

Abstract
The objective of the present investigation was to prepare novel solid dispersions (SDs) of poorly water-soluble drugs with special microstructural characteristics using electrospinning process. With the hydrophilic polymer polyvinylpyrrolidone as the filament-forming polymer and acetaminophen (APAP) as the poorly water-soluble drug model, SDs having a continuous web structure, and in the form of non-woven nanofiber membranes, were successfully prepared. The electrospun nanofiber-based SDs were compared with those prepared from three traditional SD processes such as freeze-drying, vacuum drying, and heating drying. The surface morphologies, the drug physical status, and the drug-polymer interactions were investigated by scanning electron microscopy, differential scanning calorimetry, X-ray diffraction, and attenuated total reflectance Fourier transform infrared. In vitro dissolution tests demonstrated that the electrospun nanofibers released 93.8% of the APAP content in the first 2 minutes and that the dissolution rates of APAP from the different SDs had the following order: electrospun membrane > vacuum-dried membrane ≈ freeze-dried membrane > heat-dried membrane. Electrospun nanofiber-based SDs showed markedly better dissolution-improving effects than the other SDs, mainly due to their huge surface area, high porosity resulting from web structure, and the more homogeneous distribution of APAP in the nanofiber matrix.
Key words: drug-loaded nanofiber, electrospinning, microstructural characteristics, poorly water-soluble drug, solid dispersion
INTRODUCTION
The therapeutic effectiveness of a drug depends upon the bioavailability and ultimately upon the solubility of the drug molecules. Good solubility is one of the important criteria necessary to achieve the desired concentration of drug in the systemic circulation for a pharmacological response to be shown. Currently, about 40% or more of drugs in development and about 60% of molecules obtained directly from synthesis are poorly soluble in water. The solubility behavior of poorly water-soluble drugs remains one of the most challenging aspects of formulation development (1,2).
Over the years, numerous processes and technologies have been developed to enhance the solubility of drugs, including chemical methods and physical approaches. Among all the methods, use of solid dispersions (SDs) is considered to be one of the most suitable choices to improve dissolution rates and hence the bioavailability of poorly water-soluble drugs. Through altering the drugs' physical status and particle size, SD approaches can provide the following benefits for drug delivery: enhancement of solubility and bioavailability; rapid formulation for first use in man; time reduction of process; development and scale-up; dose reduction; improved efficacy; and reduction in variability between fed and fasted conditions (3–8).
By dissolving both the active pharmaceutical ingredients and the carriers in a common solvent or mixture of solvents and then evaporating the solvent to achieve an SD is a widely used process. Differences in solvent evaporation processes are related to the solvent evaporation procedure, which usually include vacuum drying, heating of the mixture on a hot plate, slow evaporation of the solvent at low temperature, the use of a rotary evaporator, a stream of nitrogen, spray-drying, three-dimensional printing, freeze-drying, and the use of supercritical fluids (9–13).
There are two problems associated with the traditional solvent evaporation SDs. First, the advantages of SDs mainly relied on the pharmaceutical excipients, and little effort has been spent developing the microstructure of SD products, even for third-generation SDs (14). This may be related to the fact that SDs are always semi-finished products and must be transferred into concrete dosage forms for patients. Second, in the evaporation process, it is hard to remove the organic solvents from the co-precipitates to an acceptable level rapidly because the co-precipitates become more and more viscous during the “drying” process, which prevents further evaporation of the residual solvent and often results in the formation of drug crystals in the SD (15,16). Fast removal of the organic solvent during the preparation process can benefit the characteristics of SD.
Nanotechnology may provide useful strategies for overcoming these problems and result in SDs with superior performance. Today, nano-ization strategies have been widely used to enhance the dissolution properties and oral availability of numerous drugs that are poorly soluble in water, by enlarging the surface area of the drug powder or changing the crystalline form. The most commonly used methods in the literature for this purpose are mainly nanocrystal and nanoparticle technologies (17–19). However, if microstructure and nano-ization can be applied to poorly water-soluble drug SDs with macroscopic configurations, it may endow the products with both the merits possessed by “nano-” drug delivery systems in altering the biopharmaceutic and pharmacokinetic properties, together with the advantages of conventional solid dosage forms.
In the past several years, electrospinning has regained attention due in part to a surging interest in nanotechnology, as nanofibers can be easily fabricated using this top-down process (20). This process is able to combine the traditional SD approaches and nano-medicinal strategies together naturally and may show advantages over the traditional SD preparation technologies both in the preparation process and in the properties of the products.
In our previous studies, we have prepared drug-loaded nanofibers and oral fast-dissolving membranes using the electrospinning process (21,22). To further investigate the advantages of the electrospinning process, the performance of nanofiber-based SDs and for related fundamental reasons, SD from three traditional processes, i.e., freeze-drying, vacuum drying, and heating drying, were prepared and characterized for comparison here.
MATERIALS AND METHODS
Materials
Acetaminophen (APAP, purity>99%) was obtained from the 4th Pharmaceutical Factory of Weifang (Shandong, China). Kollidon K90 (Mw = 360,000, purity>95%) was obtained from BASF Corp. (Ludwigshafen, Germany). Anhydrous ethanol of analytical grade was purchased from the Sinopharm Chemical Reagent Co., Ltd. All other chemicals used were analytical grade, and water was distilled before use.
Preparation of Drug-Polymer Co-dissolving Solutions
APAP and the polymer PVP K90 were co-dissolved in 90% aqueous ethanol at ambient temperature (24°C). The concentrations of polymer polyvinylpyrrolidone (PVP) and APAP were 8.0% (w/v) and 4.0% (w/v), respectively. Mechanical stirring was applied for at least 2 h to achieve homogeneous co-dissolving solutions. The solutions were degassed with a SK5200H ultrasonator (350 W, Shanghai Jinghong Instrument Co., Ltd. Shanghai, China) for 15 min before SD preparation processes.
Preparation of SDs
Electrospinning
The electrospinning process was carried out under ambient conditions (24°C and relative humidity 65 ± 5%). The co-dissolving solutions were placed in a syringe (10 ml) with a metal needle (diameter 0.5 mm). A power supply (ZGF60KV/2 mA, Soute Co., Ltd., China) was used at a voltage of 15 kV, and the electrospun nanofibers were collected on aluminum foil at a distance of 15 cm. The flow rate was fixed at 3.0 ml/h by a syringe pump (Cole-Parmer®, USA). The thickness of the electrospun membrane can be manipulated simply by controlling the continuous electrospinning time (21).
Freeze-drying
Ten milliliters of the co-dissolving solutions was placed in a Petri-dish (12 cm in diameter) with aluminum foil on the surface and were frozen in an ultra-low temperature freezer (MDF-382E, SANYO Electric Biomedical CO., Ltd., Japan) overnight at −85 ± 1°C. Later, the frozen solutions were dried using a freeze-drying machine (LGT-10, Beijing Songyuan Huaxing Scientific CO., Ltd., China) at a temperature of −46 ± 1°C under a pressure of 900 Pa. The dried membranes were peeled off from the aluminum foil after reaching a constant weight (2,7,14).
Vacuum drying
Ten milliliters of the co-dissolving solutions was placed in a Petri-dish (12 cm in diameter) with aluminum foil on the surface and were dried in a DZF-6050 Electric Vacuum Drying Oven (Shanghai Laboratory Instrument Work Co. Ltd., Shanghai, China) to facilitate the removal of solvents at 50 ± 1°C under vacuum (320 Pa). The dried membranes were peeled off from the aluminum foil after reaching a constant weight (2,7,14).
Heating drying
Ten milliliters of the co-dissolving solutions was placed in a Petri-dish (12 cm in diameter) with aluminum foil on the surface and were dried in a ZD-85 double function gas bath constant temperature oscillator (Jintan Science Analysis Instrument Co, Ltd, China) at a temperature of 50 ± 1°C. The dried membranes were peeled from the aluminum foil after reaching a constant weight (2,14).
Characterization
A JSM-5600LV scanning electron microscope (Japan Electron Optics Laboratory Co. Ltd.) was used to observe the surface morphologies of membranes. Prior to examination, samples were gold sputter-coated under argon to render them electrically conductive. Pictures were then taken at an excitation voltage of 15 kV.
The differential scanning calorimetry (DSC) analyses were carried out using an MDSC 2910 differential scanning calorimeter (TA Instruments Co., USA). The sample pans were sealed and heated at 10°C/min from 20°C to 250°C. The nitrogen gas flow rate was 40 ml/min.
The wide-angle X-ray diffractograms were obtained on a D/Max-BR diffractometer (RigaKu, Japan) using a slit detector with Cu Ka radiation in the 2q range of 5–60° at 40 mV and 300 mA. The samples were placed on an etched glass slide and scanned at a rate of 10°/min.
The attenuated total reflectance Fourier transform infrared (ATR-FTIR) were carried out on a Nicolet-Nexus 670 Fourier transform infrared (FTIR) spectrometer (Nicolet Instrument Corporation, Madison, USA) at the scanning range of 500–4,000 cm−1 and a resolution of 2 cm−1.
Drug Contents and In Vitro Dissolution Tests
APAP contents in SDs were determined. Six disks with a diameter of 16 mm were cut from different places of the SDs for evaluating the content uniformities. The disks were dissolved in 0.1 mol/L phosphate buffer solution (PBS), and absorbance at 257 nm was used to estimate the amounts of APAP in the samples.
A nanofiber membrane was prepared for in vitro dissolution tests, by conducting the electrospinning process continuously for 4 h. The 3 × 3 cm2 samples were cut from the dried membranes for in vitro dissolution tests. Thicknesses were measured by an electronic digital caliper (Shanghai Shenhan Measuring tools Co., Ltd., Shanghai, China) and weights by an electronic balance (Sartorius, Göettingen, Germany).
In vitro dissolution studies were conducted according to the Chinese Pharmacopeia (2005 ED.) Method II, which is a paddle method using a RCZ-8A dissolution apparatus (Tianjin University Radio Factory, China) at 50 rpm in 900 ml 0.1 mol/L PBS at 37°C, sink conditions C < 0.2Cs. At predetermined time intervals, samples of 5.0 ml were withdrawn from the dissolution medium and replaced with fresh medium by means of injectors to maintain constant volume. After filtration through a 0.22-µm membrane (Millipore, USA) and appropriate dilution with PBS, the sample solutions were analyzed at 257 nm by a UV spectrophotometer (Unico Instrument Co. Ltd., Shanghai, China). Pure APAP particles (<100 μm) were tested as control. The amount of APAP present in the sample was calculated with the help of an appropriate calibration curve. All the measurements were carried out six times.
RESULTS AND DISCUSSION
Morphology
The surface morphologies of membranes from different processes are shown in Fig. 1. It is evident that there are many drug particles on the surface of membranes prepared by heat-drying (Fig. 1a) and vacuum drying (Fig. 1b), whereas there are no visible particles on the surface of membranes prepared by freeze-drying (Fig. 1c) and by electrospun nanofibers (Fig. 1d).
Fig. 1.

SEM images of membranes. Magnifications are ×2,000 for all the images, and ×10,000 for the further magnified images in the upper right-hand corners. Images are of membranes formed by a heat-drying, b vacuum drying, c freeze-drying, and d electrospinning
The surface of the heat-dried membrane was compact with phase-separated particles having a relatively regular shape and similar size, about 0.2 to 1.2 μm. The surface of the vacuum-dried membrane was uneven with phase-separated particles having an irregular shape and a large range of size distributions. During the heat-drying process, the mixed solvents ethanol and water were evaporated slowly, the co-dissolving solutions gradually became more and more viscous, and the whole drying process took a long time. In this process, it is the drug molecular mobility (a key factor governing the stability of amorphous phases) that led to the nucleation of APAP and growth of phase-separated particles from the bulk membranes as the co-dissolving solutions were transformed into solid state (23).
Vacuum drying at elevated temperature bears the risk of phase separation because the mobility of drug and matrix decrease slowly (24). Figure 1b clearly demonstrates this phenomenon. This may be associated with the continuous change of the solvents' evaporation rate under vacuum and the moderate heating conditions, which resulted in a change in the interfacial tension and the corresponding formation of particles on the membrane surface from phase separation. In comparison, in the heat-drying process, the evaporation rates of solvents are relatively constant, resulting in a relatively smooth surface and particles with more regular shape and less range of size distributions than particles on vacuum-dried membranes.
Freeze-drying is a promising and suitable technique to incorporate drug substances in stabilizing matrices. The most important advantage of freeze-drying is that the risk of phase separation is minimized as soon as the solution is vitrified (25). The freeze-dried membrane had many furrows on it but no visible phase separation particles, which is a consequence of the fast vitrification and sublimation of the solvents during the freeze-drying process.
There were no particles visible on the surface of nanofibers, which had an estimated diameter of 300–800 nm. The electrospinning process is very similar to the solvent methods for preparing SD, as far as the removing of organic solvents is concerned. During the electrospinning process, when the polymer jet is ejected and accelerated toward the collector, the solvent evaporates rapidly as the high surface area jet travels to the target (26). As an outcome of rapid evaporation of solvent, non-woven filament mats are formed rapidly, and this leads to a decrease in drug mobility. When solvent evaporation is complete, drug molecules are often “frozen” in the polymer fibers matrix comparable to the liquid solutions. All these processes are completed in several decades of milliseconds. This left no time for the drug molecules to form crystal lattices, and thus the drug complexed with the polymer on a molecular scale in the nanofiber membrane.
Differential Scanning Calorimetry
The DSC thermograms of pure APAP (Fig. 2e) exhibited a single endothermic response corresponding to the dissolution of the APAP at 169.8°C (ΔHf 118.45 J/g). The filament-forming matrix polymer PVP did not show any fusion peak or phase transition, apart from a broad endotherm due to dehydration, which lies between 60–120°C with a peak at 86.1°C. (Fig. 2f; 27).
Fig. 2.

DSC thermograms, representing the first scan with a scan rate of 10°C/min, of membranes formed by a freeze-drying, b vacuum drying, c heat-drying, d electrospinning, and of e pure APAP, and f PVP K90
DSC thermograms of the freeze-dried membrane (Fig. 2a), vacuum-dried membrane (Fig. 2b), and electrospun nanofiber membrane (Fig. 2d) had almost the same behavior with a broad endotherm, where the glass transition temperature was lower than 84°C, and there was no melting peak of the drug. This suggested that APAP was no longer present as a crystalline material, but had been converted into an amorphous state. APAP has been found to act as a plasticizer when blended to different types of polymers. Structural analogs of NSAIDs, responsible for the anti-inflammatory effect, also contribute to the common plasticizing effects (28,29). Here, APAP molecules served to plasticize the PVP, leading to a reduction of the glass transition temperature. For the heat-dried membrane, the DSC curve had an additional temperature peak at 131.2°C besides another peak at 84.5°C. This suggested that the SDs in the heat-dried membrane were different from those prepared by the other processes, in which almost all the drug existed in the membranes in an amorphous status. However, there were still some APAP crystals in the heat-dried membrane, which had different crystalline behavior compared with the pure APAP particles due to the influence of PVP on the nucleation and growth processes of APAP re-crystallization (30). The particles on the surface of heat-dried membranes may have a heterogeneous structure with mixed properties of amorphous particles and nano-/micro-crystal particles. Whereas the particles on the vacuum-dried membrane were totally amorphous complexes of drug and PVP separated from the membrane body, as indicated in Fig. 2b. On the other hand, the disruption of the crystalline structure of APAP by the polymeric carrier PVP during the re-crystallization process resulted in a lower melting temperature, i.e., from 169.8°C of pure drug crystals to 131.2°C of crystal particles of acetaminophen–polymer polyvinylpyrrolidone (APAP–PVP).
As far as freeze-dried membranes and electrospun nanofiber membranes are concerned, there is no phase separation phenomenon leading to particles on the surface. The reason for the former is that quickly freezing limited the mobility of the drug and polymer molecules, and the reason for the latter is that the solvent evaporation was too fast for drug molecules to have enough time to undergo nucleation and crystal growth.
X-ray Diffraction
The X-ray diffraction patterns of the APAP, PVP, and of membranes prepared by the different processes are compared in Fig. 3. The presence of numerous distinct peaks (Fig. 3a) indicated that APAP was present as a crystalline material with characteristic diffraction peaks appearing at a diffraction angle of 2θ at 12.04°, 15.40°, 20.28°, 24.24°, and 32.68°. The PVP diffraction (Fig. 3b) exhibits a diffused background pattern with two diffraction halos, which means that the polymer is amorphous.
Fig. 3.

X-ray diffraction patterns using a slit detector and with a scan rate of 10°/min of a APAP, b PVP K90, and membranes prepared by c electrospinning, d heat-drying, e vacuum drying, and f freeze-drying
In the spectra of membranes, all the characteristic peaks of APAP completely disappeared, but characteristic humps of amorphous forms were observed at 20.40° for the heat-dried membrane (Fig. 3d), 20.48° for the vacuum-dried membrane (Fig. 3e), 20.98° for the freeze-dried membrane (Fig. 3f), and 22.06° for the electrospun nanofiber membrane (Fig. 3c).
In the XRD diffraction patterns of the four membranes, the shape and the position of the amorphous halo are similar but different. The incorporation and distributions of drug molecules in the solid PVP membranes are mainly based on the hydrogen bonding interactions between the APAP and PVP molecules, which could alter the orientation, conformation, and organization of polymer chains in the amorphous phase and thus change the amorphous packing density of polymer chains, resulting in the difference of the widths and positions of the amorphous halo in the XRD spectra (31). As the packing density increased, the amorphous halo became wider and shifted to higher angles correspondingly. Thus, the following order of amorphous packing density is clear and reasonable: heat-drying < vacuum drying < freeze-drying < electrospinning.
The DSC results demonstrated that there were some APAP crystalline particles in the heat-dried membrane, which could not be detected from the XRD diffraction patterns. This may be because the X-ray diffraction peaks were covered by the high amorphous halo of the PVP–APAP complex or that PVP absorbed to the APAP crystals formed during the slow heat-drying process attenuated the X-ray diffraction (30).
ATR-FTIR Spectroscopy
FTIR spectroscopy is often employed to study the interactions in SD (32). ATR-FTIR spectra of pure APAP, PVP K30, and membranes prepared by the different processes are shown as Fig. 4.
Fig. 4.

ATR-FTIR spectra: membranes prepared by a vacuum drying, b freeze-drying, c heat-drying, d electrospinning, e PVP K90, f APAP
The FTIR spectrum of pure APAP is shown in Fig. 4f. The peak at 3,325 cm−1 is assigned to the N–H stretching vibration, and the peaks around 3164 cm−1 might be due to the OH stretching vibration plus other combination bands. The peaks at 1,655, 1,565, and 1,260–1,228 cm−1 are assigned to the C═O stretching vibration of amide I band, N–H in-plane bending and/or C–N stretching of amides II and III vibrational bands, respectively. The peaks at 1,611, 1,507, and 1,443 cm−1 is due to the C–C bond stretching of aromatic benzene ring (33–35). The spectrum of PVP K30 (Fig. 4e) showed important bands at 2,953 cm−1 (C–H stretch) and 1661 cm−1 (C═O) (36).
The vacuum-dried membrane (Fig. 4a), freeze-dried membrane (Fig. 4b), and electrospun membrane (Fig. 4d) had similar FTIR spectra. The sharp peak of 3,325 cm−1 and peaks around 3,164 cm−1 in the pure APAP spectrum have changed to broad but weak bands in the membranes, i.e., 3,251, 3,261, and 3,268 cm−1 for electrospun membrane, freeze-dried membrane, and vacuum-dried membrane, respectively. The peaks at 1,611, 1,507, and 1,443 cm−1 corresponding to the C═C stretching of the aromatic benzene ring attenuated, shifted, or even disappeared in the membrane spectra. In the fingerprint regions, almost all of the peaks were shifted, decreased in intensity, or even disappeared from the spectra of the three membranes. All these were attributed to the hydrogen bonding between the hydroxyl group of APAP and the carboxyl group of PVP in the solid membrane, which has also been demonstrated in other reports (32,37). In theory, PVP is capable of forming a hydrogen bond either through the nitrogen or carbonyl group on the pyrrole ring. However, steric hindrance precludes the involvement of nitrogen atom in intermolecular interactions, thus making the carbonyl group more favorable for hydrogen bonding (38), shown in Fig. 5b.
Fig. 5.

Hydrogen bonding: a APAP–APAP, b APAP–PVP K90
The FTIR spectra of the heat-dried membrane still showed a small peak of –NH valence vibration at 3,325 cm−1. The wave number peaks are similar to the pure APAP but with a shift to lower wave numbers and with weaker peak intensity. All these suggest that there were some APAP crystals in the heat-dried membrane in addition to the APAP–PVP H-bonding complex, which concurs with the results of DSC.
When the co-dissolving solutions of APAP and PVP K30 were drying under a constant moderate temperature, the drug APAP gradually became supersaturated in the co-dissolving solutions as the solvent evaporated. The drug molecular mobility, hydrogen bonding between APAP molecules (Fig. 5a), and the nature of interactions between PVP and APAP are physical and reversible (38) and lead to nucleation and crystal growth to some extent during the drying process, although PVP is found to strongly reduce the crystal growth rate by several orders of magnitude. On the other hand, during the later time period of the heat-drying process, the co-precipitates became more and more viscous and prevent further evaporation of the residual solvent, leaving a long time for the nucleation and crystal growth. De Villiers et al. reported similar results that there was still 1.99 ± 0.3% crystalline APAP in SDs with ethanol as the co-solvent of APAP and PVP K30 and a drug content of 30.10% by weight (39).
Drug Contents and In Vitro Dissolution Tests
The average values of drug contents in six disks were 10.321 ± 0.008, 11.044 ± 0.009, 10.963 ± 0.008, and 10.892 ± 0.010 mg, meaning the relative standard deviations were 0.78%, 0.81%, 0.73%, and 0.92%, for membranes prepared from electrospinning, freeze-drying, vacuum drying, and heat-drying processes, respectively. Drug content uniformity can be ensured because all the SDs were prepared from totally co-dissolving solutions and APAP and PVP had good compatibility.
The 3 × 3 cm2 patches of vacuum-dried membrane, freeze-dried membrane, and the heat-dried membrane had an average thickness of 0.161 ± 0.011, 0.157 ± 0.013, and 0.152 ± 0.010 mm and had an average weight of 146.9 ± 1.1, 148.7 ± 1.4, and 145.9 ± 1.2 mg, respectively. The 3 × 3 cm2 electrospun patches had an average thickness of 0.871 ± 0.042 and an average weight of 138.5 ± 2.1 mg. Correspondingly, the densities were 1.014, 1.052, 1.067, and 0.1767 g/cm3 for membranes prepared from vacuum-dried, freeze-dried, heat-dried, and electrospinning processes. All the sample patches had very similar amounts of drug, thus ensuring compatibility of measurements, for in vitro dissolution tests. The electrospun membranes had a very low density due to their highly porosity, but their thickness could be increased easily by prolonging the electrospinning time to manipulate the drug amount in the cut patches.
The dissolution profiles of membranes from different processes and the pure APAP particles are shown in Fig. 6, from which the following order of dissolution rate was found: electrospun membrane > freeze-dried membrane ≈ vacuum-dried membrane > heat-dried membrane > APAP particles. As shown in Fig. 6a, for the drug-loaded nanofiber membranes prepared by electrospinning 93.8% of APAP was free out in the dissolution medium in the first 2 min, and all APAP was exhausted in 5 min.
Fig. 6.

APAP dissolution profiles from membranes prepared by a electrospinning, b freeze-drying, c vacuum drying, d heat-drying, e profile of pure APAP particles
The time taken for the freeze-dried membrane (Fig. 6b) and vacuum-dried membrane (Fig. 6c) to release all the APAP was about 20 min. The regressed linear equations of the in vitro dissolution profiles were Y = 4.6387X + 2.8661 (R = 0.9854) and Y = 4.4675X + 3.9348 (R = 0.9832) for freeze-dried membrane and vacuum-dried membrane, respectively, suggesting that APAP in them was released linearly. The heat-dried membrane took 30 min to dissolve and release all the drug. The regressed linear equations of the in vitro dissolution profiles were Y = 3.2541X + 2.2659 (R = 0.9780), suggesting an approximately linear release manner (Fig. 6d). The APAP particles had a fast release rate at the beginning, but this was followed later by an unfavorable prolonged tailing-off of release toward completion, shown in Fig. 6e.
All the membranes were able to completely avoid the tailing-off of drug release due to the even distribution of APAP in the PVP matrix, and most of the drug was in an amorphous state. This is very useful for the reproducibility and robustness of drug release profiles. In comparison, the conventional tablets often have release tailing-off effects often caused by the large size of the drug particles (40,41).
The vacuum-dried membrane, freeze-dried membrane, and the heat-dried membrane release the incorporated APAP with zero-order kinetics. This may be because the membranes had almost a constant drug release surface area, due to the thin film structure, and hence the drug release was controlled by the dissolution and erosion process of the polymer. There are two stages involved in the polymer dissolution process: in the first place, the amorphous, glassy polymer PVP absorbs solvent to form a swollen gel, and next the swollen polymer chain disentangles and disperses into the solution (42,43). Thus, there are three factors that determine the dissolution rate of the loaded drug: (1) the polymer–solvent interactions, (2) the structure and the porosity of the solid membrane, and (3) the physical status of the drug distributed in the polymer matrix.
Accordingly, the fact that the electrospun membrane had the fastest dissolution rate can be attributed to the following reasons: (1) PVP has hygroscopic and hydrophilic properties, and the polymer–solvent interactions are higher than the polymer–polymer attraction forces, so that the polymer chain can absorb solvent molecules rapidly, causing an increase in the volume of the polymer matrix, and the polymer chains loosen out from their coiled shape; (2) the three-dimensional continuous web structure of the membrane can offer a huge surface area for the PVP to absorb water molecules, greater porosity for the water molecules to diffuse into the inner part of the membrane, and void space for the polymer to be swollen, disentangle, and for the dissolved APAP molecules to disperse into the bulk dissolution medium; (3) the drug and the matrix polymer formed composites at a molecular level; thus, the APAP molecules can dissolve simultaneously with PVP molecules.
The vacuum-dried membrane and the freeze-dried membrane had some void space formed during the drying process due to the vacuum condition leading to the fast evaporation of solvent, but the porosity cannot be matched with that of the electrospun membrane. The heat-dried membrane had a compact structure resulting from the slow evaporation of solvents, which would retard the diffusion of water molecules in the membrane dissolution process. In addition, there were crystalline APAP particles in the membrane, for which an additional time period was needed for APAP molecules to leave the crystal lattice progressively. Thus, the heat-dried membrane had an even slower dissolution rate than the others. The drug particles in the vacuum-dried membrane were a complex of PVP–APAP and the drug was totally amorphous, thus the drug particles had little influence on the synchronous dissolution of drug and PVP molecules.
For pure APAP particles, the dominant factor that controls their dissolution rate is their dimensions. Two characteristics were clear for APAP particles release profiles: (1) 75% of the drug was released in the first quarter and (2) the exhaustion time was over an hour. The former resulted from the smaller APAP particles, and the latter long tailing-off time periods was unavoidable due to the presence of bigger APAP particles.
Methods for improving drug solubility or dissolution rate continue to be highly sought after (44,45). Generally, the search for such methods has been guided by the Noyes–Whitney equation (46,47), which defines dissolution rate (dX/dt) as follows:
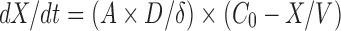 |
Where X is the amount of drug in solution, t is time, A is the effective surface area, D is the diffusion coefficient of the drug, δ is the effective diffusion boundary layer, C0 is the saturation solubility of the drug, and V is the volume of the dissolution medium. Based on this equation, the reasons that the electrospinning membrane exhibited more effective results in improving drug dissolution rate than the other membranes are listed in Table I.
Table I.
Comparison of Factors that Improve Drug Dissolution Rate
| SDs | A (surface area) | δ (diffusion layer thickness) | (C0-X/V)* |
|---|---|---|---|
| Electrospinning | Very large | Nano-scale | Big |
| Freeze-dried | Limited | Normal | Normal |
| Vacuum-dried | Limited | Normal | Normal |
| Heat-dried | More limited | Normal | Normal |
SDs solid dispersions
*Drug concentration gradients in the dissolution medium
The rate of dissolution is directly proportional to the surface area of the drug and to the drug concentration gradients in the dissolution medium, but is inversely proportional to the diffusion layer thickness at the solid–liquid interface. With regard to electrospinning membranes, first, their three-dimensional continuous nanoweb structure, and the molecular distributions of drug in them can provide tremendous surface area for the drug to be exposed to the dissolution media, and certainly, the great porosity facilitated the fast mass transformation of solvent and drug molecules from or to the dissolution medium. Secondly, a combination of the nano-scale diameters of the nanofibers which greatly decreases the diffusional layer thickness with the uniform PVP–APAP complex in the nanofibers, can greatly improve the drug wettability. Thirdly, the fast dissolution of the drug-loaded PVP nanofibers in the inner part of the electrospun membranes could augment the concentrations of supersaturated drug solution, especially with the presence of PVP, and thus correspondingly increase the drug gradients for rapid diffusion of drug molecules. In a word, it is the synergistic effect of nanosizing of fibers, web structure of the membranes, and composites of drug with the filament-forming matrix that endowed this novel type of SD with the marked effect of improving the dissolution rate of poorly water-soluble drugs.
The other three membranes had very limited surface areas, not enough porosity for quickly transferring solvent and drug molecules, and thus they dissolved gradually by the erosion of the surface of the whole membrane one layer after another layer. Especially for heat-dried membranes, they had a compact structure and some crystalline drug particles, resulting in an even poorer effect on improving the drug dissolution rate. Nevertheless, the SD membranes gave more stable drug release profiles than the crude drug particles. It can also be expected that the SD membranes from traditional processes would show better improving effects if the drug was more poorly soluble than APAP or if the excipient PVP had a smaller molecular weight, such as PVP K30.
Certainly, there are some other advantages of the nanofiber-based SD, such as easiness for formulations development, good stability due to the stereo web structure against the aging of SD, which is under investigation now. On the other hand, some researches are now focusing on the mass preparation of nanofibers for scale-up in production (48).
CONCLUSIONS
Nanofiber-based SD of APAP in the hydrophilic polymer PVP were prepared successfully using electrospinning process and were compared with those SD prepared by traditional SD processes, such as freeze-drying; vacuum drying, and heat-drying. SEM observations showed that there were microparticles on the heat-dried and vacuum-dried membranes due to phase separation during the drying process. There was no obvious particles on the surface of freeze-dried membranes and electrospun nanofibers. The very fast drying process of electrospinning and the fast vitrification and sublimation of freeze-drying process resulted in totally amorphous state of APAP in the PVP matrix membranes. The in vitro dissolution tests illustrated that the electrospun nanofibers released 93.8% of the containing APAP in the first 2 min and that the dissolution rates of APAP from the different SD had the following order: electrospun > vacuum-dried ≈ freeze-dried > heating-dried. The freeze-dried membrane had a similar drug release rate to the vacuum-dried membrane, and both were far slower than release from the electrospun nanofibers. What plays a determining role here is that the electrospun nanofiber membranes had a three-dimensional continuous web microstructure, which endowed them with excellent dissolution-improving effects for APAP. Novel electrospun nanofiber-based SD with microstructural characteristics exhibited excellent performance compared with SD prepared from conventional processes in improving dissolution rate of poorly water-soluble drugs.
Acknowledgement
This work was financially supported by the UK-CHINA Joint Laboratory for Therapeutic Textiles, China Postdoctoral Science Foundation (Special Grade No. 200902195) and Grant 08JC1400600 from Science and Technology Commission of Shanghai Municipality.
References
- 1.Chen H, Wan J, Wang Y, Mou D, Liu H, Xu H, et al. A facile nanoaggregation strategy for oral delivery of hydrophobic drugs by utilizing acid–base neutralization reactions. Nanotechnology. 2008;19:375104. doi: 10.1088/0957-4484/19/37/375104. [DOI] [PubMed] [Google Scholar]
- 2.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 3.Goddeeris C, Willems T, Den Mooter GV. Formulation of fast disintegrating tablets of ternary solid dispersions consisting of TPGS 1000 and HPMC 2910 or PVPVA 64 to improve the dissolution of the anti-HIV drug UC 781. Eur J Pharm Sci. 2008;34:293–302. doi: 10.1016/j.ejps.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Shah TJ, Amin AF, Parikh JR, Parikh RH. Process optimization and characterization of poloxamer solid dispersions of a poorly water-soluble drug. AAPS PharmSciTech. 2007;8(2):Article 29. doi: 10.1208/pt0802029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Six K, Daems T, De Hoon J, Hecken AV, Depre M, Bouche MP, et al. Clinical study of solid dispersions of itraconazole prepared by hot-stage extrusion. Eur J Pharm Sci. 2005;24:179–186. doi: 10.1016/j.ejps.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Zahedi P, Lee PI. Solid molecular dispersions of poorly water-soluble drugs in poly(2-hydroxyethyl methacrylate) hydrogels. Eur J Pharm Biopharm. 2007;65:320–328. doi: 10.1016/j.ejpb.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 7.Serajuddin AM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems and recent break-through. J Pharm Sci. 1999;88:1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 8.Liu L, Wang X. Improved dissolution of oleanolic acid with ternary solid dispersions. AAPS PharmSciTech. 2007;8(4):Article113. doi: 10.1208/pt0804113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chokshi RJ, Zia H, Sandhu HK, Shah NH, Malick WA. Improving the dissolution rate of poorly water-soluble drug by solid dispersion and solid solution—pros and cons. Drug Deliv. 2007;14:33–45. doi: 10.1080/10717540600640278. [DOI] [PubMed] [Google Scholar]
- 10.Goddeeris C, Den Mooter GV. Free flowing solid dispersions of the anti-HIV drug UC 781with Poloxamer 407 and a maximum amount of TPGS 1000: Investigating the relationship between physicochemical characteristics and dissolution behaviour. Eur J Pharm Sci. 2008;35:104–113. doi: 10.1016/j.ejps.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 11.Sun NY, Wei XL, Wu BJ, Chen J, Lu Y, Wu W. Enhanced dissolution of silymarin/polyvinylpyrrolidone solid dispersion pellets prepared by a one-step fluid-bed coating technique. Powder Technol. 2007;182:72–80. doi: 10.1016/j.powtec.2007.05.029. [DOI] [Google Scholar]
- 12.Yu DG, Zhu LM, Branford-White C, Yang XL. Three-dimensional printing in pharmaceutics—promises and problems. J Pharm Sci. 2008;97:3666–3690. doi: 10.1002/jps.21284. [DOI] [PubMed] [Google Scholar]
- 13.Pasquali I, Bettini R, Giordano F. Supercritical fluid technologies: an innovative approach for manipulating the solid-state of pharmaceuticals. Adv Drug Deliv Rev. 2008;60:399–410. doi: 10.1016/j.addr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 14.Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12:1068–1075. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto T, Zografi G. Physical properties of solid molecular dispersions of indomethacine with PVP and PVPVA in relation to indomethacine recrystallization. Pharm Res. 1999;16:1722–1728. doi: 10.1023/A:1018906132279. [DOI] [PubMed] [Google Scholar]
- 16.Den Mooter GV, Augustijns P, Blaton N. Physico-chemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm. 1998;164:67–80. doi: 10.1016/S0378-5173(97)00401-8. [DOI] [Google Scholar]
- 17.Rosen H, Abribat T. The rise and rise of drug delivery. Nat Rev Drug Discov. 2005;4:381–385. doi: 10.1038/nrd1721. [DOI] [PubMed] [Google Scholar]
- 18.Muller RH, Keck CM. Challenges and solutions for the delivery of biotech drugs - a review of drug nanocrystal technology and lipid nanoparticles. J Biotechnol. 2004;113:151–170. doi: 10.1016/j.jbiotec.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Moshfeghi AA, Peyman GA. Micro- and nanoparticulates. Adv Drug Deliv Rev. 2005;57:2047–2052. doi: 10.1016/j.addr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Huang ZM, Zhang YZ, Kotaki M, Ramakrishna S. A review on polymer nanofibers by electrospinning applications in nanocomposites. Composites Sci Technol. 2003;63:2223–2253. doi: 10.1016/S0266-3538(03)00178-7. [DOI] [Google Scholar]
- 21.Yu DG, Shen XX, Branford-White C, White K, Zhu LM, Bligh SWA. Oral fast-dissolving drug delivery membranes prepared from electrospun PVP ultrafine fibers. Nanotechnology. 2009;20:055104. doi: 10.1088/0957-4484/20/5/055104. [DOI] [PubMed] [Google Scholar]
- 22.Yu DG, Zhang XF, Shen XX, Branford-White C, Zhu LM. Ultrafine ibuprofen-loaded polyvinylpyrrolidone fiber mats using electrospinning. Polym Int. 2009;58:1010–1013. doi: 10.1002/pi.2629. [DOI] [Google Scholar]
- 23.Zhou D, Grant DJW, Zhang GGZ, Law D, Schmitt EA. A calorimetric investigation of thermodynamic and molecular mobility contributions to the physical stability of two pharmaceutical glasses. J Pharm Sci. 2007;96:71–83. doi: 10.1002/jps.20633. [DOI] [PubMed] [Google Scholar]
- 24.Langer M, Höltje M, Urbanetz NA, Brandt B, Höltje HD, Lippold BC. Investigations on the predictability of the formation of glassy solid solutions of drugs in sugar alcohols. Int J Pharm. 2003;252:167–179. doi: 10.1016/S0378-5173(02)00647-6. [DOI] [PubMed] [Google Scholar]
- 25.Dhirendra K, Lewis S, Udupa N, Atin K. Solid dispersions: a review. Pak J Pharm Sci. 2009;22:234–246. [PubMed] [Google Scholar]
- 26.Dzenis Y. Spinning continuous fibres for nanotechnology. Science. 2004;304:1917–1919. doi: 10.1126/science.1099074. [DOI] [PubMed] [Google Scholar]
- 27.Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272:1–10. doi: 10.1016/j.ijpharm.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 28.Rawas-Qalaji MM, Simons FR, Simons KJ. Fast-disintegrating sublingual tablets: effect of epinephrine load on tablet characteristics. AAPS PharmSciTech. 2006;7(2):Article 41. doi: 10.1208/pt070241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricci M, Blasi P, Giovagnoli S, Rossi C, Macchiarulo G, Luca G, et al. Ketoprofen controlled release from composite microcapsules for cell encapsulation: effect on post-transplant acute inflammation. J Control Release. 2005;107:395–407. doi: 10.1016/j.jconrel.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 30.Lindfors L, Forssen S, Westergren J, Olsson U. Nucleation and crystal growth in supersaturated solutions of a model drug. J Colloid Interface Sci. 2008;325:404–413. doi: 10.1016/j.jcis.2008.05.034. [DOI] [PubMed] [Google Scholar]
- 31.Murthy NS, Minor H, Bednarczyk C. Structure of the amorphous phase in oriented polymers. Macromolecules. 1993;26:1712–1721. doi: 10.1021/ma00059a034. [DOI] [Google Scholar]
- 32.Tantishaiyakul V, Kaewnopparat N, Ingkatawornwong S. Properties of solid dispersions of piroxicam polyvinylpyrrolidone. Int J Pharm. 1999;181:143–151. doi: 10.1016/S0378-5173(99)00070-8. [DOI] [PubMed] [Google Scholar]
- 33.Mills T, III, Price WN, Price PT, Roberson JC. Editors. Acetaminophen instrumental data for drug analysis. Elsevier, New York; 1982. p. 2.
- 34.Lin SY, Wang SL, Cheng YD. Thermally induced structural changes of acetaminophen in phase transition between the solid and liquid states monitored by combination analysis of FT-IR/DSC microscopic system. J Phys Chem Solids. 2000;61:1889–1893. doi: 10.1016/S0022-3697(00)00062-7. [DOI] [Google Scholar]
- 35.Sethia S, Squillante E. Physicochemical characterization of solid dispersions of carbamazepine formulated by supercritical carbon dioxide and conventional solvent evaporation method. J Pharm Sci. 2002;91:1948–1957. doi: 10.1002/jps.10186. [DOI] [PubMed] [Google Scholar]
- 36.Van den Mooter G, Augustijns P, Blaton N, Kinget R. Physico-chemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm. 1998;164:67–80. doi: 10.1016/S0378-5173(97)00401-8. [DOI] [Google Scholar]
- 37.Garekani HA, Sadeghi F, Ghazi A. Increasing the aqueous solubility of acetaminophen in the presence of polyvinylpyrrolidone and investigation of the mechanisms involved. Drug Dev Ind Pharm. 2003;29:173–179. doi: 10.1081/DDC-120016725. [DOI] [PubMed] [Google Scholar]
- 38.Sekizaki H, Danjo K, Eguchi H, Yonezawa Y, Sunada H, Otsuka A. Solid-state interaction of ibuprofen with polyvinylpyrrolidone. Chem Pharm Bull. 1995;43:988–993. [Google Scholar]
- 39.De Villiers MM, Wurster DE, Van der Watt JG, Ketkar A. X-ray powder diffraction determination of the relative amount of crystalline acetaminophen in solid dispersions with polyvinylpyrrolidone. Int J Pharm. 1998;163:219–224. doi: 10.1016/S0378-5173(97)00367-0. [DOI] [Google Scholar]
- 40.Rigbya SP, Van der Walle CF, Raistrick JH. Determining drug spatial distribution within controlled delivery tablets using MFX imaging. J Control Release. 2004;96:97–100. doi: 10.1016/j.jconrel.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Kim C. Compressed doughnut-shaped tablets with zero-order release kinetics. Pharm Res. 2004;12:1045–1048. doi: 10.1023/A:1016218716951. [DOI] [PubMed] [Google Scholar]
- 42.Miller-Chou BA, Koenig JL. A review of polymer dissolution. Prog Polym Sci. 2003;28:1223–1270. doi: 10.1016/S0079-6700(03)00045-5. [DOI] [Google Scholar]
- 43.Konno H, Handa T, Alonzo DE, Taylor LS. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm. 2008;70:493–499. doi: 10.1016/j.ejpb.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 44.Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–934. doi: 10.1021/ja02086a003. [DOI] [Google Scholar]
- 45.Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur J Pharm Sci. 2003;18:113–120. doi: 10.1016/S0928-0987(02)00251-8. [DOI] [PubMed] [Google Scholar]
- 46.Wurster DE, Taylor PW. Dissolution rates. J Pharm Sci. 1965;54:169–175. doi: 10.1002/jps.2600540202. [DOI] [PubMed] [Google Scholar]
- 47.Verreck G, Chun I, Peeters J, Rosenblatt J, Brewster ME. Preparation and characterization of nanofibers containing amorphous drug dispersion generated by electrostatic spinning. Pharm Res. 2003;20:810–817. doi: 10.1023/A:1023450006281. [DOI] [PubMed] [Google Scholar]
- 48.Varabhas JS, Chase GG, Reneker DH. Electrospun nanofibers from a porous hollow tube. Polymer. 2008;49:4226–4229. doi: 10.1016/j.polymer.2008.07.043. [DOI] [Google Scholar]