

Abstract
In response to DNA damage, the Fanconi anemia (FA) core complex functions as a signaling machine for monoubiquitination of FANCD2 and FANCI. It remains unclear whether this complex can also participate in subsequent DNA repair. We have shown previously that the FANCM constituent of the complex contains a highly conserved helicase domain and an associated ATP-dependent DNA translocase activity. Here we show that FANCM also possesses an ATP-independent binding activity and an ATP-dependent bi-directional branch-point translocation activity on a synthetic four-way junction DNA, which mimics intermediates generated during homologous recombination or at stalled replication forks. Using an siRNA-based complementation system, we found that the ATP-dependent activities of FANCM are required for cellular resistance to a DNA-crosslinking drug, mitomycin C, but not for the monoubiquitination of FANCD2 and FANCI. In contrast, monoubiquitination requires the entire helicase domain of FANCM, which has both ATP dependent and independent activities. These data are consistent with participation of FANCM and its associated FA core complex in the FA pathway at both signaling through monoubiquitination and the ensuing DNA repair.
INTRODUCTION
Fanconi anemia (FA) is a rare genetic disorder characterized by genomic instability, bone marrow failure, developmental abnormalities and cancer predisposition (1,2). A hallmark feature of FA is the hypersensitivity of patient cells to drugs that induce DNA interstrand crosslinks (ICLs), including mitomycin C (MMC) and cisplatin (1–3). This feature suggests that FA cells are defective at one or more steps in the repair pathway of the ICLs. Recently, the interest in FA has intensified as three genes implicated in the disease, FANCD1, FANCN and FANCJ, were found to be breast cancer susceptibility (BRCA) genes, BRCA2, PALB2 and BRIP1 (4–12). Moreover, FA gene products are found to act as signaling and DNA-processing molecules in a DNA damage response network (FA–BRCA network). The network includes many proteins that help to maintain genome integrity, particularly BRCA1, ATR and BLM (3). Thus, studies of FA proteins should reveal part of mechanisms of genome maintenance.
FA has at least 13 complementation groups (FANC- A, B, C, D1, D2, E, F, G, I, J, L, M and N), each of which is associated with mutation in an eponymous gene. The FA gene products can be classified into three groups, which function at different stages of the FA pathway (3). One group comprises eight FA proteins (FANC-A, B, C, E, F, G, L and M), which are all components of the FA core complex (13–15). This complex contains a ubiquitin ligase, FANCL (13), and was suggested to function as a molecular machine to monoubiquitinate FANCD2 and FANCI in response to DNA damage or replication signals via the ATR pathway (15–17). A second group consists of FANCD2 and FANCI, which form the FA ‘ID’ complex acting downstream of the core complex (16,17). The ubiquitinated ID complex associates with chromatin and colocalizes with BRCA1 and γH2AX at DNA damage sites (18,19). The third group includes FANCD1 (BRCA2), FANCN (PALB2) and FANCJ (BRIP1) (4–9). These proteins are not involved in monoubiquitination of the ID complex and could function either downstream or in a pathway parallel to that of the other FA proteins.
Among the open questions are how do FA proteins interact with DNA, and what is the functional significance of these interactions? For the FA core complex, interactions with DNA are likely mediated by FANCM and FAAP24, the only known components with DNA-interacting domains and activities. FANCM has helicase and ERCC4-like endonuclease domains (15,20). It forms a heterodimer with FAAP24, which also contains an ERCC4-like domain (21). The FANCM-FAAP24 heterodimer belongs to the endonuclease family of XPF(ERCC4)-ERCC1, Mus81-EME1 and archaeal Hef [which forms homodimers and also has helicase activity (22,23)]. However, neither endonuclease nor helicase activity has been detected for human FANCM, the FANCM-FAAP24 heterodimer or the FA core complex (15,21). The fact that FANCM lacks the DNA-processing activities observed in XPF-ERCC1 and Hef suggests that FANCM functions differently compared with the other members of this family.
To date, FANCM has been shown to have an ATP-dependent DNA translocase property (15), whereas FAAP24 has a DNA-binding activity and can target FANCM to single-stranded DNA (ssDNA) or Y-shaped DNA, which mimics intermediates generated during replication or repair (21). Because both FANCM and FAAP24 are required for FANCD2 monoubiquitination, it was suggested that their interactions with DNA recruit the FA core complex to damaged sites to allow ubiquitination to occur (15,21). Here we present evidence that FANCM has additional activities that suggest its direct participation in DNA repair.
RESULTS
The FANCM helicase domain has specific binding affinity to four-way junction and fork DNA
We have shown previously that FANCM has no detectable helicase activity to unwind multiple DNA structures, including a linear DNA with both single and double-stranded regions, a replication fork and a four-way junction DNA [4WJ; also termed as Holliday Junction (HJ) in previous work] (15). Instead, we noticed that both the wildtype and the ATPase-point mutant (K117R) of FANCM reduced the gel mobility of the fork and 4WJ, but not linear DNA, in an ATP-independent manner [Supplementary Fig. 4 in reference (15)]. This implies that FANCM has a specific DNA-binding affinity to fork and 4WJ. Consistent with these observations, Constantinou and colleagues (24) have recently shown that a recombinant baculoviral FANCM protein has a similar binding specificity.
FANCM has two known DNA-interacting motifs: N-terminal helicase and C-terminal endonuclease domains. Because the endonuclease domain has low DNA-binding activity, and its association with FAAP24 formed a heterodimer which preferentially binds to ssDNA and Y-shaped DNA, but not fork and 4WJ DNA (21), we infer that the observed binding specificity in FANCM is likely derived from the helicase domain.
We expressed and purified a recombinant baculoviral protein containing only the N-terminal helicase domain of FANCM (rFANCM-Hel) (Fig. 1A). The recombinant protein displayed several features that mimic those of full-length FANCM purified from EBNA-293 cells, including an ATPase activity that can be stimulated by ssDNA and dsDNA, with absence of detectable helicase activity to linear, fork, 4WJ and other types of DNA (Supplementary Material, Fig. S1a–d). Moreover, rFANCM-Hel also exhibited ATP-independent binding affinity to fork and 4WJ structures, but not linear DNA containing both single and double-stranded regions (Supplementary Material, Fig. S1b–d). These findings support the notion that the helicase domain is responsible for the observed binding specificity of FANCM.
Figure 1.

FANCM helicase domain has high binding affinity for 4WJ and fork DNA. (A) Coomassie-stained SDS–gel showing the baculoviral recombinant protein containing the helicase domain of FANCM (rFANCM-Hel). (B) A gel-shift assay showing DNA-binding preference of rFANCM-Hel using a variety of synthetic DNA substrates illustrated at the bottom. The p32-labeled probe is denoted with an asterisk. The arrow indicates DNA-bound rFANCM-Hel. [(C) and (D)] Gel-shift assays showing binding of rFANCM-Hel to fork (C) and 4WJ (D) at increasing protein concentrations. (E) A competition experiment showing rFANCM-Hel has higher affinity to 4WJ than fork or dsDNA. The p32-labeled 4WJ DNA was used at 0.6 nm, and the non-labeled competitor DNA concentration was as high as about 200-fold in excess (125 nm). The quantifications of (C)–(E) are shown in Supplementary Material, Figure S2.
Screening of more DNA structures showed that rFANCM-Hel has a preference to bind 4WJ (Fig. 1B). A titration experiment also revealed that rFANCM-Hel has higher affinity to 4WJ than to fork DNA (Fig. 1C and D; see quantification in Supplementary Material, Fig. S2a). Moreover, a competition experiment was performed by adding increasing concentrations of unlabeled 4WJ, fork or dsDNA to the radio-labeled 4WJ DNA probe in the gel-shift assay. The results showed that 4WJ is the strongest competitor, whereas dsDNA is the weakest, with fork in between (Fig. 1E; see quantification in Supplementary Material, Fig. S2b). These data suggest that 4WJ is a preferred binding substrate for FANCM helicase domain.
FANCM has a branch-migration activity for 4WJ in both directions
Recently, RAD54, a helicase-domain-containing protein involved in homologous recombination (HR), was shown to possess a branch-migration (BM) activity for a movable 4WJ (or HJ) substrate (25). This new 4WJ was designed to have terminal DNA branches homologous to each other, so that the junction can move freely in one direction and allow the two DNA duplexes to separate without any need for a helicase activity [The 4WJ used in our previous study does not have homologous terminal branches and it cannot be dissociated by FANCM (15)]. Interestingly, RAD54 has several features that resemble those of FANCM (see Discussion). The similarity between the two proteins raised the possibility that FANCM may also possess BM activity resembling that of RAD54. Indeed, Constantinou and colleagues (24) have demonstrated that a recombinant baculoviral FANCM protein possesses BM activity for a movable 4WJ. They termed this BM activity of FANCM as ‘branch point translocation’ (BPT). To be consistent, we will also use their terminology in what follows. One advantage of using BPT, but not BM, is to avoid the inference that FANCM is involved in HR, as the latter term has often been associated to the migration of the HJ (see Discussion).
Using the movable 4WJ substrate described for RAD54 (25), we found that the recombinant FANCM from human EBNA-HEK293 cells (15) (Fig. 2A) also displayed BPT activity (Fig. 2B). This activity is ATP-hydrolysis dependent, because a point mutant that inactivates the ATPase activity of FANCM (K177R) (15) has no detectable activity. These results are consistent with the findings by Constantinou and colleagues using the baculoviral protein.
Figure 2.

FANCM has branch-migration (BM) activity for movable 4WJ DNA in both directions. (A) A silver-stained SDS–gel showing the Flag-tagged FANCM of either wildtype (WT) or the K117R ATPase point mutant isolated from EBNA-HEK293 cells as described previously (15). A mock purification was done using EBNA-HEK293 cells that do not express any tagged protein. (B) A BM assay showing wildtype FANCM can branch-migrate a movable 4WJ (25), but the point mutant (K117R) cannot. The 4WJ substrate (lane 3), a duplex intermediate used to assemble the 4WJ (lane 2), and the final BM product (lane 1) are illustrated on the left. This 4WJ has a 1 bp mismatch to inhibit spontaneous BM, which is illustrated as two angles in the BM product. The spontaneous BM was monitored using the DNA substrate without any added protein (none). The labeled ssDNA is marked with an asterisk. [(C) and (D)] BM assays showing that FANCM can branch-migrate partial 4WJ substrates in directions of both 5′ to 3′ (C) or 3′ to 5′ (D). The substrates have either a 5′ (C) or a 3′ (D) protruding ssDNA, and only BM in the aforementioned direction (marked by an arrow) can yield the expected BM product. A possible binding activity between FANCM and the substrates was observed (marked by an arrowhead). The unassembled duplex refers to a DNA intermediate used to assemble the partial 4WJ substrate. We were unable to fully convert these intermediates into the final substrates despite repeated efforts. Control experiments showed that these duplex intermediates should not interfere with the BM assay, because they cannot be dissociated by FANCM to produce ssDNA that has mobility similar to the BM product (data not shown).
RAD54 also has BM activity for partial 4WJ DNA in which one of the four arms of 4WJ is an ssDNA (25). It can catalyze BM on substrates that have either a 5′ or a 3′ protruding end, which indicates that the direction of BPT could be either 5′ to 3′ or 3′ to 5′, relative to the displaced ssDNA (25). We found that FANCM promoted BPT for the same two partial 4WJ substrates used in the RAD54 study, indicating that the BPT activity of FANCM could also be in either direction (Fig. 2C and D).
We failed to detect BPT or dsDNA translocase activity (using triple-helix unwinding assay) in rFANCM-Hel protein (data not shown). Regions of FANCM deleted in rFANCM-Hel are likely needed for these activities because Constantinou and colleagues (24) have observed BPT activity in full-length FANCM isolated from baculovirus.
An siRNA-based system to test for the function of FANCM in vivo
To investigate the function of the FANCM helicase domain in vivo, we first attempted to complement FA-M patient-derived cell line (15) or the chicken DT40 cells that are inactivated of the FANCM gene (20). These approaches have all failed, most likely because these cells have adapted to the absence of FANCM, so that they can no longer tolerate re-introduction of exogenous FANCM.
We next established an siRNA-based complementation system using HeLa cells that can tolerate exogenous FANCM (Fig. 3A). Briefly, HeLa cells were transfected with expression vectors for either wildtype FANCM (FANCM-WT) or a helicase domain-deletion mutant (FANCM-ΔHel) or the ATPase point mutant (FANCM-K117R) (Fig. 3A and B). Clones that stably express these proteins at comparable levels were selected (data not shown) and transfected with an siRNA oligo targeting the 3′-untranslated region (3′-UTR) of FANCM. Because exogenous FANCM constructs lack the 3′-UTR, this siRNA should deplete only FANCM derived from the endogenous DNA. The depletion was verified by using a clone that expresses FANCM-ΔHel protein, in which the endogenous full-length and exogenous truncated FANCM can be distinguished (Fig. 3E). As a positive control, an siRNA targeting the open-reading frame (ORF) of the FANCM gene was also used. It should deplete FANCM derived from both endogenous and exogenous DNA, as confirmed using the clone expressing FANCM-ΔHel (Fig. 3E). Both 3′-UTR and ORF siRNA oligos have comparable efficiency in depleting FANCM and reducing FANCD2 monoubiqutination in HeLa cells (Fig. 3C; see the quantification for FANCD2 monoubiquitination below the figure). Thus, the difference in signals generated by 3′-UTR siRNA (which depletes only endogenous FANCM but does not affect the exogenous FANCM) and ORF siRNA (which depletes both endogenous and exogenous FANCM) should reflect the effect of complementation by the exogenous FANCM.
Figure 3.
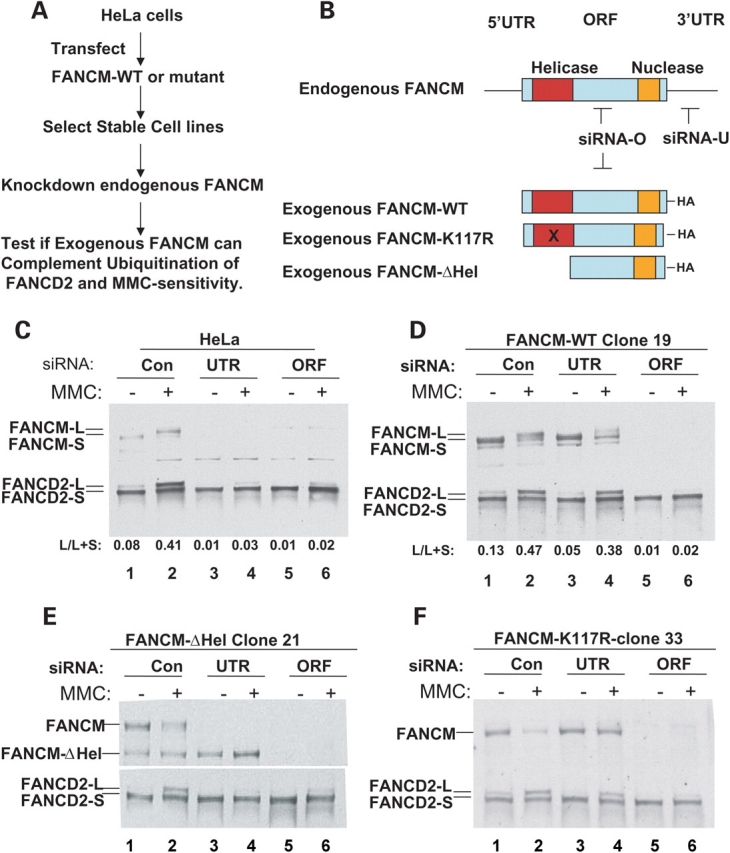
The FANCM helicase domain, but not its associated ATP-dependent activity, is required for FANCD2 monoubiquitination. (A) A flow-chart shows an siRNA-based complementation system to study FANCM function in HeLa cells. (B) A graphic presentation showing the various FANCM constructs and siRNA used in the study. Notably, siRNA-O targets the open-reading frame (ORF) and should deplete both endogenous and exogenous FANCM. In contrast, siRNA-U targets the 3′-untranslated region (3′-UTR) and should deplete only the endogenous, but not the exogenous, FANCM, because this region is absent in the latter. [(C)–(F)] Immunoblotting shows the effects of FANCM depletion on FANCD2 monoubiquitination in HeLa cells (C), and HeLa-derived cell lines that stably expressing FANCM-WT (D), its helicase deletion mutant (FANCM-ΔHel) (E) and the K117R ATPase point mutant (F). The monoubiquitinated and unubiquitinated forms of FANCD2 are marked by FANCD2-L and FANCD2-S, respectively. The ratio between FANCD2-L and the total FANCD2 (both L and S) was obtained by using TotalLab100 image analysis software and shown at the bottom of each figure (L/L+S). FANCM-L and FANCM-S represent hyper- and hypo-phosphorylated forms of this protein (15). The absence or presence of MMC is indicated. A control siRNA (con) was included.
FANCM helicase domain is required for FANCD2 monoubiquitination
In a clone expressing wildtype FANCM, ORF-siRNA efficiently depleted total FANCM, resulting in strong reduction of FANCD2 monoubiquitination (Fig. 3D) and MMC hypersensitivity (Fig. 4A) when compared with cells treated with a control siRNA. In contrast, UTR-siRNA failed to deplete FANCM derived from the exogenous FANCM construct (Fig. 3D, lanes 3 and 4). As a result, FANCD2 monoubiquitination (Fig. 3D) and MMC sensitivity (Fig. 4A) were significantly rescued. These data are consistent with previous findings that FANCM is required for FANCD2 monoubiquitination and cellular resistance to drugs that induce ICLs (15,20).
Figure 4.
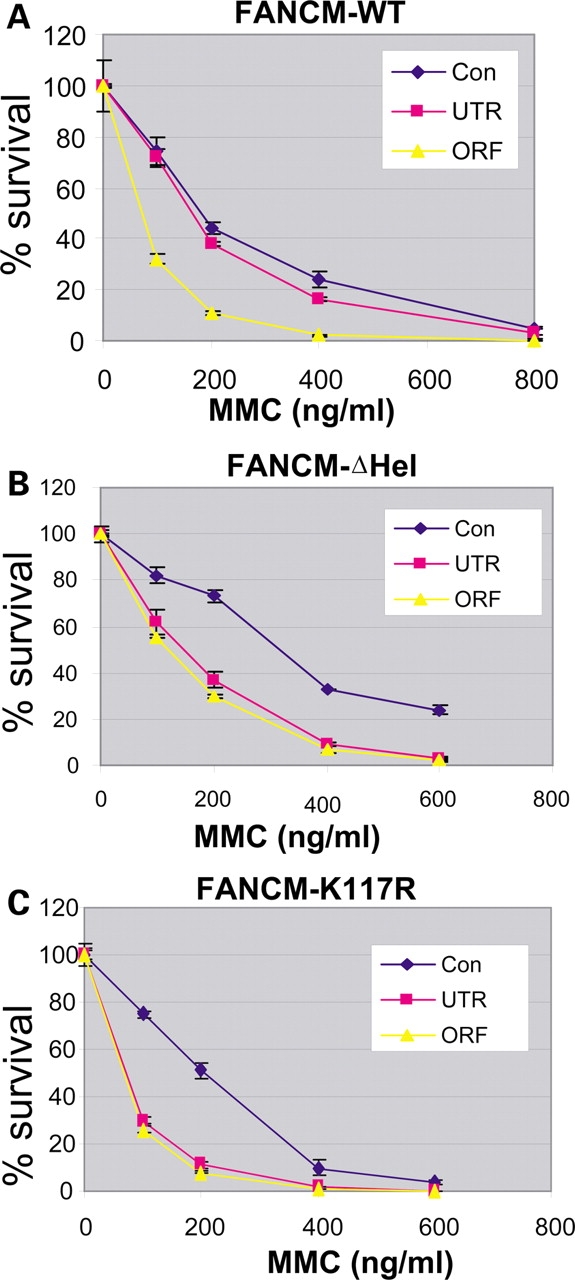
The ATP-dependent function of FANCM is required for cellular resistance for MMC. [(A)–(C)] Graphs showing that the MMC hypersensitivity of the HeLa cells depleted of endogenous FANCM can be complemented by exogenous wildtype FANCM (WT) (A), but not its helicase deletion mutant (FANCM-ΔHel) (B), or the K117R ATPase mutant (FANCM-K117R) (C). Cells treated with control siRNA (con), the siRNA targeting 3′-untranslated region (UTR), or the open-reading frame (ORF) of FANCM are indicated. Notably, the ORF siRNA depletes both endogenous and exogenous FANCM, whereas the UTR siRNA depletes only the former but not the latter (Fig. 3). Thus, the difference between the results of two siRNAs should reflect the effect of the exogenous FANCM. The assay was done in duplicate, and three independent assays were done for each clone. We noticed that the control-siRNA-treated HeLa cells expressing FANCM-ΔHel mutant are more resistant to MMC than those expressing full-length FANCM protein. This is likely due to reduced toxicity of FANCM protein after its helicase domain is deleted. Perhaps, expression of FANCM from the exogenous viral promoter confers certain growth disadvantage or toxicity to their hosts. This may help to explain our failure to obtain stable expression of FANCM in FANCM-deleted patient or DT40 cells.
We noticed that while the rescue of the MMC hypersensitivity was near complete, the rescue of the FANCD2 monoubiquitination was not, as the ratio between the monoubiquitinated (FANCD2-L) and total FANCD2 in cells treated with the 3′-UTR oligo was always lower than that of cells treated with a control siRNA oligo (compare lanes 3 and 4 with 1 and 2 in Fig. 3D). This may represent a technical limitation of this system, because the endogenous FANCM constitutes a significant percentage of total FANCM (∼50%; Fig. 3E), and its depletion could not be fully compensated by the exogenous FANCM.
When the same analysis was performed on a HeLa clone expressing the FANCM-ΔHel mutant, neither FANCD2 monoubiquitination (Fig. 3E) nor MMC hypersensitivity (Fig. 4B) was rescued, indicating that the helicase domain is required for the steps of the FA pathway before monoubiquitination.
The ATP-dependent activity of FANCM is dispensable for FANCD2 monoubiquitination
We also analyzed a HeLa clone expressing the K117R ATPase point mutant of FANCM and found that this mutant rescued FANCD2 monoubiquitination to a level similar to that achieved by the wildtype protein (Fig. 3F; also see Fig. 5). The result suggests that the ATP-dependent activities of FANCM, including translocase and BPT activities, are dispensable for FANCD2 monoubiquitination. Interestingly, the same mutant failed to correct MMC hypersensitivity (Fig. 4C). The data suggest that although ATP-dependent activities of FANCM are not essential for FANCD2 monoubiquitination, they are required to repair or bypass the ICLs. Thus, these activities should function after FANCD2 monoubiquitination in the FA pathway.
Figure 5.

The FANCM helicase domain, but not its associated ATP-dependent activity, is required for monoubiquitination of both FANCI and FANCD2. (A) Immunoblotting shows that monoubiquitination of FANCI and FANCD2 is reduced in HeLa cells depleted of FANCM by siRNA. The ubiquitinated and non-ubiquitinated forms of these two proteins are marked by L (for long) and S (for short), respectively. The long- and short-form of FANCM represent hyper- and hypo-phosphorylated version of this protein, respectively (15). The presence or absence of mytomycin C (MMC) and hydroxyurea (HU) are indicated. Notably, the induction of monoubiquitination is higher in the presence of HU than MMC (compare lane 2 with 6). The ORF siRNA was used for FANCM depletion in this experiment. A non-specific polypeptide crossreactive with FANCM antibody is indicated with an asterisk. [(B)–(E)] Immunoblotting shows the effects of FANCM depletion on FANCI and FANCD2 monoubiquitination in HeLa cells (B), and HeLa-derived cell lines that stably expressing FANCM-WT (C), its helicase deletion mutant (FANCM-ΔHel) (D) and the K117R ATPase point mutant (E). A control siRNA (con) was included. The 3′-UTR and ORF siRNA oligos are described in Figure 3.
The FANCM helicase domain, but not its associated ATP-dependent activity, is also required for FANCI monoubiquitination
Recently, a new FA protein, FANCI, was found to be monoubiquitinated in response to DNA damage, and its monoubiquitination is defective in FA cells deficient in FA core complex components FANCA and FANCG (16,17). We found that in FANCM-depleted HeLa cells, the level of monoubiquitinated FANCI is reduced concomitantly with the monoubiquitinated FANCD2 (Fig. 5A). These findings are consistent with both FANCI and FANCD2 as ubiquitination substrates of the FA core complex (16,17).
We analyzed the dependence of FANCI monoubiquitination on FANCM helicase domain using the siRNA-based complementation system. We used hydroxyurea (HU) to induce stronger FANCI monoubiquitination than is seen with MMC (Fig. 5A). In agreement with the results from FANCD2 studies (Fig. 3), we found that the ectopic expression of wildtype FANCM largely rescued the defective monoubiquitination of both FANCD2 and FANCI (Fig. 5B and C; see quantification below each figure). Moreover, the helicase deletion mutant of FANCM failed to rescue monoubiquitination of either FANCD2 or FANCI (Fig. 5D), whereas the K117R mutant largely rescued monoubiquitination of both (Fig. 5E). The data suggest that monoubiquitination of FANCD2 and FANCI requires the FANCM helicase domain but not its ATP-dependent activities.
Deletion of the FANCM helicase domain does not affect the assembly of the FA core complex
FANCM is essential for the assembly of the FA core complex, and its absence causes instability and failed nuclear localization of several components of the complex (15,20). To exclude the possibility that the failure of FANCM-ΔHel to complement FANCM-depleted HeLa cells is due to defective assembly of the core complex, we performed immunoprecipitation-coupled Western analyses. We found that the FANCM-ΔHel protein associated well with other core complex components as did full-length wildtype FANCM (FANCM-WT-FL) (Fig. 6A and B). Apparently, deletion of the helicase domain does not affect the assembly of the FA core complex.
Figure 6.

FANCM can form homodimers through its C-terminal domain. (A) Graphic presentations of HA-tagged FANCM constructs used in establishing stable HeLa cell lines and co-immunoprecipitation analysis. The full-length wildtype FANCM (WT-FL), the helicase deletion mutant (ΔHel), the C-terminal deletion mutant (WT-ΔC) and the K117R ATPase point mutant in the context of C-terminal deletion (K117R-ΔC) are shown. (B) Immunoblotting analysis of the polypeptides that co-immunoprecipitate with different HA-tagged FANCM proteins as described in (A). The asterisk indicates the endogenous FANCM that co-immunoprecipitates with FANCM-ΔHel. Notably, very little endogenous FANCM was detected in the immunoprecipitate of FANCM-WT-ΔC or FANCM-K117R-ΔC mutants, suggesting that the C-terminal domain of FANCM is important for FANCM dimerization. (C) Graphs to illustrate different GST- or 6-histidine-tagged (His) FANCM and FAAP24 proteins used in the pull-down assays in (D). These bi-cistronic vectors have been used previously to show that bacterially expressed GST-FANCM can specifically associate with FAAP24 (21). (D) The GST-pull-down assay shows that GST-FANCM not only stably associates with His-FAAP24, as described previously (21), but also can associate with His-FANCM. As a control, the GST protein alone fails to pull down His-FANCM. The number of the amino acid residues in the FANCM fusion proteins is indicated as subscript, and both FANCM fusion proteins include the entire ERCC4 endonuclease and helix–hairpin–helix domains. The degradation products are marked with a bracket. We found that these degradation products have higher affinity to the glutathione beads than the GST-FANCM fusion proteins, suggesting that FANCM reduces the binding of GST to the beads.
FANCM can form homodimers through its C-terminal region
Some endogenous FANCM co-immunoprecipitated with the exogenous FANCM-ΔHel, but was undetectable in the negative control (Fig. 6, lane 3; also see Supplementary Material, Fig. S3a, lane 2, for better resolution). The results implied association between endogenous and exogenous FANCM molecules in vivo. In the presence of ethidium bromide (EtBr), which can disrupt DNA–protein contacts and has often been used to demonstrate DNA-independent protein–protein interactions (26), we again observed the association (Supplementary Material, Fig. S3b). These data imply that FANCM protein may form stable homodimers in vivo.
The homodimerization of FANCM is reminiscent of its archaeal ortholog, Hef, which forms homodimers through two interfaces—one in the endonuclease, and the other one in the helix–hairpin–helix domain (22,23). Both domains are conserved in FANCM and FAAP24 and are required for the heterodimerization between the two proteins observed in co-immunoprecipitation experiments (21). We repeated the experiments using cells expressing FANCM mutants lacking the dimerization domains (FANCM-WT-ΔC or FANCM-K117R-ΔC) and found that they associated with less endogenous FANCM than did the helicase deletion mutant (Fig. 6B; Supplementary Material, Fig. S3a), suggesting that the C-terminal region is important for FANCM homodimerization. The C-terminal deletion mutants also associated with lower levels of FAAP24 than did full-length FANCM or the helicase deletion mutant (Fig. 6B), suggesting that the C-terminal region is required for both homo- and hetero-dimerization of FANCM in vivo.
To examine whether the C-terminal regions of two FANCM molecules could directly interact, we repeated the GST-pull down assay used previously to show direct interactions between the FANCM C-terminal region and FAAP24 (21). We found that the GST-fusion protein containing the C-terminal region of FANCM (GST-FANCM1799–2048) not only pulled down 6-histidine-tagged FAAP24 (His-FAAP24) (Fig. 6C and D, lanes 2 and 5), but also the C-terminal region of FANCM (His-FANCM1727–2048) (Fig. 6D, lanes 3 and 6). In controls, the GST protein alone failed to pull down any detectable level of His-FANCM1727–2048 (Fig. 6D, lanes 1 and 4). These data suggest that the C-terminal region of FANCM can mediate its own dimerization.
DISCUSSION
Whether the FA core complex participates in DNA repair reactions has remained unclear. The only module within the core complex that possesses an enzymatic activity on DNA is the helicase domain of FANCM, suggesting that this domain could be the primary component through which the complex copes with the damaged DNA. However, biochemical actions of this domain remain largely unknown. Our data now suggest an essential role of this module in both signaling and repair reactions, as discussed in what follows.
FANCM possesses binding and translocation activity for movable 4WJ DNA
Although the sequence of the FANCM helicase domain is highly homologous to its yeast and archaeal orthologs, MPH1 and Hef, FANCM displays no detectable helicase activity possessed by its ancient ancestors (15). Instead, evidence thus far suggests that some features of FANCM are more similar to those of RAD54. Both FANCM and RAD54 lack detectable helicase activity (15,27), but exhibit ATP-dependent dsDNA translocase activity (15,28–31). In addition, both proteins could form homodimers (32) (this study), and have higher binding affinity to 4WJ DNA structures (24,25) (this study). Moreover, both FANCM and RAD54 display ATP-dependent bi-directional BM or BPT activity for movable 4WJ DNA, but not for non-movable 4WJ (24,25) ( this study). This last feature also distinguishes these two proteins from several helicases that can migrate non-movable 4WJ, such as RuvAB (33), RecG (34), BLM (35) and WRN (36).
The mechanism through which FANCM translocate the movable 4WJ may be different from that of the other helicases that migrate non-movable 4WJ. In the latter case, the helicases could not only bind and melt the branch point of the junction, but also translocate on ssDNA and unwind one strand from the other. In contrast, FANCM lacks the ability of the helicase to translocate on ssDNA and unwind the double-helix. Instead, FANCM may bind and catalyze the local melting of the branch point of the 4WJ DNA. Because the homologous DNA branches in the movable 4WJ can spontaneously migrate without need of extra energy, the melting of the branch point itself may contribute to the observed BPT activity. Moreover, the ATP-dependent translocase activity of FANCM may also contribute to the BPT activity, by carrying the branch-point with it during its translocation on dsDNA.
FANCM may participate in DNA repair at stalled replication forks
The movable 4WJ DNA can form either at the HJ during HR or at stalled replication forks which regress to form the ‘chicken foot’ structures (37,38). Although RAD54 has been suggested to promote BM of HJ during HR (25), current evidence argues against a role of FANCM in this process. For example, yeast cells mutated in the ortholog of FANCM (MPH1) are not defective in general HR and are deficient only in a branch of HR-dependent error-free bypass pathway (39). Similarly, chicken DT40 cells inactivated for FANCM are proficient in HR-dependent repair of double-strand breaks induced by I-SceI restriction enzyme, as well as in HR-dependent gene conversion at the immunoglobin locus (20). In contrast, RAD54-knockout DT40 cells are deficient in both processes (40,41). These differences argue that unlike RAD54, FANCM is not involved in general HR.
On the other hand, FA proteins have been hypothesized to help stabilize or restart blocked replication forks (reviewed in 42). The evidence includes that the FA cells are highly sensitive to ICL drugs that block DNA replication, and that the FA pathway is specifically activated by ATR, the checkpoint kinase responding to replication stress. In the context of this hypothesis, we favor the possibility that FANCM functions at stalled replication forks, possibly by binding and translocating the 4WJ DNA generated by fork regression. Consistent with this notion, FANCM-deficient DT40 cells display an elevated level of spontaneous sister-chromatid exchange (SCE) (20), which are similar to cells deficient of the BLM helicase. BLM could mediate fork regression to generate a movable 4WJ and thus facilitate the re-start of blocked forks through a mechanism that avoids SCE (43). FANCM has been shown to associate with BLM in a complex (15), so that it may cooperate with BLM in this process by translocating 4WJ during either fork regression or the subsequent processing of 4WJ, or both. This action of FANCM may be needed for repair or bypass of the fork block [see repair models in reviews (37,38)].
BPT and other ATP-dependent activities of FANCM are required after monoubiquitination
Using an siRNA-based complementation system, we showed that BPT and other ATP-dependent activities of FANCM are dispensable for the signaling events prior to the monoubiquitination. Rather, they are required for later steps in the FA pathway to repair or bypass the ICLs. The notion that the FA core complex could function after FANCD2 monoubiquitination is supported by a genetic study in DT40 cells. The study predicted that the FA core complex should have two additional functions after FANCD2 monoubiquitination: one is to recruit the FANCD2 to chromatin, and the other is at an unknown step after recruiting FANCD2 to chromatin (44). Our data suggest that this unknown step could be mediated by ATP-dependent DNA-processing activities of FANCM.
The helicase domain of FANCM acts both before and after monoubiquitination
We also showed that deletion of the FANCM helicase domain, which disrupts both its ATP-independent and -dependent activities, complemented neither the defective monoubiquitination nor the MMC hypersensitivity of the FANCM-depleted cells. These data, together with those of the ATP-hydrolysis mutant, suggest that the helicase domain of FANCM could act both before and after the ubiquitination step of the FA pathway: its ATP-independent DNA-binding activity acts before, whereas its ATP-dependent BM and translocase activity of FANCM works after. However, it should be cautioned that other important functions of the helicase domain may be inactivated by the deletion. For example, FANCM is known to be hyperphosphorylated in response to DNA damage (15), and deletion of the helicase domain may remove important phosphorylation sites, resulting in defective monoubiquitination. Future studies using other point mutants within the helicase domain should help assess these possibilities.
FANCM can form homodimers in addition to heterodimerization with FAAP24
Previous studies have shown that FANCM forms a heterodimer with FAAP24 through its C-terminal-conserved region (21). Here we show that FANCM can also form homodimers through the same region (Fig. 6). Our data imply the existence of two groups of FA core complexes in cells: one with FANCM-FAAP24 heterodimer, and the other with FAMCM homodimer. Because FAAP24 can target FANCM to DNA and is required for FANCD2 monoubiquitination (21), the first group may act before monoubiquitination in the FA pathway. Because many helicase proteins work as homodimers or oligomers (45), the second group may use the ATP-dependent activities of FANCM to participate in DNA repair after monoubiquitination.
The feature that FANCM can act after monoubiquitination makes it functionally similar to two other FA proteins which possess DNA-processing activities but are not part of the FA core complex. Those two proteins, BRCA2/FANCD1 and FANCJ, are dispensable for the monoubiquitination and directly participate in DNA repair. Speculatively, FANCM could utilize its ATP-dependent DNA-processing activity to work coordinately with BRCA2 and FANCJ in DNA repair, ultimately leading to the removal of the damaged DNA.
MATERIALS AND METHODS
Cell lines and the antibodies
HeLa and EBNA-293 cells were cultured in DMEM supplemented with 10% fetal calf serum.
Polyclonal antibodies against FANCM, FANCD2, FANCL, FAAP100 and FAAP24 antibodies have been described previously (13,15,21,46). The FANCA antibody and FANCE antibodies were kindly provided by Drs M. Hoatlin and K.J. Patel. A rabbit FANCI polyclonal antibody was made against a chimeric protein containing a region of FANCI (amino acids 632–725) fused to the maltose-binding protein (New England Biolabs). This chimeric protein was purified following the manufacturer’s protocol and used for immunization of rabbits and for affinity purification of the antibody. The Flag antibody and the conjugated beads were from Sigma. The antibodies against the tags of HA, 6-histindine and GST are from Applied Biological Materials.
Plasmid construction and protein purification
The Flag-tagged full-length FANCM and its K117R mutant in pCEP4 vectors, their expression and purification from EBNA-293 cells were described previously (15).
The baculoviral vector expressing the FANCM helicase domain (rFANCM-Hel) was constructed by cloning a region of FANCM (amino acid 1–754) into XhoI and KpnI sites of pFBav2-HK vector (ProteinOne, Inc.). This vector contains a 6×Histine tag, which was used for subsequent purification through metal–ion affinity columns. The protein was expressed in insect cells and purified by the manufacturer (ProteinOne, Inc). The identity of the protein was confirmed by mass spectrometry.
The HA-tagged FANCM vectors used for siRNA-based complementation assays were made by subcloning either the wildtype FANCM or its K117R mutant cDNA (15) into the pIRESneo3 vector (CloneTech Laboratories, Inc.) by using standard molecular biology techniques. The HA-tag was linked to the C-terminus of FANCM. The helicase deletion mutant (FANCM-ΔHel) was made by removing the region between amino acid 74–507. The C-terminal deletion mutants (FANCM-WT-ΔC and FANCM-K117R-ΔC) removed the region between amino acid 1800 and 2048, which includes the entire ERCC4-like endonuclease domain and the helix–hairpin–helix motif. Transfection of plasmids used Lipofectamine 2000 and followed the manufacturer’s protocols (Invitrogen). The immunoprecipitation with HA antibody followed a previous protocol (13). In some experiments, EtBr (25 µg/ml final concentration) was added to nuclear extract prior to immunoprecipitation.
siRNA-based complementation
Two FANCM siRNA oligos were purchased from Dharmacon. The sense strand sequences are AAAGACCUCUCACAAUAUU (3′-UTR) and AGACAUCGCUGAAUUUAAA (ORF). Transfection of siRNA oligos to HeLa cells used oligofectamine and followed the manufacturer’s protocols (Invitrogen). Twenty-four hours post-transfection, cells were either untreated or treated with MMC (60 ng/ml) for 20 h or HU (1 mm) for 24 h, before harvested for immunoblotting analysis. Alternatively, 60 h post-transfection, cells were pulsed-treated with MMC at the indicated concentrations (Fig. 4) and subjected to the colony-survival assay as described (15).
HeLa cells were first transfected with HA-tagged FANCM expression vectors (pIRESneo3) and grown in the presence of G418. The G418-resistent clones were selected and screened by immunoblotting with both HA and FANCM antibodies. Those that stably express exogenous FANCM proteins were chosen for subsequent analyses. Three independent FANCM-WT clones, three FANCM-K117R mutant clones and four FANCM-ΔHel clones were analyzed by siRNA-based complementation assay using FANCD2 monoubiquitination as the readout, and the results are comparable. Two independent clones from each construct were also analyzed by the colony survival assay in the presence of MMC. The quantification of the FANCD2 and FANCI monoubiquitination used TotalLab100 image analysis software (Nonlinear Dynamics) to quantitate the immunoblotting signals on the X-ray films.
We found that the degree of complementation roughly correlates with the level of the exogenous FANCM-WT or FANCM-K117R proteins: the high expression clones complement better than the low expression clones. In addition, some clones lost the expression of the exogenous proteins over a period of time, and their complementation capacity became reduced or diminished. Thus, monitoring the expression levels of the exogenous FANCM protein is required.
The DNA-binding, helicase and BM assays
The DNA oligos are purchased from Invitrogen and are all gel-purified. The sequences for those used for gel-shift and helicase assays are listed in Supplementary Material, Table S1. The DNA-binding reactions were performed in a buffer containing 50 mm Tris.HCl (pH 7.5), 4 mm MgCl2, 50 mm NaCl, 5 mm DTT, 0.1 mg/ml BSA. The final concentration of the DNA substrate was 1 nm, unless indicated in the figure legend. Most reactions (Fig. 1A–E; Supplementary Material, Fig. S1) contain 0.44 µm of recombinant FANCM-Hel, unless otherwise indicated in the figures (Fig. 1F and G). The reaction was performed at room temperature for 30 min.
For competition experiment, the p32-labeled 4WJ DNA (0.6 nm final concentration) was mixed with increasing concentrations of unlabeled competitor DNA (4WJ, fork or dsDNA) prior to the binding assay. The results were quantified using a phosphor-imager.
The helicase assay used the same buffer, except that 1 mm ATP was included in the buffer.
The ATPase assay has been described previously (15).
The sequences for the branch migration assays are identical to those used in the RAD54 study (25). The oligo sequences for the movable 4WJ are number 22, 11M, 21 and 10. They contain two mismatches to prevent spontaneous migration. The oligo sequences for the 5′ partial 4WJ are 12T, 11, 10M and 13M, whereas those for the 3′ partial 4WJ are 13T, 10, 11M and 12M. The buffer and other reaction conditions are also identical to the RAD54 study (25). The final concentration of FANCM wildtype and mutant proteins is about 5 nm.
GST-pull down assay
The original bi-cistronic expression vector expressing GST-FANCM1799–2048 and 6-Histidine-tagged FAAP24 have been described previously (21). To investigate FANCM homodimerization, we replaced the FAAP24 with the C-terminal region of FANCM containing both the ERCC4 endonuclease domain and the helix–hairpin–helix region (amino acid 1727–2048). To generate the control bi-cistronic vector expressing GST and His-FANCM1727–2048, the FANCM part was deleted from GST-FANCM1799–2048 region of the original vector. The expression of Escherichia coli recombinant proteins and the GST-pull down assay followed the same protocol which was used to show heterodimerization between FANCM and FAAP24 (21). We found that compared with the GST-FANCM and His-FAAP24 heterodimers, the bacterial GST-FANCM1799–2048 and His-FANCM1727–2048 homodimer proteins are less stable and bind poorly to the glutathione beads. The bacterial culture was grown at room temperature until log phase. IPTG was then added to the culture and the incubation was continued at 14°C for additional 4 h.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institute of Health, and by the Fanconi Anemia Research Foundation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs A. Constantinou for communicating results, suggestions on the branch migration assay and advice on the manuscript-writing; A.R. Meetei and K.J. Patel for communicating unpublished results; A.V. Mazin for advice on the branch migration assay; M. Hoatlin for FANCA antibody; K.J. Patel for FANCE antibody; A. Ciccia and S. West for bacterial expression vectors of FANCM and FAAP24; D. Schlessinger and R. Nagaraja for critical reading of the manuscript; D. Xu for advice and suggestions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Joenje H., Patel K.J. The emerging genetic and molecular basis of Fanconi anaemia. Nat. Rev. Genet. 2001;2:446–459. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy R.D., D’Andrea A.D. The Fanconi anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 3.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anemia and BRCA proteins. Nat. Rev. Genet. 2007;8 doi: 10.1038/nrg2159. doi:10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 4.Howlett N.G., Taniguchi T., Olson S., Cox B., Waisfisz Q., De Die-Smulders C., Persky N., Grompe M., Joenje H., Pals G., et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 5.Levitus M., Waisfisz Q., Godthelp B.C., de Vries Y., Hussain S., Wiegant W.W., Elghalbzouri-Maghrani E., Steltenpool J., Rooimans M.A., Pals G., et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 6.Levran O., Attwooll C., Henry R.T., Milton K.L., Neveling K., Rio P., Batish S.D., Kalb R., Velleuer E., Barral S., et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat. Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 7.Litman R., Peng M., Jin Z., Zhang F., Zhang J., Powell S., Andreassen P.R., Cantor S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Xia B., Dorsman J.C., Ameziane N., de Vries Y., Rooimans M.A., Sheng Q., Pals G., Errami A., Gluckman E., Llera J., et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 9.Reid S., Schindler D., Hanenberg H., Barker K., Hanks S., Kalb R., Neveling K., Kelly P., Seal S., Freund M., et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 10.Rahman N., Seal S., Thompson D., Kelly P., Renwick A., Elliott A., Reid S., Spanova K., Barfoot R., Chagtai T., et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tischkowitz M., Xia B., Sabbaghian N., Reis-Filho J.S., Hamel N., Li G., van Beers E.H., Li L., Khalil T., Quenneville L.A., et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc. Natl Acad. Sci. USA. 2007;104:6788–6793. doi: 10.1073/pnas.0701724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erkko H., Xia B., Nikkila J., Schleutker J., Syrjakoski K., Mannermaa A., Kallioniemi A., Pylkas K., Karppinen S.M., Rapakko K., et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 13.Meetei A.R., de Winter J.P., Medhurst A.L., Wallisch M., Waisfisz Q., van de Vrugt H.J., Oostra A.B., Yan Z., Ling C., Bishop C.E., et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 14.Meetei A.R., Levitus M., Xue Y., Medhurst A.L., Zwaan M., Ling C., Rooimans M.A., Bier P., Hoatlin M., Pals G., et al. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 2004;36:1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 15.Meetei A.R., Medhurst A.L., Ling C., Xue Y., Singh T.R., Bier P., Steltenpool J., Stone S., Dokal I., Mathew C.G., et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R., III, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D’Andrea A.D., et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sims A.E., Spiteri E., Sims R.J., III, Arita A.G., Lach F.P., Landers T., Wurm M., Freund M., Neveling K., Hanenberg H., et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Higuera I., Taniguchi T., Ganesan S., Meyn M.S., Timmers C., Hejna J., Grompe M., D’Andrea A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 19.Bogliolo M., Lyakhovich A., Callen E., Castella M., Cappelli E., Ramirez M.J., Creus A., Marcos R., Kalb R., Neveling K., et al. Histone H2AX and Fanconi anemia FANCD2 function in the same pathway to maintain chromosome stability. EMBO J. 2007;26:1340–1351. doi: 10.1038/sj.emboj.7601574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosedale G., Niedzwiedz W., Alpi A., Perrina F., Pereira-Leal J.B., Johnson M., Langevin F., Pace P., Patel K.J. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 2005;12:763–771. doi: 10.1038/nsmb981. [DOI] [PubMed] [Google Scholar]
- 21.Ciccia A., Ling C., Coulthard R., Yan Z., Xue Y., Meetei A.R., Laghmani el H., Joenje H., McDonald N., de Winter J.P., et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Komori K., Hidaka M., Horiuchi T., Fujikane R., Shinagawa H., Ishino Y. Cooperation of the N-terminal helicase and C-terminal endonuclease activities of archaeal Hef protein in processing stalled replication forks. J. Biol. Chem. 2004;279:53175–53185. doi: 10.1074/jbc.M409243200. [DOI] [PubMed] [Google Scholar]
- 23.Nishino T., Komori K., Ishino Y., Morikawa K. X-ray and biochemical anatomy of an archaeal XPF/Rad1/Mus81 family nuclease: similarity between its endonuclease domain and restriction enzymes. Structure (Camb.) 2003;11:445–457. doi: 10.1016/s0969-2126(03)00046-7. [DOI] [PubMed] [Google Scholar]
- 24.Gari K., Decaillet C., Stasiak A.Z., Stasiak A., Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 25.Bugreev D.V., Mazina O.M., Mazin A.V. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006;442:590–593. doi: 10.1038/nature04889. [DOI] [PubMed] [Google Scholar]
- 26.Lai J.S., Herr W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl Acad. Sci. USA. 1992;89:6958–6962. doi: 10.1073/pnas.89.15.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petukhova G., Sung P., Klein H. Promotion of Rad51-dependent D-loop formation by yeast recombination factor Rdh54/Tid1. Genes Dev. 2000;14:2206–2215. doi: 10.1101/gad.826100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Komen S., Petukhova G., Sigurdsson S., Stratton S., Sung P. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell. 2000;6:563–572. doi: 10.1016/s1097-2765(00)00055-1. [DOI] [PubMed] [Google Scholar]
- 29.Ristic D., Wyman C., Paulusma C., Kanaar R. The architecture of the human Rad54-DNA complex provides evidence for protein translocation along DNA. Proc. Natl Acad. Sci. USA. 2001;98:8454–8460. doi: 10.1073/pnas.151056798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaskelioff M., Van Komen S., Krebs J.E., Sung P., Peterson C.L. Rad54p is a chromatin remodeling enzyme required for heteroduplex DNA joint formation with chromatin. J. Biol. Chem. 2003;278:9212–9218. doi: 10.1074/jbc.M211545200. [DOI] [PubMed] [Google Scholar]
- 31.Amitani I., Baskin R.J., Kowalczykowski S.C. Visualization of Rad54, a chromatin remodeling protein, translocating on single DNA molecules. Mol. Cell. 2006;23:143–148. doi: 10.1016/j.molcel.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Mazina O.M., Rossi M.J., Thoma N.H., Mazin A.V. Interactions of hRad54 protein with branched DNA molecules. J. Biol. Chem. 2007;282:21068–21080. doi: 10.1074/jbc.M701992200. [DOI] [PubMed] [Google Scholar]
- 33.Tsaneva I.R., Muller B., West S.C. ATP-dependent branch migration of Holliday junctions promoted by the RuvA and RuvB proteins of E. coli. Cell. 1992;69:1171–1180. doi: 10.1016/0092-8674(92)90638-s. [DOI] [PubMed] [Google Scholar]
- 34.Lloyd R.G., Sharples G.J. Dissociation of synthetic Holliday junctions by E. coli RecG protein. EMBO J. 1993;12:17–22. doi: 10.1002/j.1460-2075.1993.tb05627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karow J.K., Constantinou A., Li J.L., West S.C., Hickson I.D. The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl Acad. Sci. USA. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Constantinou A., Tarsounas M., Karow J.K., Brosh R.M., Bohr V.A., Hickson I.D., West S.C. Werner’s syndrome protein (WRN) migrates Holliday junctions and co- localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGlynn P., Lloyd R.G. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002;18:413–419. doi: 10.1016/s0168-9525(02)02720-8. [DOI] [PubMed] [Google Scholar]
- 38.Wu L., Hickson I.D. DNA helicases required for homologous recombination and repair of damaged replication forks. Annu. Rev. Genet. 2006;40:279–306. doi: 10.1146/annurev.genet.40.110405.090636. [DOI] [PubMed] [Google Scholar]
- 39.Schurer K.A., Rudolph C., Ulrich H.D., Kramer W. Yeast MPH1 gene functions in an error-free DNA damage bypass pathway that requires genes from homologous recombination, but not from postreplicative repair. Genetics. 2004;166:1673–1686. doi: 10.1534/genetics.166.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bezzubova O., Silbergleit A., Yamaguchi-Iwai Y., Takeda S., Buerstedde J.M. Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell. 1997;89:185–193. doi: 10.1016/s0092-8674(00)80198-1. [DOI] [PubMed] [Google Scholar]
- 41.Essers J., Hendriks R.W., Swagemakers S.M., Troelstra C., de Wit J., Bootsma D., Hoeijmakers J.H., Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997;89:195–204. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- 42.Thompson L.H. Unraveling the Fanconi anemia–DNA repair connection. Nat. Genet. 2005;37:921–922. doi: 10.1038/ng0905-921. [DOI] [PubMed] [Google Scholar]
- 43.Ralf C., Hickson I.D., Wu L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006;281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- 44.Matsushita N., Kitao H., Ishiai M., Nagashima N., Hirano S., Okawa K., Ohta T., Yu D.S., McHugh P.J., Hickson I.D., et al. A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Mol. Cell. 2005;19:841–847. doi: 10.1016/j.molcel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 45.Mackintosh S.G., Raney K.D. DNA unwinding and protein displacement by superfamily 1 and superfamily 2 helicases. Nucleic Acids Res. 2006;34:4154–4159. doi: 10.1093/nar/gkl501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ling C., Ishiai M., Ali A.M., Medhurst A.L., Neveling K., Kalb R., Yan Z., Xue Y., Oostra A.B., Auerbach A.D., et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007;26:2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.