

Abstract
Stability of formulations over shelf-life is critical for having a quality product. Choice of excipients, manufacturing process, storage conditions, and packaging can either mitigate or enhance the degradation of the active pharmaceutical ingredient (API), affecting potency and/or stability. The purpose was to investigate the influence of processing and formulation factors on stability of levothyroxine (API). The API was stored at long-term (25°C/60%RH), accelerated (40°C/75%RH), and low-humidity (25°C/0%RH and 40°C/0%RH) conditions for 28 days. Effect of moisture loss was evaluated by drying it (room temperature, N2) and placed at 25°C/0%RH and 40°C/0%RH. The API was incubated with various excipients (based on package insert of marketed tablets) in either 1:1, 1:10, or 1:100 ratios with 5% moisture at 60°C. Commonly used ratios for excipients were used. The equilibrium sorption data was collected on the API and excipients. The API was stable in solid state for the study duration under all conditions for both forms (potency between 90% and 110%). Excipients effect on stability varied and crospovidone, povidone, and sodium laurel sulfate (SLS) caused significant API degradation where deiodination and deamination occurred. Moisture sorption values were different across excipients. Crospovidone and povidone were hygroscopic whereas SLS showed deliquescence at high RH. The transient formulation procedures where temperature might go up or humidity might go down would not have major impact on the API stability. Excipients influence stability and if possible, those three should either be avoided or used in minimum quantity which could provide more stable tablet formulations with minimum potency loss throughout its shelf-life.
Key words: excipients, formulation, levothyroxine sodium pentahydrate, moisture sorption, stability
INTRODUCTION
Levothyroxine sodium pentahydrate is the sodium salt of the levo-isomer of thyroxine, an active physiological substance found in the thyroid gland. Synthetic version is primarily used in the treatment of hypothyroidism and as a thyroid stimulating hormone suppressant, in the treatment of various types of euthroid goiters (1). Since its first inception in the market, there have been various recalls for these drug products from various manufacturers. The primary reason for these recalls is due to sub-potency before the expiration date of the drug product because of stability failures (2).
Although expiration dating is based on the scientific data at normal and stressed conditions, not all the batches and strengths undergo stability testing and for this reason understanding the factors that affects the product stability is critical. Levothyroxine has been a subject of advisory committee meetings at FDA due to its potency and stability issues (3). Levothyroxine has a complex stability profile and is sensitive to various environmental factors such as light, air, and humidity, among others (4–8). Effects of some excipients have been studied by Patel et al. (8) and Gupta et al. (9). However, in both the reports, the impurities remain largely unidentified. Thus stability of levothyroxine has not been systematically characterized in the presence of processing and formulation factors by a stability-indicating method which can identify majority of its degradation products.
Hydrate formation and the dehydration of the product may occur during the manufacturing or storage of pharmaceuticals and this phenomenon of hydration and dehydration is known to be is very sensitive and variable (10). Since biological effects of hydrates sometimes differ from that of anhydrates, it is important that these behavioral differences be known for the development of stable pharmaceutical formulations. As the phase transition that occurs on hydration or dehydration is accompanied by a change in the physicochemical properties, it is important to understand the mechanisms of these transitions under various conditions (11–13). Currently, there are no studies that investigate the stability of levothyroxine solid state under hydrous and anhydrous conditions.
The compatibility of drug and its excipients is extremely critical in the formulation of a quality drug product. The formulation of a stable and effective dosage form requires careful selection of excipients used to facilitate administration, promote consistent release and bioavailability of the drug, and to promote the active moiety from the environment. Although often regarded as ‘inert’, excipients can in fact readily interact with drugs (14). Lack of compatibility between levothyroxine and its excipients can lead to stability problems and inadequate bioavailability/bioequivalence issues that are critical for patients in need of treatment for hypothyroidism. It is important to know that the product development phase of a drug and its excipients is just as significant as the stability properties of the finished product. Environmental conditions of 40°C/75%RH are typically used for 3–6 months as accelerated conditions. This is a lengthy process when trying to evaluate the stability of formulations in a more efficient and timely manner. Another method that is commonly employed for evaluating the drug–excipient compatibility is isothermal stress testing of binary drug–excipient mixtures. The method involves storing the drug–excipient blends with or without moisture at high temperature and determining the drug content (15–17).
The current research was aimed to fill the knowledge gaps which still exist in understanding the stability of this active pharmaceutical ingredient (API) by conducting stability studies by a stability-indicating method with degradation impurities under hydrous and anhydrous conditions. Also, the effect of commonly used formulation excipients in the presence of moisture was studied to understand the stability profile of the drug substance.
MATERIALS AND METHODS
Materials
l-Thyroxine sodium (l-T4) was obtained from KV Pharmaceutical (St Louis, MO, USA). 3,3′,5-tri-iodo-l-thyronine; 3,5-diiodo-l-thyronine; 3,5-diiodo-l-tyrosine; 3-iodo-l-tyrosine; l-thyronine; l-tyrosine; 3,3′,5-tri-iodo-l-thyroacetic acid; 3,3′,5,5′-tetra-iodo-l-thyroacetic acid, corn starch (CS), acacia, sucrose, magnesium stearate (MS), mannitol (M), and sodium laurel sulfate (SLS) were purchased from Sigma-Aldrich [St. Louis, MO, USA]. Microcrystalline cellulose (MCC) and croscarmellose sodium (CCS) were obtained from FMC Biopolymer [Philadelphia, PA, USA]. Lactose monohydrate (LM) [Kerry BioScience, Chicago, IL, USA], povidone (P) [BASF, Florham Park, NJ, USA], talc [Spectrum Chemicals, Gardena, CA, USA], crospovidone (CP) [ISP Technologies Inc, Wayne, NJ, USA], sodium starch glycolate (SSG) [Explotab, Patterson, NY, USA], colloidal silicon dioxide (CSD) [Aerosil, Evonik Degussa, Orange, CA, USA], and confectioner’s sugar (CFS) [Domino’s sugar, Baltimore, MD, USA] were used as received. Methanol, acetonitrile, 0.01 M NaOH, 0.1% trifluoroacetic acid (TFA), theophylline reagents, and Fisherbrand low adhesion specialty tips (21-381-83) were purchased from Fisher Scientific (Suwanee, GA, USA). For all studies, distilled and deionized water was used.
Stability of Levothyroxine Sodium
Stability of Pentahydrate Form
The pure drug was weighed and placed in open amber glass containers to equilibrate with temperature and humidity conditions. The stability conditions were as follows: 25°C/0%RH and 40°C/0%RH (Fisher Scientific, St. Louis, MO, USA) and 25°C/60%RH and 40°C/75%RH (HotPak, Baltimore, MD, USA). For conditions of 25°C/0%RH and 40°C/0%RH, the samples were placed in desiccators using indicating Drierite anhydrous calcium sulfate dessicants and positioned under the appropriate temperature condition. Humidity values were measured using a hygrometer (Fisher Sci, Suwanee, GA, USA).
To determine the chemical changes of levothyroxine sodium hydrate, the samples were analyzed at predetermined days (0, 3, 6, 10, 14, and 28). Samples were weighed (10 mg) in three 100-mL amber glass volumetric flasks followed by the addition of 20 mL of sample diluent (10 mM NaOH–MeOH; 1:1 v/v), sonicated for 10 min, and filled to volume with sample diluent. One milliliter of each sample was then transferred to individual 10-mL amber volumetric flasks. One milliliter of theophylline internal standard (0.1 mg/mL) was then added and solution filled to volume with sample diluent. The contents of each solution were transferred to an automatic injector for HPLC analysis. The determination of the API was carried out by a previously determined validated method (18).
Stability of Dehydrated Form
The API was weighed (50 mg) on a calibrated balance and placed in eight 1.5-mL amber glass septum vials for each stability condition (25°C/0%RH and 40°C/0%RH). Samples were then placed in vacuum/gas compartments containing Fisher Scientific thermo-hygro readers (11-661-13, Suwanee, GA, USA) and color-indicating desiccants. A vacuum was used to remove the air from each compartment followed by a purge of nitrogen in each compartment. Compartments were then closed and placed in their temperature chambers, respectively.
The samples were analyzed at predetermined days (0, 3, 6, 10, 14, and 28). For each temperature condition, a vial is removed from the vacuum/gas chamber and immediately purged with a low flow of nitrogen to assure the prevention of moisture entering the vials. Vial is then sealed with septum vial screw caps containing Tuf-Bond disks for secured closure and placed in a desiccator. The desiccator is taken to a compact glove box (Plaslabs, Lansing, MI, USA) where nitrogen is slowly being purged in the closed atmosphere. It is here that the weighing is performed for HPLC, differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA) studies. Samples are then placed back in desiccator and removed from compact glove box for their respective analyses.
Drug–Excipient Mixtures Studies
The API was weighed on a calibrated balance and was then transferred to 15-mL glass culture test tubes. Excipients were weighed in either 1:1, 1:10, or 1:100 w/w ratios to the drug, ideally depending upon the normal strength in which they are used in the formulations. Deionized water was added to each test tube to obtain a 5% moisture content and vortexed (20 s) for a more homogenous mixture of components. Samples were then closed using screw top closures and transferred to a stability chamber (Fisher Scientific, St. Louis, MO, USA) set at an accelerated test condition of 60°C ± 1°C (drug–excipient ratios are included in Table I). It is known that levothyroxine degrades rapidly at higher temperature (5). In present study, the objective was to screen various formulation excipients for their effects on stability of levothyroxine. If the tight conditions were not maintained, it would be challenging to understand whether the degradation was caused by the excipient or due to slightly elevated temperature. Therefore, a very tight range was used to perform the study.
Table I.
Drug–Excipient Ratios
| Excipient | Abbreviations | Drug–excipient ratio | Amount of water (mL) |
|---|---|---|---|
| Colloidal silicon dioxide | CSD | 1:1 | 1 |
| Crospovidone | CP | 1:10 | 5.5 |
| Magnesium stearate | MS | 1:10 | 5.5 |
| Mannitol | M | 1:10 | 5.5 |
| Microcrystalline cellulose | MCC | 1:10 | 5.5 |
| Povidone | P | 1:1 | 1 |
| Sodium lauryl sulfate | SLS | 1:1 | 1 |
| Sucrose | S | 1:10 | 5.5 |
| Acacia | A | 1:1 | 1 |
| Confectioner’s sugar | CFS | 1:10 | 5.5 |
| Lactose monohydrate | LM | 1:100 | 50.5 |
| Talc | T | 1:10 | 5.5 |
| Croscarmellose sodium | CCS | 1:1 | 1 |
| Corn starch | CS | 1:10 | 5.5 |
| Sodium starch glycolate | SSG | 1:1 | 1 |
The samples were analyzed at predetermined times for a total of 28 (0, 1, 2, 5, 9, 13, and 28) days to observe the chemical change. Those results were compared with samples collected at time zero. For sample analysis, the contents were washed into individual 100-mL amber volumetric flasks, sonicated for 10 min, and filled to volume with sample diluent (10 mM NaOH–MeOH; 1:1 v/v). One milliliter of each sample was then transferred to individual 10-mL amber volumetric flasks. One milliliter of theophylline internal standard (0.1 mg/mL) was then added and solution filled to volume with sample diluent. The contents of each solution were transferred to an automatic injector for HPLC analysis. The determination of the API was determined by a previously determined validated method.
HPLC System
A validated HPLC method was used to analyze the samples (18). The HP 1100 HPLC equipment from Agilent (Wilmington, DE, USA) consisted of a quaternary pump, an automatic injector, a diode array detector, and a column oven. Data was collected using Agilent ChemStation software. Separation was achieved with a reverse phase Inertsil ODS 2 column (250 × 4.6 mm, 5 µm, 150 A) with an Inertsil ODS Security Guard cartridge (4.0 × 3.0 mm, 10 µm). It provided baseline separation with gradient conditions with 0.1% TFA (A) and acetonitrile (B) from 92% to 8% A in 25 min, at 8% A from 25 to 30 min, from 8% to 92% A from 30 to 35 min run time of 40 min in a single chromatographic run. The flow rate was 0.8 mL/min, column temperature was 25°C, and the injection volume was 50 µL. The UV detection wavelength was set at 215, 223, 228, 232, and 240 nm. However, all calculations were performed at 223 nm.
Differential Scanning Calorimetry
Approximately 1–2 mg of sample was weighed and placed in an aluminum pan (no seal) with an empty open aluminum pan as a reference. The sample was scanned at a heating rate of 10°C/min from 25–300°C (TA Instruments Model 2920). The cell was purged with nitrogen during the run.
Thermogravimetric Analysis
Approximately 5–10 mg of sample was heated in an open platinum pan from 25–300°C (TA Instruments Model 2950) at 10°C/min. The cell was purged with nitrogen during the run.
Equilibrium Moisture Content
Equilibrium moisture content of l-T4 and individual excipients listed in Table I at different relative humidity (RH) levels was determined gravimetrically at 25°C on a Symmetrical Gravimetric Analyzer (Model SGA-100, VTI Corporation, Hialeah, FL, USA). Sorption profiles were collected by increasing the instrument RH from 5% to 95%, while desorption profiles were collected by decreasing the RH from 95% to 5% for the same sample. At each RH level, the sample was allowed to reach moisture equilibrium before the RH was changed to the next level. The equilibrium criteria was based on the hygroscopicity of the sample but was always tighter than <0.05% weight change in 20 min. The sample size also varied between 3 and 30 mg for the same reason. All samples were dried in the instrument at 40/60°C for 1 h prior to water sorption experiment, except l-T4 and MS which were dried at 25°C since these two materials undergo physical/chemical changes at higher temperature.
RESULTS AND DISCUSSION
Influence of Processing Factors on Stability of Levothyroxine Sodium Pentahydrate
Levothyroxine sodium is available in pentahydrate form and is used in the tablet dosage form. Influence of processing factors on moisture level has not yet been determined systematically to determine degradation products by a stability indicating method. Therefore, it was investigated at various conditions in the current study. The drug substance was placed in stability chamber under various conditions as described in the “MATERIALS AND METHODS” section. In addition to long-term and accelerated stability conditions, samples were also exposed at 0% RH at 25°C and 40°C. Levothyroxine sodium pentahydrate loses water molecules at those temperatures when the RH is below 30% (Fig. 1). This is consistent with the report in the literature (8). Whether loss of moisture is related to destabilization of the crystal lattice and thus affecting its potency is not yet known and therefore, in the present work, there was an effort to determine the effect of moisture loss on the stability of the drug.
Fig. 1.

Moisture sorption isotherm of levothyroxine sodium pentahydrate
Potency of levothyroxine was determined at various time points under various stability conditions and the results are listed in Table II. It was seen that the pentahydrate form was stable in all the conditions for the duration of the study. The moisture content was analyzed by TGA and the moisture loss (%) data for the samples are presented in Table III. There was no moisture loss when the samples were left at higher humidity values; however, there was about 3% moisture loss when the humidity was low. DSC thermogram for the drug substance at 0 time point is shown in Fig. 2. The DSC thermogram showed that levothyroxine sodium pentahydrate showed two exotherms (at 120°C and 159°C) and an endotherm at 209°C. The two exotherms might correspond to the moisture loss from the hydrate form. The TGA profile showed the weight loss of 10% initially which corresponds to the total amount of moisture including hydrated water amount in the levothyroxine sodium pentahydrate. The moisture content was in close agreement with the value obtained from Karl-Fisher analysis which was about 9.8%.
Table II.
Stability of Levothyroxine Sodium Pentahydrate and Dehydrated Form Under Various Stability Conditions
| Conditions | Days | |||||
|---|---|---|---|---|---|---|
| 0 | 3 | 6 | 10 | 14 | 28 | |
| Pentahydrate form | ||||||
| 25°C/60%RH | 102.88 ± 0.77 | 99.75 ± 5.39 | 97.42 ± 5.81 | 101.59 ± 0.39 | 101.23 ± 1.23 | 101.48 ± 1.00 |
| 25°C/0%RH | 105.04 ± 0.75 | 100.90 ± 0.54 | 103.52 ± 2.68 | 100.91 ± 0.36 | 102.80 ± 0.83 | 95.13 ± 0.84 |
| 40°C/75%RH | 102.88 ± 0.77 | 101.77 ± 1.54 | 101.39 ± 1.01 | 99.23 ± 2.17 | 102.76 ± 1.27 | 100.68 ± 2.03 |
| 40°C/0%RH | 102.88 ± 0.77 | 102.80 ± 3.02 | 98.97 ± 0.64 | 97.67 ± 3.41 | 101.43 ± 0.38 | 99.44 ± 1.09 |
| Dehydrated form | ||||||
| 25°C/0%RH | 92.32 ± 2.03 | 99.00 ± 0.99 | 98.11 ± 4.58 | 104.59 ± 6.81 | 96.82 ± 1.42 | 96.20 ± 0.80 |
| 40°C/0%RH | 92.32 ± 2.03 | 99.85 ± 4.55 | 91.30 ± 2.47 | 96.21 ± 3.95 | 95.36 ± 2.20 | 95.43 ± 2.40 |
The results are expressed as average ± SD for n = 3
Table III.
Moisture Loss (%) Data by TGA for Levothyroxine Sodium Pentahydrate and Dehydrated Form Under Various Stability Conditions
| Conditions | Days | |||||
|---|---|---|---|---|---|---|
| 0 | 3 | 6 | 10 | 14 | 28 | |
| Pentahydrate form | ||||||
| 25°C/60%RH | 9.77 ± 0.07 | 9.69 ± 0.09 | 9.77 ± 0.16 | 8.81 ± 1.61 | 9.59 ± 0.50 | 9.48 ± 0.67 |
| 25°C/0%RH | 9.77 ± 0.07 | 8.31 ± 2.05 | 9.30 ± 0.24 | 6.06 ± 0.71 | 7.64 ± 1.12 | 8.52 ± 0.19 |
| 40°C/75%RH | 9.77 ± 0.07 | 10.12 ± 0.18 | 10.26 ± 0.17 | 9.43 ± 1.13 | 10.16 ± 0.37 | 9.96 ± 0.50 |
| 40°C/0%RH | 9.77 ± 0.07 | 8.86 ± 0.24 | 8.01 ± 0.48 | 8.21 ± 0.33 | 7.35 ± 0.16 | 7.24 ± 0.33 |
| Dehydrated forma | ||||||
| 25°C/0%RH | 7.21 | 3.06 | N/Ab | N/Ab | 3.91 | 2.62 |
| 40°C/0%RH | 7.21 | 2.25 | 4.53 | 4.65 | 4.77 | 2.99 |
The results are expressed as average ± SD for n = 3
aFor dehydrated form n = 1
bData not available due to instrument error
Fig. 2.

DSC thermogram a and TGA profile b of levothyroxine sodium pentahydrate
The study was also conducted by placing the pentahydrate form into the anaerobic chamber where all the air was removed and the chamber was flushed with nitrogen. The samples were then removed from the desiccator, weighed in the chamber respectively for both TGA and DSC, and transferred back to a desiccator. Samples were then taken out of chamber for moisture content determination. This was used as a starting material as the dehydrated form and the samples were kept in low-humidity conditions at two temperatures (25°C and 40°C). The data for potency and moisture loss are shown in Tables II and III, respectively. It was found that there was a significant moisture loss for these samples at the end of study duration. For dehydrated form, only one sample was evaluated for moisture content at various time points. It was mainly done to see whether the moisture content changed at various time points. The data is highly variable but is mainly due to sample preparation techniques. As soon as dehydrated form comes in contact with the moisture, it absorbs moisture. Care was taken not to expose material to environment before analyzing them by TGA; however, it was beyond control once the sample is on the TGA pan and on the instrument when the thermostat will close the system. The data although variable, gave a qualitative estimate that the moisture levels were very low throughout the study period. However, the potency was not affected, thus confirming that removal of moisture from the molecule does not give rise to an unstable preparation. Moreover, the HPLC chromatograms did not indicated the presence of any impurity. Many of the manufacturing procedures such as drying, milling, and compression operations might give rise to low-humidity conditions or increase in the temperature. If the duration of exposure is short enough it might not cause any degradation of the drug substance, which was shown by the current results.
Influence of Formulation Factors on Stability of Levothyroxine Sodium Pentahydrate
Using accelerated conditions gives the opportunity to conduct stability studies in a shorter period of time. Commonly, conditions such as 40°C/75%RH are used as accelerated and last between 3 and 6 months. Increasing the temperature to 60°C places l-thyroxine in its threshold temperature which causes rapid degradation. Screening of excipients was performed at a higher temperature of 60°C as opposed to accelerated stability studies which are usually performed at 40°C for 6 months. The higher temperature would accelerate the degradation of levothyroxine as it was observed in another study (5) that levothyroxine degrades rapidly at higher temperatures. Thus, a short term of 28 days was used in the study in order to determine the excipients which caused instability of levothyroxine. The combination of drug and excipient in this temperature environment allows for expedited drug degradation studies. Moreover, the mechanism of degradation does not change at higher temperatures (5). Another purpose of conducting this study was to evaluate manufacturing conditions such as wet granulation followed by drying which is done at higher temperatures, although it did not last for more than a few hours. Thus, conducting a short-term stability study at higher temperature would give an evidence of instability during manufacturing conditions.
Conventional isothermal stress testing approaches to drug–excipient compatibility evaluation typically involve challenging binary drug–excipient mixtures (in realistic ratios) with moisture as the majority of drug degradation reactions involve moisture (19). Increasing the moisture content may be accomplished by increasing the relative humidity of the environment, or by adding a fixed amount of water. With exposure of the samples to high humidities, the excipient–drug interaction depends upon the free moisture present and relative hygroscopicities (16). Drug degradation thus may vary depending on the hygroscopicity of the excipients (16,20). Therefore, it is suggested that a constant amount of water be added to facilitate interactions between the excipient and drug, and to surround undissolved particles with an aqueous layer saturated with drug, excipient, and any impurities present, as well as the microenvironmental pH. Literature values range from 5% to 20% added water (16,19–22). In the current study, 5% moisture was used for all the drug–excipient mixtures.
Although levothyroxine was found to be stable in solid state at accelerated temperature conditions, it degraded up to 40% in the presence of moisture when exposed to higher temperature. It is sensitive to moisture which causes its degradation rapidly as compared to its dry form. Different excipients influenced the stability of levothyroxine sodium pentahydrate in slurries to varying extents (Fig. 3a and b). The smallest amount of degradation, i.e., API remaining at the end of 28 days >50% was observed with CSD (61.3%), MS (90.1%), A (76.3%), LM (96.4%), CCS (71.6%), CS (65.0%), and SSG (65.8%). The following excipients displayed close to 50% degradation: M (50.0%), MCC (51.5%), and CFS (53.8%). Unfavorable degradation, i.e., API remaining at the end of 28 days <20% was shown with CP (0%), P (3.2%), and SLS (14.1%). In another study also, CP was found to affect stability of levothyroxine sodium pentahydrate at 50°C after 1 month (8). Figures 4, 5, and 6 shows the degradation products of levothyroxine API in presence of CP, P, and SLS, respectively. The identification was based on the retention times of these impurities on chromatograms and comparing with the chromatogram of the API with eight different impurities (18).
Fig. 3.

Levothyroxine pentahydrate stability in presence of 5% moisture and excipient. (a) colloidal silicon dioxide, crospovidone, magnesium stearate, mannitol, microcrystalline cellulose, povidone, sodium laurel sulfate, no excipient. (b) sucrose, acacia, confectioner's sugar, lactose monohydrate, talc, croscarmellose sodium, corn starch, sodium starch glycolate
Fig. 4.

Stability of levothyroxine sodium pentahydrate in presence of crospovidone
Fig. 5.

Stability of levothyroxine sodium pentahydrate in presence of povidone
Fig. 6.

Stability of levothyroxine sodium pentahydrate in presence of sodium laurel sulfate
Half-lives were calculated for levothyroxine for all the drug–excipient mixtures (Table IV). The datapoint from 0 to 5 h were used which followed a first-order kinetics. Since levothyroxine undergoes biphasic degradation, only the first phase was used for the half-life calculations. This was due to faster degradation rate than the second phase. Degradation rate constant (k) was calculated and the following equation was used to calculate the half-life.
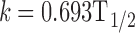 |
1 |
Table IV.
Degradation Half-Lives for Drug–Excipient Mixture
| Drug with excipient | Degradation half-life (days) |
|---|---|
| CSD | 32 |
| CP | 3 |
| MS | 33 |
| M | 23 |
| MCC | 18 |
| P | 3 |
| SLS | 3 |
| Control | 26 |
| S | 21 |
| A | 25 |
| CFS | 40 |
| LM | 93 |
| T | 26 |
| CCS | 27 |
| SSG | 34 |
CSD colloidal silicon dioxide, CP crospovidone, MS magnesium stearate, M mannitol, MCC microcrystalline cellulose, P povidone, SLS sodium laurel sulfate, S sucrose, A acacia, CFS confectioner's sugar, LM lactose monohydrate, T talc, CCS croscarmellose sodium, SSG sodium starch glycolate
This suggests that these three excipients should either be avoided, replaced, or their use can be minimized in solid dosage forms of levothyroxine sodium. Alternatively, care should be taken to avoid exposure to moisture.
The equilibrium moisture studies were carried out to understand hygroscopicity of the excipients. The equilibrium moisture varied from one excipient to the other at all RH levels (Fig. 7a and b). The highest moisture uptake was observed with P, CP, SSG, A, CCS, and CS, all of which showed >10% moisture sorption above 50% RH. The lowest moisture uptake was observed with LMH, M, S, T, SLS, and MS, all of which showed <1% moisture sorption up to 75% RH. Povidone and crospovidone are very hygroscopic and therefore they have a tendency to absorb any moisture present, either from environment or from API. Thus it might act as a catalyst if the moisture is present and might accelerate the degradation reaction as was seen in stability samples. SLS, on the other hand, is not very hygroscopic at low RH. But at very high RH, it showed deliquescence. This might have caused acceleration in its potential to degrade levothyroxine. Thus, it is a combination of factors, such as processing as well as formulation, which might be very important to consider while formulating levothyroxine sodium pentahydrate tablets as some of them influences negatively on its stability.
Fig. 7.

Moisture sorption isotherms of excipients used in levothyroxine sodium tablets. (a) povidone, crospovidone, sodium starch glycolate, acacia, croscarmellose sodium, corn starch, colloidal silicon dioxide, microcrystalline cellulose and (b) magnesium stearate, sodium laurel sulfate, talc, sucrose, mannitol, lactose monohydrate
The type of excipients used in the manufacturing of levothyroxine sodium pentahydrate tablets will influence its stability. Certain excipients which caused rapid degradation of levothyroxine in this study should be avoided to have a more stable preparation. Thus excipients should be judiciously selected which will not cause any degradation of levothyroxine. The primary degradation pathway in solid-state levothyroxine was identified as deiodination and deamination (5). These findings are consistent with the previous findings of Patel et al., Andre et al., and Kazemifard et al. (6,8,23).
Thus influence of processing and formulation factors should be carefully evaluated before formulating the tablets. Lot-to-lot variability, potency, and stability issues could be limited by choosing the factors which do not cause or catalyze the degradation of levothyroxine sodium. The main purpose of the manuscript was to evaluate the effect of formulation and manufacturing variables on the stability of levothyroxine, specifically, to understand the causes of potency loss in some marketed tablet formulations as compared to initial potency, and to screen the excipients for their protective effects. It would be of interest to see the effect of tabletting, but it is beyond the scope of the present study.
CONCLUSIONS
Levothyroxine sodium pentahydrate drug substance was stable at long-term as well as accelerated stability conditions for the study period of 28 days. It loses some of its moisture rapidly when placed in a dry environment; however, it is stable in the dehydrated form as well as seen from the studies conducted by placing a dried form under the stability conditions of 0% RH at 25°C and 40°C. The processing conditions while formulating it in tablet forms might encounter such transient conditions and, therefore, it was important to evaluate effects of these factors on the stability of levothyroxine. Formulation factors such as excipients were also found to influence its stability. Crospovidone, povidone, and sodium laurel sulfate were found to be unsuitable excipients when formulated in the presence of moisture as they cause degradation of levothyroxine. The degradation pathways for levothyroxine in presence of these excipients were deiodination and deamination. Therefore, a careful selection of excipients might prevent potency loss over the shelf-life of the tablets which could be a significant issue for some of the formulations of levothyroxine sodium pentahydrate.
Acknowledgments
Disclaimer
The opinions expressed in this work are only of the authors, and do not necessarily reflect the policy and statements of the FDA.
References
- 1.Goodman L, Gilman A. The pharmacological basis of therapeutics. 10. New York: McGraw-Hill; 2001. [Google Scholar]
- 2.Rhodes C. Regulatory aspects of the formulation and evaluation of levothyroxine tablets. Clin Res Regul Aff. 1998;15:173–186. doi: 10.3109/10601339809109194. [DOI] [Google Scholar]
- 3.FDA. Regulatory history and current issues. Health & Human Services; 2006.
- 4.Post A, Warren R. Sodium levothyroxine. In: Florey K, editor. Analytical profiles of drug substances. New York: Academic Press; 1976. pp. 226–281. [Google Scholar]
- 5.Won CM. Kinetics of degradation of levothyroxine in aqueous solution and in solid state. Pharm Res. 1992;9:131–137. doi: 10.1023/A:1018952415732. [DOI] [PubMed] [Google Scholar]
- 6.Kazemifard AG, Moore DE, Aghazadeh A. Identification and quantitation of sodium-thyroxine and its degradation products by LC using electrochemical and MS detection. J Pharm Biomed Anal. 2001;25:697–711. doi: 10.1016/S0731-7085(01)00370-3. [DOI] [PubMed] [Google Scholar]
- 7.Wortsman J, Papadimitriou DC, Borges M, Defesche CL. Thermal inactivation of l-thyroxin. Clin Chem. 1989;35:90–92. [PubMed] [Google Scholar]
- 8.Patel H, Stalcup A, Dansereau R, Sakr A. The effect of excipients on the stability of levothyroxine sodium pentahydrate tablets. Int J Pharm. 2003;264:35–43. doi: 10.1016/S0378-5173(03)00387-9. [DOI] [PubMed] [Google Scholar]
- 9.Das Gupta V, Odom C, Bethea C, Plattenburg J. Effect of excipients on the stability of levothyroxine sodium tablets. J Clin Pharm Ther. 1990;15:331–336. doi: 10.1111/j.1365-2710.1990.tb00393.x. [DOI] [PubMed] [Google Scholar]
- 10.Burger A, Griesser UJ. Physical stability, hygroscopicity and solubility of succinyl-sulfathiazole. Eur J Pharm Biopharm. 1991;37:18–24. [Google Scholar]
- 11.Morris KR, Griesser UJ, Eckhardt CJ, Stowell JG. Theoretical approaches to physical transformations of active pharmaceutical ingredients during manufacturing processes. Adv Drug Deliv Rev. 2001;48:91–114. doi: 10.1016/S0169-409X(01)00100-4. [DOI] [PubMed] [Google Scholar]
- 12.Otsuka M, Hasegawa H, Matsuda Y. Effect of polymorphic transformation during the extrusion-granulation process on the pharmaceutical properties of carbamazepine granules. Chem Pharm Bull. 1997;45:894–898. [Google Scholar]
- 13.Phadnis NV, Suryanarayanan R. Polymorphism in anhydrous theophylline—implications on the dissolution rate of theophylline tablets. J Pharm Sci. 1997;86:1256–1263. doi: 10.1021/js9701418. [DOI] [PubMed] [Google Scholar]
- 14.Monkhouse DC. Stability aspects of preformulation and formulation of solid pharmaceuticals. Drug Dev Ind Pharm. 1984;10:1373–1412. doi: 10.3109/03639048409039058. [DOI] [Google Scholar]
- 15.Kandarapu R, Grover V, Chawla HPS, Garg S. Evaluation of the compatibility of ketorolac tromethamine with selected polymers and common tablet excipients by thermal and isothermal stress testing. S T P Pharma Sci. 2001;11:449–457. [Google Scholar]
- 16.Serajuddin AT, Thakur AB, Ghoshal RN, Fakes MG, Ranadive SA, Morris KR, Varia SA. Selection of solid dosage form composition through drug-excipient compatibility testing. J Pharm Sci. 1999;88:696–704. doi: 10.1021/js980434g. [DOI] [PubMed] [Google Scholar]
- 17.Gu L, Strickley RG, Chi LH, Chowhan ZT. Drug-excipient incompatibility studies of the dipeptide angiotensin-converting enzyme inhibitor, moexipril hydrochloride: dry powder vs wet granulation. Pharm Res. 1990;7:379–383. doi: 10.1023/A:1015871406549. [DOI] [PubMed] [Google Scholar]
- 18.Shah RB, Bryant A, Collier J, Habib MJ, Khan MA. Stability indicating validated HPLC method for quantification of levothyroxine with eight degradation peaks in the presence of excipients. Int J Pharm. 2008;360:77–82. doi: 10.1016/j.ijpharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 19.Kopelman SH, Augsburger LL. Excipient compatibility study of Hypericum perforatum extract (St. John’s Wort) using similarity metrics to track phytochemical profile changes. Int J Pharm. 2002;237:35–46. doi: 10.1016/S0378-5173(02)00025-X. [DOI] [PubMed] [Google Scholar]
- 20.Patel N, Patel I, Cutie A, Wadke D, Monkhouse D, Reier G. The effect of selected direct compression excipients on the stability of aspirin as a model hydrolysable drug. Drug Dev Ind Pharm. 1998;14:77–98. doi: 10.3109/03639048809151962. [DOI] [Google Scholar]
- 21.van Dooren AA, Duphar BV. Design for drug–excipient interaction studies. Drug Dev Ind Pharm. 1983;9:43–55. doi: 10.3109/03639048309048544. [DOI] [Google Scholar]
- 22.Kowalski J, Kalb O, Joshi YM, Serajuddin ATM. Application of melt granulation technology to enhance stability of a moisture sensitive immediate-release drug product. Int J Pharm. 2009;381:56–61. doi: 10.1016/j.ijpharm.2009.05.043. [DOI] [PubMed] [Google Scholar]
- 23.Andre A, Domanig R, Riemer E, Moser H, Groeppelin A. Identification and thermal degradation products of l-triiodothyroxine sodium (liothyroxine sodium) by reversed phase HPLC with photodiode-array UV and mass spectrometric detection. J Chromatogr A. 1996;725:287–294. doi: 10.1016/0021-9673(95)01007-6. [DOI] [Google Scholar]