upon knockdown of atlastin-2 or -3, there was a clear difference in proportion of cells displaying each phenotype in each atlastin siRNA condition. Thus, the majority of atlastin-2 siRNA cells exhibited more elongated Golgi tubules, whereas the majority of atlastin-3 siRNA cells had a more fragmented ‘mini-stack’ Golgi morphology.
We considered the possibility that, in our siRNA treatment conditions, the fragmented GM130- and YFP-Golgi-positive membranes may reflect Golgi proteins retained at ER exit sites, and consequently that the more elongated, tubular structures might represent Golgi proteins trapped within the ER. However, these GM130- and YFP-Golgi-labeled compartments were BFA sensitive, clearly demonstrating that they are formed after proteins have trafficked from the ER in the form of VTCs. It is likely that once VTCs have formed from the ER, atlastins are important in the movement of these vesicles along microtubules towards the MTOC (microtubule organizing center), the fusion of vesicular tubular complex (VTC) membranes, or perhaps both. The fact that these structures are BFA-sensitive is also consistent with our results showing that all atlastin siRNA cells examined were still able to traffic VSVG to the plasma membrane, with only a very small subset of cells showing a delay in trafficking. Indeed, secretory protein trafficking does not require a stacked Golgi morphology, nor does there need to be a centralized location of the Golgi apparatus (29). This trafficking, however, may not be as efficient as in control cells, potentially accounting for the delay in VSVG trafficking seen in a small subset of atlastin-2 and -3 siRNA cells.
Thus, our results suggest that while depletion of atlastins by siRNAs and overexpression of dominant-negative, GTPase-deficient atlastin proteins have substantial effects on the morphology of the Golgi apparatus and the reticular ER structure, respectively, protein trafficking in the secretory pathway does not seem to be greatly affected. This is particularly interesting in light of a recent study that found that mutations in proteins critical for ER-to-Golgi transport in the secretory pathway affected development of dendrites far more than axons in Drosophila (30). Conversely, since SPG3A is a long axonopathy, and loss of atlastin-1 causes effects predominantly on the development of axons in cultured cortical neurons (13), atlastin-1, and possibly atlastin-2 and -3, may be important in other intracellular trafficking pathways, perhaps those involved in the establishment and maintenance of specific polarized membranes or membrane processes. Indeed, Sannerud et al. (31) have provided evidence for such a ‘Golgi-bypass’ pathway defined by Rab1 that is linked to the dynamics of the smooth ER as well as pre-Golgi intermediate compartments and functions in the delivery of membranes to developing neurites.
Mechanistically, the atlastin GTPases may serve an important role in fusion of ER membranes to form the more widely-spread ER reticulum by way of three-way junctions or else by influencing the association of ER with microtubules. Alternatively, atlastins may also play a role in VTC or Golgi ‘mini-stack’ movement along microtubules towards the MTOC as well as the formation of the normal stacked Golgi structure. In this regard, the interaction of atlastin-1, but not atlastin-2 or -3, with the microtubule-severing AAA ATPase spastin that is mutated in the most common HSP, SPG4, is particularly intriguing. It will be interesting to determine whether atlastin-2 and -3 interact with other microtubule-severing proteins, or whether this interaction with microtubule-severing enzymes is specific to atlastin-1 and represents an important functional specialization with relevance for the pathogenesis of the HSPs.
Importantly, spastin is also present at the ER, and this distribution is increased upon overexpression of atlastin-1. Furthermore, overexpression of SPG4 mutant spastin disrupts ER morphology in conjunction with microtubule abnormalities (21), and overexpression of wild-type spastin results in increased lateral mobility of the translocon complexes that are part of membrane-bound polysomes (32). Thus, proper ER morphology may be an important determinant of axon development and maintenance. Lastly, at least one additional HSP protein, the SPG17 protein seipin, localizes prominently to ER membranes (33). Thus, in addition to proteins implicated in endocytosis that are mutated in a number of HSPs (4), regulation of ER and Golgi morphology and possibly novel trafficking pathways important for membrane addition or dynamics at axons may also be a major theme in HSP disease pathogenesis.
MATERIALS AND METHODS
Eukaryotic DNA expression constructs
The pGW1 construct expressing Myc-tagged human atlastin-1 and the pRK5 construct expressing the untagged protein have been described previously (12). The full coding sequences of human atlastin-2a (GenBank accession number NM_022374) and atlastin-2b (GenBank accession number AAM97342) cDNAs were cloned into the EcoRI sites of the pGW1-Myc expression vector for production of N-terminal Myc-tagged proteins as well as the pRK5 expression vector for production of untagged proteins. The full coding sequence for human atlastin-3 (GenBank accession number AK097588) preceded by a Kozak consensus sequence, was cloned into the XmaI sites of pGW1-Myc and pRK5. Where indicated for atlastin-3, another candidate initiator Met residue (as in GenBank accession number NM_015459; Supplementary Material, Fig. S1) was used. Atlastin deletion constructs were generated by PCR using Pfu Turbo, and site-directed mutagenesis was performed using the QuikChange method (Stratagene). The YFP-Golgi expression construct was purchased from BD Biosciences Clontech and the ts045 VSVG-GFP construct was provided by Dr J. Lippincott-Schwartz (34).
Antibodies
Polyclonal antibodies raised in rabbits and guinea pigs against atlastin-1 (No. 5409; residues 1–18) have been described previously (12,13). Affinity-purified polyclonal antibodies were generated commercially (Quality Controlled Biochemicals) in rabbits and goats against atlastin-2b (No. 6955; residues 561–578; acetyl-CIKAGLTDQVSHHARLKTD-OH) and atlastin-3 (No. 7053; residues 550–567; acetyl-CATVRDAVVGRPSMDKKAQ-OH) as well as to a region common to atlastin-2a and -2b (No. 6327; residues 1–17; MAEGDEAARGQQPHQGLC-OH) and a region identical in atlastin-1, -2 and -3 (No. 6944; residues 341–358 in atlastin-1; acetyl-LPHPKSMLQATAEANNLAC-OH), with terminal cysteine residues added to facilitate coupling to keyhole limpet hemocyanin. Other antibodies used include mouse monoclonal anti-GM130 (IgG1, clone 35; BD Transduction Laboratories), anti-Myc-epitope (IgG1, clone 9E10; Santa Cruz Biotechnology), PLCγ-1 (mixed IgGs, #05-163, Millipore), and anti-BiP/Grp78 (IgG2a, clone 40; BD Transduction Laboratories) as well as rabbit polyclonal anti-calnexin (H-70; Santa Cruz Biotechnology).
Tissue preparation, fractionation and protease digestion
Human tissue homogenates were obtained from BD Biosciences Clontech. Membrane association, Triton X-114 phase partitioning, and protease digestion assays were performed as described previously (12).
Protein interaction studies
Chemical cross-linking studies were performed as described previously (12). Co-immunoprecipitation studies were performed using affinity-purified goat anti-atlastin-2b (5 µg) and guinea pig anti-atlastin-1 (5 µg) antibodies. Immune complexes were collected using protein A-Sepharose, washed, resolved by SDS–PAGE, and immunoblotted with rabbit polyclonal antibodies against atlastin-1, -2b, and -3. FPLC Gel-exclusion FPLC was performed as described previously (12). Yeast two-hybrid assays were conducted in the yeast strain AH109 using a pGBKT7-spastin bait construct (22) and atlastin-1, -2a, -2b -and -3 prey in pGAD10 or pGADT7 vectors. Strength of interaction was assessed by nutritional selection on histidine-lacking media as described previously (35,36).
Cell culture and transfection
HeLa cells were maintained in Dulbecco’s MEM supplemented with 10% fetal bovine serum (Gibco). For immunostaining experiments, HeLa cells plated on coverslips in 6-well plates were transfected with 1 µg of plasmid DNA using Lipofectamine (Invitrogen). Twenty-four hours later, cells were washed in phosphate-buffered saline [PBS(phosphate-buffered saline); pH 7.4] and fixed using 4% formaldehyde. Cells to be used for immunoblot analysis were washed with PBS and lysed with 0.1% Triton X-100 in PBS.
For siRNA transfections, HeLa cells were plated at 50% confluency and transfected the next morning with 100 nM siRNA oligonucleotides using Oligofectamine (Invitrogen) for 4 h. Cells in the double knockdown conditions were treated with 150 nM of each siRNA oligonucleotide duplex. Cells were then scraped for immunoblot analysis or fixed for immunocytochemical analysis 48 and 72 h after transfection. Specific siRNA oligonucleotides (Invitrogen) for atlastin-2 were targeted against the following sequences: #1, GGAGCUAUCCUUAUGAACAUUCAUA; #2, UCCUGGUCUUAAAGUUGCAACUAAU; and #3, GAGAGCUUCGAAAUCUGGUUCCAUU. Atlastin-3-specific siRNAs (Invitrogen) were as follows: #1, GCCCUGACUUUGAUGGGAAAUUAAA; #2, GGGCUACAUCAGGUAUUCUGGUCAA; #3, GGUUAGAGAUUGGAGUUUCCCUUAU. The control siRNA oligonucleotide was obtained from Ambion.
Confocal microscopy
All cells were imaged using a Zeiss LSM510 confocal microscope with a 63 × 1.4 NA Plan-APOCHROMAT lens. Acquisition was performed using LSM 510 version 3.2 SP2 software (Carl Zeiss Microimaging), and data were processed using Adobe Photoshop 7.0 and Adobe Illustrator CS2 software. For quantification of cell biology studies, at least 100 cells were counted per experimental group, with experiments conducted at least three times. Cells with fragmented or else tubular Golgi not restricted to tight cisternae within the perinuclear region were considered ‘disrupted’.
BFA treatment and VSVG-GFP trafficking
HeLa cells plated in two-well glass bottom chambers were transfected with 0.5 µg of YFP-Golgi DNA, and the following day they were placed in growth media containing 5 µg/ml BFA (Epicentre). An image was acquired before addition of the BFA and subsequent images were taken every 30 s after addition of BFA.
For VSVG-GFP trafficking studies, HeLa cells grown on coverslips were transfected with the ts045 VSVG-GFP construct and immediately placed in a 40°C incubator. Sixteen hours after transfection, HeLa cells were moved to 32°C to allow for VSVG trafficking from ER to Golgi, and then fixed and processed for immunocytochemical analysis 0, 30, 60, 90, 120 and 180 min after the temperature change.
Immunogold electron microscopy
For atlastin-2 immunogold electron microscopy, HeLa cells were fixed in 2% paraformaldehyde and 0.1% glutaraldehyde in PBS for 1 h. For atlastin-3 immunogold staining, HeLa cells were fixed in 4% paraformaldehyde in PBS for 1 h. Cells were then processed for electron microscopic analysis and imaged as described previously (12,13).
Statistical analysis
Statistical significance was assessed using two-tailed, unpaired Student’s t-tests, assuming unequal variance.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health.
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. Nagle and D. Kaufmann (National Institute of Neurological Disorders and Stroke DNA Sequencing Facility) for DNA sequencing, Dr J.-H. Tao-Cheng and V. Tanner-Crocker (National Institute of Neurological Disorders and Stroke EM Facility) for assistance with electron microscopy, Drs B. Lauring and J. Lippincott-Schwartz for providing materials, and Dr C.-R. Chang for experimental assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Crosby A.H., Proukakis C. Is the transportation highway the right road for hereditary spastic paraplegia? Am. J. Hum. Genet. 2002;71:1009–1016. doi: 10.1086/344206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reid E. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J. Med. Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 4.Soderblom C., Blackstone C. Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol. Ther. 2006;109:42–56. doi: 10.1016/j.pharmthera.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Züchner S. The genetics of hereditary spastic paraplegia and implications for drug therapy. Expert Opin. Pharmacother. 2007;8:1433–1439. doi: 10.1517/14656566.8.10.1433. [DOI] [PubMed] [Google Scholar]
- 6.Zhao X., Alvarado D., Rainier S., Lemons R., Hedera P., Weber C.H., Tukel T., Apak M., Heiman-Patterson T., Ming L., et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 7.Dalpozzo F., Rossetto M.G., Boaretto F., Sartori E., Mostacciuolo M.L., Daga A., Bassi M.T., Martinuzzi A. Infancy onset hereditary spastic paraplegia associated with a novel atlastin mutation. Neurology. 2003;61:580–581. doi: 10.1212/01.wnl.0000078189.73611.df. [DOI] [PubMed] [Google Scholar]
- 8.Wilkinson P.A., Hart P.E., Patel H., Warner T.T., Crosby A.H. SPG3A mutation screening in English families with early onset autosomal dominant hereditary spastic paraplegia. J. Neurol. Sci. 2003;216:43–45. doi: 10.1016/s0022-510x(03)00210-7. [DOI] [PubMed] [Google Scholar]
- 9.Abel A., Fonknechten N., Hofer A., Dürr A., Cruaud C., Voit T., Weissenbach J., Brice A., Klimpe S., Auburger G., Hazan J. Early onset autosomal dominant spastic paraplegia caused by novel mutations in SPG3A. Neurogenetics. 2004;5:239–243. doi: 10.1007/s10048-004-0191-2. [DOI] [PubMed] [Google Scholar]
- 10.Dürr A., Camuzat A., Colin E., Tallaksen C., Hannequin D., Coutinho P., Fontaine B., Rossi A., Gil R., Rousselle C., et al. Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch. Neurol. 2004;61:1867–1872. doi: 10.1001/archneur.61.12.1867. [DOI] [PubMed] [Google Scholar]
- 11.Namekawa M., Ribai P., Nelson I., Forlani S., Fellmann F., Goizet C., Depienne C., Stevanin G., Ruberg M., Dürr A., Brice A. SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology. 2006;66:112–114. doi: 10.1212/01.wnl.0000191390.20564.8e. [DOI] [PubMed] [Google Scholar]
- 12.Zhu P.-P., Patterson A., Lavoie B., Stadler J., Shoeb M., Patel R., Blackstone C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 13.Zhu P.-P., Soderblom C., Tao-Cheng J.-H., Stadler J., Blackstone C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- 14.Praefcke G.J.K., McMahon H.T. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 15.Namekawa M., Muriel M.-P., Janer A., Latouche M., Dauphin A., Debeir T., Martin E., Duyckaerts C., Prigent A., Depienne C., et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis. Mol. Cell. Neurosci. 2007;35:1–13. doi: 10.1016/j.mcn.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y., Paik D., Bang S., Kang J., Chun B., Lee S., Bae E., Chung J., Kim J. Loss of spastic paraplegia gene atlastin induces age-dependent death of dopaminergic neurons in Drosophila. Neurobiol. Aging. 2008;29:84–94. doi: 10.1016/j.neurobiolaging.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Cole N.B., Sciaky N., Marotta A., Song J., Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirschberg K., Miller C.M., Ellenberg J., Presley J.F., Siggia E.D., Phair R.D., Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu P.-P., Patterson A., Stadler J., Seeburg D.P., Sheng M., Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J. Biol. Chem. 2004;279:35967–35974. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
- 20.Muglia M., Magariello A., Nicoletti G., Patitucci A., Gabriele A.L., Conforti F.L., Mazzei R., Caracciolo M., Ardito B., Lastilla M., et al. Further evidence that SPG3A gene mutations cause autosomal dominant hereditary spastic paraplegia. Ann. Neurol. 2002;51:794–795. doi: 10.1002/ana.10185. [DOI] [PubMed] [Google Scholar]
- 21.Sanderson C.M., Connell J.W., Edwards T.L., Bright N.A., Duley S., Thompson A., Luzio J.P., Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum. Mol. Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans K., Keller C., Pavur K., Glasgow K., Conn B., Lauring B. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc. Natl Acad. Sci. USA. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez C., Fujita H., Hubbard A., Sztul E. ER to Golgi transport: requirement for p115 at a pre-Golgi VTC stage. J. Cell Biol. 1999;147:1205–1221. doi: 10.1083/jcb.147.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dailey M.E., Bridgman P.C. Dynamics of the endoplasmic reticulum and other membranous organelles in growth cones of cultured neurons. J. Neurosci. 1989;9:1897–1909. doi: 10.1523/JNEUROSCI.09-06-01897.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banno T., Kohno K. Conformational changes of the smooth endoplasmic reticulum are facilitated by L-glutamate and its receptors in rat Purkinje cells. J. Comp. Neurol. 1998;402:252–263. [PubMed] [Google Scholar]
- 26.Lee C., Chen L.B. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- 27.Waterman-Storer C.M., Salmon E.D. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 1998;8:798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
- 28.Vedrenne C., Hauri H.-P. Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic. 2006;7:639–646. doi: 10.1111/j.1600-0854.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 29.Burkhardt J.K. The role of microtubule-based motor proteins in maintaining the structure and function of the Golgi complex. Biochim. Biophys. Acta. 1998;1404:113–126. doi: 10.1016/s0167-4889(98)00052-4. [DOI] [PubMed] [Google Scholar]
- 30.Ye B., Zhang Y., Song W., Younger S.H., Jan L.Y., Jan Y.N. Growing dendrites and axons differ in their reliance on the secretory pathway. Cell. 2007;130:717–729. doi: 10.1016/j.cell.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sannerud R., Marie M., Nizak C., Dale H.A., Pernet-Gallay K., Perez F., Goud B., Saraste J. Rab1 defines a novel pathway connecting the pre-Golgi intermediate compartment with the cell periphery. Mol. Biol. Cell. 2006;17:1514–1526. doi: 10.1091/mbc.E05-08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikonov A.V., Hauri H.-P., Lauring B., Kreibich G. Climp-63-mediated binding of microtubules to the ER affects the lateral mobility of translocon complexes. J. Cell Sci. 2007;120:2248–2258. doi: 10.1242/jcs.008979. [DOI] [PubMed] [Google Scholar]
- 33.Windpassinger C., Auer-Grumbach M., Irobi J., Patel H., Petek E., Hörl G., Malli R., Reed J.A., Dierick I., Verpoorten N., et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 34.Presley J.F., Cole N.B., Schroer T.A., Hirschberg K., Zaal K.J., Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 35.Blackstone C., Roberts R.G., Seeburg D.P., Sheng M. Interaction of the deafness-dystonia protein DDP/TIMM8a with the signal transduction adaptor molecule STAM1. Biochem. Biophys. Res. Commun. 2003;305:345–352. doi: 10.1016/s0006-291x(03)00767-8. [DOI] [PubMed] [Google Scholar]
- 36.Meijer I.A., Dion P., Laurent S., Dupré N., Brais B., Levert A., Puymirat J., Rioux M.F., Sylvain M., Zhu P.-P., et al. Characterization of a novel SPG3A deletion in a French-Canadian family. Ann. Neurol. 2007;61:599–603. doi: 10.1002/ana.21114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.