

Abstract
We have identified a mouse recessive mutation that leads to attenuated and hyperpermeable retinal vessels, recapitulating some pathological features of familial exudative vitreoretinopathy (FEVR) in human patients. DNA sequencing reveals a single nucleotide insertion in the gene encoding the low-density lipoprotein receptor-related protein 5 (LRP5), causing a frame shift and resulting in the replacement of the C-terminal 39 amino acid residues by 20 new amino acids. This change eliminates the last three PPP(S/T)P repeats in the LRP5 cytoplasmic domain that are important for mediating Wnt/β-catenin signaling. Thus, mutant LRP5 protein is probably unable to mediate its downstream signaling. Immunostaining and three-dimensional reconstructions of retinal vasculature confirm attenuated retinal vessels. Ultrastructural data further reveal that some capillaries lack lumen structure in the mutant retina. We have also verified that LRP5 null mice develop similar alterations in the retinal vasculature. This study provides direct evidence that LRP5 is essential for the development of retinal vasculature, and suggests a novel role played by LRP5 in capillary maturation. LRP5 mutant mice can be a useful model to explore the clinical manifestations of FEVR.
INTRODUCTION
The low-density lipoprotein (LDL) receptor-related protein 5 (LRP5) is a member of the LDL receptor family and belongs to a subfamily consisting of its mammalian homolog LRP6 and the Drosophila protein Arrow (1). LRP5 is a multifunctional cell surface receptor and functions as an indispensable element of the canonical Wnt/β-catenin signaling pathway by acting as a Wnt co-receptor (2). The Wnt signaling pathway plays crucial roles in development and disease (3–5). A receptor complex, composed of a member of the Frizzled family of serpentine receptors (6) and the coreceptor LRP5 or LRP6 (2), mediates Wnt/β-catenin signaling in vertebrates. In the presence of a Wnt ligand, the stabilized β-catenin can associate with nuclear T-cell factor/lymphoid enhancer factor (TCF/LEF), thus controlling the activation of Wnt-responsive genes.
Genetic studies have elucidated the involvement of mutant LRP5 proteins in various human diseases. The LRP5 gene, mapped to human chromosome 11q13 and mouse chromosome 19, encodes a single-span transmembrane protein with 1615 amino acid residues in humans and 1614 amino acid residues in mice (7). The human and mouse LRP5 proteins share 95% sequence identity (7). Mutations of LRP5 have been found to cause bone mass disorders (8) and ocular abnormalities in both humans and mice. Human LRP5 loss-of-function mutations cause autosomal-recessive osteoporosis-pseudoglioma syndrome (OPPG) with osteoporosis as well as blindness, due to the remnants of embryonic hyaloid vessels in the eye (9). Gain-of-function mutations of human LRP5 lead to high bone density (10,11). Mouse genetic studies have confirmed the importance of LRP5 in the regulation of bone mass density. LRP5-deficient mice have been generated by two research groups (12,13). One group has shown that a lack of LRP5 causes low bone mass and the persistence of embryonic hyaloid vessels in the eye, recapitulating human OPPG (12). The other group has further suggested the involvement of LRP5 in cholesterol metabolism and in the modulation of glucose-induced insulin secretion (13).
Recently, mutations of LRP5 have been linked to familial exudative vitreoretinopathy (FEVR) in humans (14). FEVR is a genetically heterogeneous eye disease characterized by incomplete vascularization of the peripheral retina in human patients. Mutations of the Wnt-receptor Frizzled 4 (FZD4) and its ligand Norrie have been demonstrated to cause autosomal-dominant FEVR in some patients (15), while the mutations of LRP5 have been linked to autosomal-dominant or autosomal-recessive FEVR in humans (14,16). However, the role of LRP5 in retinal vasculature development has not been carefully examined in experimental model systems.
Here, we report a new N-ethyl-N-nitrosourea (ENU)-induced recessive mouse mutant line (r18) that carries a frame shift mutation in the Lrp5 gene. Our studies of both r18- and LRP5-deficient mice demonstrate that these mutant mice develop abnormal retinal vasculature, recapitulating some symptoms of FEVR. Moreover, the extreme C-terminus is essential for the function of LRP5 in vivo. These LRP5 mutant mice provide a valuable experimental model for exploring the clinical manifestations of FEVR and for evaluating potential intervention strategies.
RESULTS
Identification of ENU-induced r18 mouse mutation
In an effort to find new genetic models for human eye diseases, we identified mouse r18 mutant founder from the G3 generation of ENU-mutagenized mice by fundus examination using an indirect ophthalmoscope. Subsequent mouse breeding and fundus examination confirmed that r18 is an autosomal recessive mutation. A normal fundus was observed in the heterozygous mutant retina (Fig. 1A) whereas the fundus image of homozygous mutant retina revealed pale and attenuated retinal arteries and mild hypopigmentation (Fig. 1B). Moreover, some of the r18 homozygous mutant mice developed retinal hemorrhage at weaning age, and the hemorrhage disappeared as the mice aged without any treatment (Fig. 1C, indicated by an arrow). Both heterozygous and homozygous mutant mice were viable and fertile without obvious gross abnormalities.
Figure 1.

Attenuated retinal vessels in ENU-induced r18 mutant mice. (A–C) Fundus photos of 2.5-month-old heterozygous and homozygous r18 mutant littermates. Only the homozygous r18 mutant mouse shows attenuated retinal vessels (B), and some homozygous mutant mice occasionally develop retinal hemorrhage (C, indicated by an arrow).
Homozygous mutant mice display abnormal leaky retinal vasculature
We further examined the properties of mouse retinal vessels to understand the fundus phenotypes. Fluorescein angiography was performed on 3-week-old littermates. Heterozygous mice showed normal retinal angiograms collected at different time points after the fluorescein dye injection (Fig. 2A–C). However, hyperpermeable retinal vessels were observed in homozygous mutant mice (Fig. 2D–F). The obvious leakage of fluorescein dye was detected at 60 s after dye injection, as the boundaries of retinal capillary vessels became fuzzy (Fig. 2E). At 90 s after the injection, the boundaries of capillary vessels were difficult to see and the whole retinal vasculature became indistinguishable from the hyperfluorescent background of the retina (Fig. 2F). Moreover, the density of retinal small vessels and/or capillaries in homozygous r18 mutant mice (Fig. 2D) was obviously lower than that of the heterozygous control (Fig. 2A). These data suggest that the retinal vasculature of homozygous r18 mutant mice is not fully developed, and this immature vasculature is not only leaky, but also has fewer small vessels and/or capillaries.
Figure 2.

Fluorescein angiograms of 3-week-old heterozygous and homozygous r18 mutant littermates. (A–C) The heterozygous mutant mouse shows normal retinal vasculature at 30 s (A), 60 s (B) and 90 s (C) after the injection of fluorescein dye. (D–F) The homozygous mutant mouse shows attenuated retinal vasculature and leakage of fluorescein dye in the retina detected at 60 s after the injection (E). The retinal vasculature becomes indistinguishable from the hyperfluorescent background at 90 s after the injection (F).
To further investigate the retinal vessel defect in r18 mutant mice, we performed immunostaining analysis using a specific antibody against CD31, an endothelial cell marker. In wild-type mice, a typical distribution of retinal vasculature was revealed by CD31-positive cells, with the vessels being predominantly located in the ganglion cell (GC) layer, inner plexiform layer (IPL) and outer plexiform layer (OPL) (Fig. 3A). However, in homozygous r18 mutant mice, CD31-positive cells were detected in the GC and the IPL layers, but rarely in the OPL layer (Fig. 3B). This result further suggests that the r18 mutation leads to an incomplete development of retinal vasculature, especially the vessel network of the OPL.
Figure 3.

Immunostaining of 2-week-old wild-type and r18 mutant mouse retinas. Retinal vessels are visualized with an anti-CD31 antibody (green), and cell nuclei are labeled by DAPI (blue). (A) A normal distribution of CD31-stained vessels is detected in ganglion cell (GC) layer, inner plexiform layer (IPL) and outer plexiform layer (OPL) in the wild-type retina. (B) An abnormal distribution of CD31-stained vessels is observed in the homozygous mutant retina, and fewer CD31-positive signals are observed in the outer plexiform layer. The scale bar is 50 µm.
Incomplete capillary network and lumen-less capillaries in the mutant retina
In order to obtain detailed information about the retinal vasculature in the r18 mutant mice, we examined the three-dimensional retinal vessel network using a lipophilic fluorescent dye (DiI; 1,1′-dioctadecyi-3,3,3′,3′-tetramethylindocarbocyanine perchlorate). The dye was transcardially perfused to stain vascular endothelial cells in the vessel lumen. A series of Z-stack images were collected from whole mount retinas of 6-week-old r18 littermates under a fluorescent microscope, followed by 3D reconstruction of the vessel network. Comparable regions of 3D retinal vasculature were presented to reveal the difference between heterozygous and homozygous mutant animals. A normal distribution of retinal vasculature was detected in the heterozygous mutant mice (Fig. 4A, the upper panel), and the 90°-rotated 3D image further revealed a typical three-layer vessel network in the retina (Fig. 4A, the lower panel). In contrast, the homozygous mutant retina showed an attenuated and abnormally patterned vasculature (Fig. 4B), lacking small vessels in the OPL (Fig. 4B, the lower panel). Moreover, a normal vessel network was present in the OPL of heterozygous retina, but not in that of homozygous mutant retina (Fig. 4C and D). Transmission electron microscopic (TEM) analysis revealed that all capillaries of wild-type retina formed lumens in the IPL (Fig. 5A and C) while some endothelial cells in the capillaries of the r18 homozygous mutant failed to form a lumen in the IPL (Fig. 5B and D). Also, endothelial cells were wrapped by a thicker basal lamina in homozygous mutant retina compared with heterozygous mutant retina. Thus, immature endothelial cells in lumen-less capillaries are associated with an attenuated vessel network in homozygous r18 mutant retina, as evidenced by angiogram, 3D vasculature reconstruction and TEM analysis.
Figure 4.

Three-dimensional images of retinal vasculature labeled with fluorescent dye in 6-week-old r18 littermates. (A and B) A normal three-layer vessel network is present in the heterozygous control retina (A), whereas an attenuated and disorganized vessel network is present in the homozygous r18 mutant retina (B). Upper panels are front views of 3D retinal vasculature. The lower panels are rotated 90° relative to the upper panels. The bottom, middle and upper layers represent vessels in the OPL, IPL and GC layer, respectively. (C and D) Vessel networks are located in the OPL of heterozygous (C) and homozygous (D) retinas. Note the fully established vessel network in the heterozygous mutant retina, but attenuated vessels of incomplete network in the homozygous mutant retina.
Figure 5.
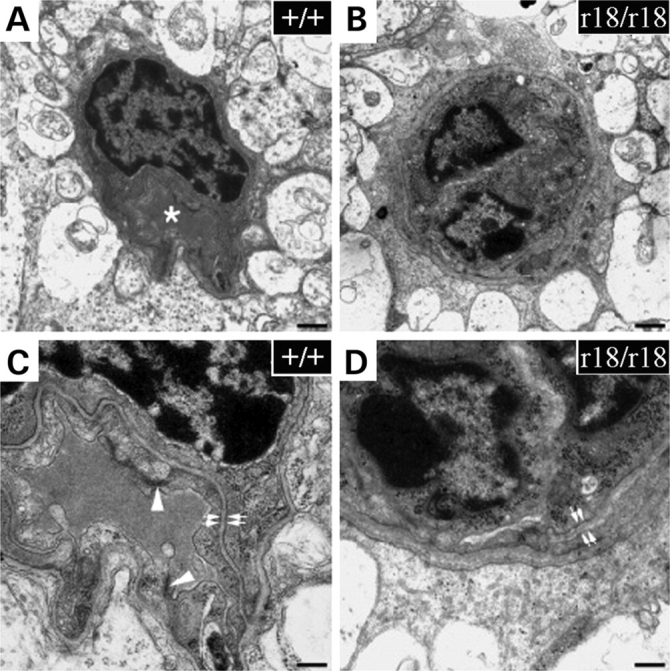
Electron microscopic images of capillaries in the retinal inner plexiform layer of 4-week-old wild-type (A and C) and homozygous r18 (B and D) mice. The lumen of a wild-type capillary is shown (A, asterisk). However, lumen-less capillaries are often observed in the r18 mutant retina (B). Compared with the control, capillaries in the r18 mutant retina show a much thicker basal lamina (D, double white arrows) and an absence of tight junctions between endothelial cells (C, arrowhead). The scale bars are 500 nm for (A) and (B), 200 nm for (C) and (D).
The r18 mutation is a single nucleotide insertion in exon 23 of the Lrp5 gene
To identify the causative mutation of the r18 phenotype, we performed whole-genome linkage analysis. Using genomic DNA samples from 35 meioses, we mapped the r18 locus to mouse chromosome 19 with a peak logarithm of the odds (LOD) score of 4.6 (Fig. 6A). Additional fine mapping was performed by genotyping 472 backcross mice with standard microsatellite markers found in Ensembl and Mouse Genome databases, and three new markers, r18–new9 (Fig. 6B, 5 Mb from the centromere; Ensembl distance), r18–new12 (3.7 Mb from the centromere; Ensembl distance) and r18–new18 (3.2 Mb from the centromere; Ensembl distance), which we identified. The causative gene was localized to an interval between the centromere and marker r18–new9 on chromosome 19 (Fig. 6B). Among all tested meioses, the genotype data for the marker D19Mit68 always matched mouse phenotypes. Lrp5 is located close to this marker based on the Ensembl Mouse Genome database. Since LRP5 mutations have been reported to affect the formation of retinal vessels in FEVR in humans (12), we performed DNA sequencing analysis of the Lrp5 gene in r18 mutant mice.
Figure 6.

Chromosome mapping and identification of the gene mutated in r18 mice. (A) Whole-genome linkage analysis suggests that the r18 mutant locus is located on chromosome 19 with a LOD score of 4.6. (B) Fine mapping localizes the r18 locus between marker r18–new9 and the centromere, near the vicinity of the marker D19Mit68, based on the Ensembl Mouse Genome Server. (C) DNA sequencing reveals a single nucleotide insertion (C, in red color) in the Lrp5 gene of the r18 mutant mice, which leads to a frame shift after the 1576th codon of the LRP5 protein. This frame shift is predicted to result in the replacement of the last 39 C-terminal amino acid residues of the LRP5 protein by a new polypeptide of 20 amino acids and a premature termination codon. Amino acid sequence alignment shows that the r18 mutation eliminates the last three of five PPP(S/T)P repeats (labeled in red) at the C-terminus of the LRP5 protein.
cDNA was amplified from total RNA isolated from homozygous r18 mutant retina using reverse transcriptase-polymerase chain reaction (RT-PCR) with specific primers covering the entire coding region (4842 bp) of Lrp5 mRNA. DNA sequencing of RT-PCR fragments revealed the addition of one C nucleotide. This single nucleotide insertion resulted in a frame shift from the 1576th codon for the amino acid proline (Fig. 6C). This frame shift was predicted to replace the last 39 amino acid residues at the C-terminus of LRP5 protein with 20 new amino acid residues and to result in premature termination at codon 1596. Hereafter, we designate the r18 mutation as Lrp5r18. The Lrp5r18 mutation disrupts the last three of the five PPP(S/T)P repeats in the C-terminal intracellular domain of LRP5 (Fig. 6C). Since the PPP(S/T)P motif is important for mediating Wnt downstream signaling, we hypothesize that this C-terminal-truncated LRP5 protein is a loss-of-function mutant and is probably unable to mediate certain signaling needed for normal development of retinal vasculature. Therefore, we further examined the retinal vasculature in LRP5 null mutant mice.
Retinal vasculature phenotypes are similar in LRP5 null and Lrp5r18 mutant mice
Fluorescein angiography was performed on 1-month-old heterozygous and homozygous null mutant littermates. Heterozygous LRP5 knockout mice showed normal retinal angiograms (Fig. 7A). However, homozygous knockout mice displayed hyperfluorescent retinal vasculature caused by hyperpermeable retinal vessels (Fig. 7B). CD31 immunostaining of heterozygous knockout retina revealed a normal distribution of retinal vessels in the GC layer, IPL and OPL (Fig. 7C). However, CD31-positive cells were predominantly detected in the GC layer and IPL, but rarely in the OPL of homozygous knockout retina (Fig. 7D). These data confirm that the LRP5 null mutation and the LRP5 truncation mutation (Lrp5r18) lead to similar defects of retinal vasculature.
Figure 7.

Characterization of the retinal vasculature in LRP5 knockout mice. (A and B) Fluorescein angiograms of LRP5 heterozygous and homozygous knockout mice taken at 90 s after the dye injection. (C and D) Immunostaining of retina sections of 3-week-old LRP5 heterozygous and homozygous knockout mice. Retinal vessels are stained with anti-CD31 antibody (green) and cell nuclei are labeled by DAPI (blue). The distribution of CD31-stained vessels appears normal in the heterozygous mutant retina (C) while fewer CD31-stained retinal vessels appear in the outer plexiform layer of the homozygous mutant retina (D). The scale bar is 50 µm.
DISCUSSION
This work demonstrates that LRP5 is required for the development of retinal vasculature, and strongly suggests that LRP5 plays a novel role in the maturation and lumen formation of capillaries in the inner and outer plexiform layers. The Lrp5r18 mutation results in a truncated LRP5 protein that lacks the last three PPP(S/T)P repeats in the C-terminus. We further confirm that LRP5 knockout mice also develop similar defects of retinal vasculature. Thus, our findings suggest that the Lrp5r18 mutation is a loss-of-function allele and that LRP5 plays important roles in the development of retinal vasculature as well as the lumen formation of capillaries.
Both Lrp5r18 mutants and LRP5 knockout mice recapitulate the incomplete vascularization of the peripheral retina observed in human FEVR patients. FEVR is a genetically heterogeneous disease. Three inheritance forms (X-linked recessive, autosomal dominant and autosomal recessive) have been described in humans. The X-linked recessive FEVR has been associated with mutations in the Norrie disease gene (17,18); autosomal dominant FEVR has been linked to mutations of the Wnt receptor FZD4 and its coreceptor LRP5 (14,15,19,20); LRP5 mutations have also been reported to cause autosomal recessive FEVR (16). Norrin, the protein product of the Norrie disease gene, has been suggested to act as a ligand for FZD4 (21). Knockout mice of either FZD4 (21) or Norrie (22,23) develop defective intraretinal vasculature. Several in vitro studies have suggested that LRP5 acts as a co-receptor for FZD4 (2,24–26). Therefore, FEVR is probably caused by altered signaling downstream of Norrie, FZD4 and LRP5.
Previous studies have shown that the N-terminal extracellular domain of LRP5 binds to Wnt ligands while its C-terminal intracellular domain is important for signaling events (2). The C-terminal intracellular domain of LRP5 has 207 amino acid residues with five PPP(S/T)P motifs that are presumably essential for its signaling (2). It has been suggested that LRP5 interacts with Axin through its C-terminal intracellular domain. In the Wnt/β-catenin signaling pathway, Axin has been proposed to function as a scaffold that forms a complex with GSK-3β and β-catenin, and to promote GSK-3β-dependent phosphorylation of β-catenin (27). Mutant LRP5 proteins lacking the extracellular domain constitutively bind to Axin and induce TCF/LEF-1 activation by destabilizing Axin and stabilizing β-catenin in vitro (28). The intracellular domains of both LRP5 and LRP6 have been shown to increase free β-catenin levels constitutively and activate TCF/LEF-1 (28,29). Mutant LRP5 proteins without the last three PPP(S/T)P motifs are unable to bind to Axin and fail to activate LEF-1 (28). Studies of LRP6 have also suggested the importance of PPP(S/T)P motif in the Wnt signaling. The transfer of a PPP(S/T)P motif from LRP6 to the LDL receptor fully activates the Wnt pathway (30), and phosphorylated PPP(S/T)P motif provides a docking site for Axin (30). Thus, the PPP(S/T)P motifs of LRP5 and LRP6 are essential for mediating β-catenin signaling in vitro.
The Lrp5r18 mutation disrupts the C-terminal 39 amino acids of LRP5, which include the last three PPP(S/T)P repeats. The absence of these PPP(S/T)P repeats probably eliminates the docking sites for Axin. Thus, LRP5 truncated mutant proteins are probably unable to bind Axin to promote subsequent stabilization of β-catenin, thereby suppressing the activation of downstream transcriptional factors and the expression of genes needed for the development of retinal vasculature. However, it is also possible that the C-terminal truncation perturbs the stability or intracellular trafficking of LRP5. Future studies of the Lrp5r18 mutant will clarify the specific role(s) of the C-terminal region and the PPP(S/T)P motifs of LRP5.
In summary, this study provides the first direct in vivo evidence of the importance of LRP5 in the development of retinal vasculature. The extreme C-terminus is essential for the function of LRP5 in vivo. We believe that the Lrp5r18 mutant will be an appropriate animal model for further elucidating the molecular mechanism that controls the development of retinal vasculature, as well as for understanding the underlying molecular mechanism of FEVR.
MATERIALS AND METHODS
ENU-induced mice
All studies and examinations were conducted in accordance with a protocol for the Use of Animals in Research, approved by the ACUC committee at University of California, Berkeley. ENU-mutagenesis and breeding of the mice were performed as previously described (31). Briefly, male wild-type C57BL/6J mice were intraperitoneally injected with ENU (90 mg/kg body weight) weekly for three times. Three months after the injection, each mouse was bred to wild-type C57BL/6J female mice to produce G1 mice, which were screened for dominant eye phenotypes. The G1 mice were mated with wild-type C57BL/6J mice to generate the G2 mice, and the G2 mice were backcrossed with the G1 mice to generate the G3 mice, which were screened for recessive eye phenotypes.
Fundus examination and fluorescein angiography
Fundus examination
Mouse pupils were dilated with a mixed ophthalmic solution containing 0.5% atropine sulfate (E. Fougera & Co) and 1.25% phenylephrine hydrochloride (Wilson Ophthamic). An indirect ophthalmoscope (Keeler) with a super vitreoFundus or super 66 stereo Fundus lenses was used for examining abnormalities of retinal vessels and retinal pigment distribution. Mouse fundus was photographed with a Kowa Genesis fundus camera for small animals (Tokyo, Japan).
Fluorescein angiography
Mouse pupils were dilated, and mice were intraperitoneally injected with 25% Angiofluor™ (Alliance Pharmaceutical, Inc.) at a dose of 0.01 ml/5 g of mouse body weight. Photos were taken at appropriate intervals with a camera containing a barrier filter for fluorescein angiography. Kodak 800 ASA color film was used for photograph. Developed negative film was scanned and processed by Photoshop for digital images.
3D images of retinal vasculature
Mice were overdosed with carbon dioxide and perfused immediately through left cardiac ventricle with a series of solutions. Phosphate-buffered saline (PBS) solution was first perfused to wash out the blood, then a solution containing 0.1 mg/ml of lipophilic dye, DiI (Sigma), and followed by a fixative solution of 4% formaldehyde in PBS. Enucleated eyeballs were collected and further fixed in 4% formaldehyde in PBS for overnight at 4°C. Flat-mounted retinas, prepared from these postfixed eyeballs, were used for collecting Z-stack fluorescent images of the retinal vasculature by Zeiss Axiovert 200 fluorescent microscope equipped with Apotome and Axiocam. Three-dimensional retinal vasculature was constructed from these Z-stack images with the Axiovision 4 MOD Inside 4D program.
Histology, electron microscopic analysis and immunohistochemistry
Histology
Mice were euthanized by CO2 asphyxiation according to the ACUC-approved animal protocol. Dissected eyes were fixed in a solution containing 2.5% gluteraldehyde, 2% paraformaldehyde in 0.1 m cacodylate buffer (pH, 7.2) for at least 24 h at 4°C. Fixed eyes were post-fixed in 1% aqueous OsO4, stained en bloc with 2% aqueous uranyl acetate, and followed by dehydration through graded acetone. Samples were embedded in Epon 12-Araldyte 502 resin (Ted Pella). For light microscopy, 1 µm sections were collected on glass slides and stained with Toluidine blue. Images were acquired via a Zeiss Axiovert 200 light microscope with a digital camera.
Electron microscopic analysis
Ultrathin sections (80 nm) were collected from re-trimmed plastic blocks containing the areas of interest. Sections were stained with Sato’s triple lead solution before being examined. Images were taken under a JEOL JEM-1200EX II electron microscope (JEOL, Tokyo, Japan).
Immunohistochemistry
Mouse eyes were fixed with 4% paraformaldehyde in PBS for 2 h at 4°C. After washing with PBS, fixed samples were placed into 30% sucrose/PBS for overnight at 4°C before embedded in Tissue-Tek O.C.T. compound. Frozen sections (∼10 µm) were stained with a rat anti-CD31 antibody (BD Pharmingen), and followed by an anti-rat FITC-labeled secondary antibody, and then mounted with DAPI VectorShield mounting medium (Vector Laboratories, Inc.). A Zeiss Axiovert 200 fluorescent microscope with Axiocam was used to examine and collect the digital images.
Genomic linkage analysis
Homozygous r18 mutant mice (in C57BL/6J strain background) were mated to wild-type C3H/HeN mice to generate heterozygous hybrid mice. Heterozygous hybrid mice were crossed to homozygous r18 mutant mice to generate backcross mice. The backcross mice were phenotyped and tailed for genotyping. A total of 59 microsatellite markers were used for genome-wide linkage analysis (31). After the chromosomal linkage was identified, we further performed fine mapping. Marker sequence information (D19Mit68, D19Mit59 and D19Mit69) was obtained from the Ensembl Mouse database. Three new mapping markers were identified for informative simple length polymorphisms amplified with the following primer pairs: r18–new18, forward primer TGACACAAAGTTCAGCATAGGTAAGAGTGGC, and reverse primer TGTGATTCTGGTCCAAGGTTATGACTGC; r18–new12, forward primer ATAATGCTGTGAGCTAGATCCTGTATAAGCCC, and reverse primer AGTGACCTCTCTGCAAGAATGGCAAAC; r18–new9, forward primer AGGCAGGTGTCTATGGAGAAGGCAG, and reverse primer AAACTTCTGCTCCCACCACTTCCAC.
DNA sequencing
Total RNA was isolated from the retinas of homozygous mutant mice by using the TRIzol® reagent (Invitrogen Life Technologies), and cDNAs were synthesized with the Superscript™ first-strand synthesis system for RT-PCR kit (Invitrogen Life Technologies). The coding region of LRP5 was amplified by various primer pairs with Platinum® pfx DNA polymerase (Invitrogen Life Technologies). PCR fragments with overlapping regions were sequenced.
FUNDING
This work was supported by grant EY013849 (X.G.) from the National Eye Institute.
ACKNOWLEDGEMENTS
We thank Dr. Lawrence Chan at Baylor College of Medicine for providing the LRP5 knockout mice and Dr. Rong Wen at University of Pennsylvania for generously sharing the lipophilic dye DiI and vessel painting technique.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hussain M.M., Strickland D.K., Bakillah A. The mammalian low-density lipoprotein receptor family. Annu. Rev. Nutr. 1999;19:141–172. doi: 10.1146/annurev.nutr.19.1.141. [DOI] [PubMed] [Google Scholar]
- 2.He X., Semenov M., Tamai K., Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663–1677. doi: 10.1242/dev.01117. [DOI] [PubMed] [Google Scholar]
- 3.Wodarz A., Nusse R. Mechanisms of Wnt signaling in development. Annu. Rev. Cell Dev. Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 4.Moon R.T. Wnt/beta-catenin pathway. Sci. STKE. 2005 doi: 10.1126/stke.2712005cm1. 2005, cm1. [DOI] [PubMed] [Google Scholar]
- 5.van Es J.H., Barker N., Clevers H. You Wnt some, you lose some: oncogenes in the Wnt signaling pathway. Curr. Opin. Genet. Dev. 2003;13:28–33. doi: 10.1016/s0959-437x(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 6.Perrimon N. Serpentine proteins slither into the wingless and hedgehog fields. Cell. 1996;86:513–516. doi: 10.1016/s0092-8674(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 7.Hey P.J., Twells R.C., Phillips M.S., Yusuke N., Brown S.D., Kawaguchi Y., Cox R., Guochun X., Dugan V., Hammond H., et al. Cloning of a novel member of the low-density lipoprotein receptor family. Gene. 1998;216:103–111. doi: 10.1016/s0378-1119(98)00311-4. [DOI] [PubMed] [Google Scholar]
- 8.Ferrari S.L., Deutsch S., Antonarakis S.E. Pathogenic mutations and polymorphisms in the lipoprotein receptor-related protein 5 reveal a new biological pathway for the control of bone mass. Curr. Opin. Lipidol. 2005;16:207–214. doi: 10.1097/01.mol.0000162326.62419.e4. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y., Slee R.B., Fukai N., Rawadi G., Roman-Roman S., Reginato A.M., Wang H., Cundy T., Glorieux F.H., Lev D., et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 10.Little R.D., Carulli J.P., Del Mastro R.G., Dupuis J., Osborne M., Folz C., Manning S.P., Swain P.M., Zhao S.C., Eustace B., et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyden L.M., Mao J., Belsky J., Mitzner L., Farhi A., Mitnick M.A., Wu D., Insogna K., Lifton R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 12.Kato M., Patel M.S., Levasseur R., Lobov I., Chang B.H., Glass D.A., II, Hartmann C., Li L., Hwang T.H., Brayton C.F., et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujino T., Asaba H., Kang M.J., Ikeda Y., Sone H., Takada S., Kim D.H., Ioka R.X., Ono M., Tomoyori H., et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc. Natl. Acad. Sci. USA. 2003;100:229–234. doi: 10.1073/pnas.0133792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toomes C., Bottomley H.M., Jackson R.M., Towns K.V., Scott S., Mackey D.A., Craig J.E., Jiang L., Yang Z., Trembath R., et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 2004;74:721–730. doi: 10.1086/383202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robitaille J., MacDonald M.L., Kaykas A., Sheldahl L.C., Zeisler J., Dube M.P., Zhang L.H., Singaraja R.R., Guernsey D.L., Zheng B., et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 2002;32:326–330. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 16.Jiao X., Ventruto V., Trese M.T., Shastry B.S., Hejtmancik J.F. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am. J. Hum. Genet. 2004;75:878–884. doi: 10.1086/425080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z.Y., Battinelli E.M., Fielder A., Bundey S., Sims K., Breakefield X.O., Craig I.W. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 1993;5:180–183. doi: 10.1038/ng1093-180. [DOI] [PubMed] [Google Scholar]
- 18.Shastry B.S., Hejtmancik J.F., Trese M.T. Identification of novel missense mutations in the Norrie disease gene associated with one X-linked and four sporadic cases of familial exudative vitreoretinopathy. Hum. Mutat. 1997;9:396–401. doi: 10.1002/(SICI)1098-1004(1997)9:5<396::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Toomes C., Downey L.M., Bottomley H.M., Scott S., Woodruff G., Trembath R.C., Inglehearn C.F. Identification of a fourth locus (EVR4) for familial exudative vitreoretinopathy (FEVR) Mol. Vis. 2004;10:37–42. [PubMed] [Google Scholar]
- 20.Qin M., Hayashi H., Oshima K., Tahira T., Hayashi K., Kondo H. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathy with mutations in the LRP5 and/or FZD4 genes. Hum. Mutat. 2005;26:104–112. doi: 10.1002/humu.20191. [DOI] [PubMed] [Google Scholar]
- 21.Xu Q., Wang Y., Dabdoub A., Smallwood P.M., Williams J., Woods C., Kelley M.W., Jiang L., Tasman W., Zhang K., et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 22.Richter M., Gottanka J., May C.A., Welge-Lussen U., Berger W., Lutjen-Drecoll E. Retinal vasculature changes in Norrie disease mice. Invest. Ophthalmol. Vis. Sci. 1998;39:2450–2457. [PubMed] [Google Scholar]
- 23.Rehm H.L., Zhang D.S., Brown M.C., Burgess B., Halpin C., Berger W., Morton C.C., Corey D.P., Chen Z.Y. Vascular defects and sensorineural deafness in a mouse model of Norrie disease. J. Neurosci. 2002;22:4286–4292. doi: 10.1523/JNEUROSCI.22-11-04286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinson K.I., Brennan J., Monkley S., Avery B.J., Skarnes W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 25.Wehrli M., Dougan S.T., Caldwell K., O’Keefe L., Schwartz S., Vaizel-Ohayon D., Schejter E., Tomlinson A., DiNardo S. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- 26.Wu C.H., Nusse R. Ligand receptor interactions in the Wnt signaling pathway in Drosophila. J. Biol. Chem. 2002;277:41762–41769. doi: 10.1074/jbc.M207850200. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda S., Kishida S., Yamamoto H., Murai H., Koyama S., Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao J., Wang J., Liu B., Pan W., Farr G.H., III, Flynn C., Yuan H., Takada S., Kimelman D., Li L., et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol. Cell. 2001;7:801–809. doi: 10.1016/s1097-2765(01)00224-6. [DOI] [PubMed] [Google Scholar]
- 29.Mi K., Johnson G.V. Role of the intracellular domains of LRP5 and LRP6 in activating the Wnt canonical pathway. J. Cell Biochem. 2005;95:328–338. doi: 10.1002/jcb.20400. [DOI] [PubMed] [Google Scholar]
- 30.Tamai K., Zeng X., Liu C., Zhang X., Harada Y., Chang Z., He X. A mechanism for Wnt coreceptor activation. Mol. Cell. 2004;13:149–156. doi: 10.1016/s1097-2765(03)00484-2. [DOI] [PubMed] [Google Scholar]
- 31.Du X., Tabeta K., Hoebe K., Liu H., Mann N., Mudd S., Crozat K., Sovath S., Gong X., Beutler B. Velvet, a dominant Egfr mutation that causes wavy hair and defective eyelid development in mice. Genetics. 2004;166:331–340. doi: 10.1534/genetics.166.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]