

Abstract
The purpose of the present study was to develop and optimize reservoir-based transdermal therapeutic system (TTS) for buspirone (BUSP), a low bioavailable drug. A three-factor, three-level Box–Behnken design was employed to optimize the TTS. Hydroxypropyl methylcellulose, d-limonene and propylene glycol were varied as independent variables; cumulative amount permeated across rat abdominal skin in 24 h, flux and lag time were selected as dependent variables. Mathematical equations and response surface plots were used to relate the dependent and independent variables. The statistical validity of polynomials was established, and optimized formulation factors were selected by feasibility and grid search. Validation of the optimization study with seven confirmatory runs indicated high degree of prognostic ability of response surface methodology. BUSP-OPT (optimized formulation) showed a flux 104.6 µg cm−2 h−1, which could meet target flux. The bioavailability studies in rabbits showed that about 2.65 times improvement (p < 0.05) in bioavailability, after transdermal administration of BUSP-OPT compared to oral solution. The ex vivo–in vivo correlation was found to have biphasic pattern and followed type A correlation. Reservoir-based TTS for BUSP was developed and optimized using Box–Behnken statistical design and could provide an effective treatment in the management of anxiety.
Key words: bioavailability, Box–Behnken, buspirone, ex vivo–in vivo correlation, reservoir TTS
INTRODUCTION
Buspirone (BUSP) is an anxiolytic drug that has dopaminergic, noradrenergic, and serotonin-modulating properties. BUSP is rapidly absorbed from gastrointestinal tract but systemic bioavailability is low (4%) because of extensive first pass metabolism (1). Most of metabolites of BUSP are inactive, although oxidative dealkylation produces an active metabolite, 1-(2–pyrimidinyl)-piperazine (which is about 20% to 25% as potent as parent drug). The major metabolite is 5-hydroxybuspirone and the metabolites are excreted mainly in urine and feces (2). The mean elimination half life of unchanged BUSP is merely 2–3 h (3). The low oral bioavailability restricts its use. Therefore, current BUSP treatment generally involves taking three daily oral doses of between 5 and 20 mg each. Due to the chronic nature of therapy required, a decrease in the number of daily doses would be desirable, as it would greatly enhance patient compliance. To overcome the problem of first pass metabolism, improve bioavailability and for effective treatment of anxiety, an alternative long-acting formulations could be beneficial. Transdermal route of administration may be a good alternative to circumvent these problems. The physical properties of BUSP such as low molecular weight (385.5), low dose (5 to 20 mg), and lipophilicity (log P 1.6) (4) are also favorable indicators for designing transdermal therapeutic system (TTS). For certain drugs, transdermal delivery offers a number of advantages with respect to oral or parenteral administration: improved patient compliance, reduced side-effects, elimination of first pass effect, interruption or termination of treatment when unnecessary, etc. (5).
There are no reports on transdermal delivery systems for BUSP. However, two reports were based on iontophoresis (4,6). Reservoir-type TTS containing a gel as a drug reservoir is selected for study. The gel serves as a vehicle for drug and needs to facilitate the controlled release of BUSP. For this study, hydroxypropyl methylcellulose K4M (HPMC) was selected as gel forming agent. Designing drug delivery systems with minimum number of experiments is very crucial for pharmaceutical scientists (7). In this study, we demonstrate the use of response surface methodology (Box–Behnken design) to optimize the reservoir-type TTS for BUSP.
The objectives of present study were to develop and optimize reservoir-type TTS for BUSP using HPMC (X1) as gel forming agent and d-limonene (DLM; X2) as penetration enhancer. Propylene glycol (PG; X3) was used as co-solvent, as it potentiates the permeation of the drug. The dependent variables are cumulative amount permeated in 24 h (Q24; Y1), flux (Y2) and lag time (Y3). The formulation was also evaluated for in vivo performance in rabbits.
MATERIALS AND METHODS
Materials
Buspirone hydrochloride and HPMC were gift samples from Dr. Reddys Laboratories, Hyderabad, India. d-limonene and Dulbecco's buffer (pH 7.4) were purchased from Himedia, Mumbai, India. Propylene glycol was purchased from Merck, Mumbai, India. All other chemicals and solvents were of analytical reagent grade.
Preparation of Reservoir-Based TTS
Weighed quantity of HPMC was placed in about 30 mL of water containing 20% v/v alcohol and was allowed for swelling for about 6 h. BUSP (250 mg) was solubilized in about 10 mL of water containing 20% v/v alcohol and was added to the polymeric solution. Measured quantities of PG and DLM were added with constant stirring (400 rpm) for 2 h and the quantity was adjusted to 50 gm with the addition of water.
Experimental Design
A three-factor, three-level Box–Behnken statistical design was used to optimize the formulation factors and evaluate main, interaction, and quadratic effects on Q24, flux and lag time. Box–Behnken design was used to explore quadratic response surfaces and constructing second-order polynomial models with Design Expert (Version 7.1, Stat-Ease Inc., Minneapolis, MN). The design was specifically selected since it requires fewer runs than central composite design in cases of three or four variables. This cubic design is characterized by set of points lying at midpoint of each edge and center point of the multidimensional cube (8). A design matrix comprising of 13 experimental runs was constructed. The non-linear computer generated quadratic model is given as 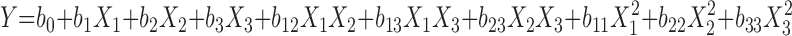 where Y is the measured response; b0 is an intercept; b1 to b33 are regression coefficients computed from observed experimental values of Y; and X1, X2, and X3 are the coded levels of independent variables. The terms
where Y is the measured response; b0 is an intercept; b1 to b33 are regression coefficients computed from observed experimental values of Y; and X1, X2, and X3 are the coded levels of independent variables. The terms 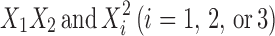 represent the interaction and quadratic terms, respectively. The dependent and independent variables selected were shown in Tables I and II along with their low, medium and high levels.
represent the interaction and quadratic terms, respectively. The dependent and independent variables selected were shown in Tables I and II along with their low, medium and high levels.
Table I.
Variables and Observed Responses in Box–Behnken Design for Buspirone Transdermal Therapeutic Systems
| Formulation | Independent variables | Dependent variables | Assay (%) | G I | Kp x 10−3 (cm−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| X 1 (gm) | X 2 (gm) | X 3 (mL) | Y 1 (μg) | Y 2 (μg cm−2 h−1) | Y 3 (h) | ||||
| BUSP1 | −1 | 0 | −1 | 7,191.8 ± 136.2 | 84.3 ± 0.67 | 0.30 ± 0.05 | 101.8 | 1.40 | 8.28 |
| BUSP2 | 1 | 0 | −1 | 3,124.4 ± 280.6 | 29.6 ± 1.01 | 0.34 ± 0.03 | 100.4 | 2.55 | 2.95 |
| BUSP3 | 0 | 0 | 0 | 7,633.9 ± 60.6 | 88.8 ± 0.98 | 0.18 ± 0.02 | 98.3 | 2.24 | 9.03 |
| BUSP4 | −1 | 1 | 0 | 5,354.9 ± 175.6 | 59.0 ± 0.74 | 0.22 ± 0.04 | 99.4 | 1.23 | 5.94 |
| BUSP5 | 0 | 0 | 0 | 7,704.5 ± 257.5 | 92.1 ± 0.21 | 0.29 ± 0.05 | 99.5 | 2.32 | 9.26 |
| BUSP6 | 0 | 1 | −1 | 4,977.1 ± 374.8 | 53.5 ± 0.35 | 0.45 ± 0.07 | 100.1 | 1.75 | 5.34 |
| BUSP7 | 1 | 0 | 1 | 2,854.3 ± 294.0 | 27.8 ± 0.72 | 0.34 ± 0.10 | 102.3 | 2.75 | 2.72 |
| BUSP8 | 0 | −1 | −1 | 8,231.4 ± 385.5 | 100.2 ± 1.83 | 0.64 ± 0.02 | 100.3 | 2.15 | 9.99 |
| BUSP9 | −1 | −1 | 0 | 8,921.6 ± 275.8 | 108.4 ± 2.61 | 0.45 ± 0.05 | 99.2 | 0.73 | 10.93 |
| BUSP10 | 1 | 1 | 0 | 2,344.2 ± 110.5 | 20.4 ± 0.13 | 0.44 ± 0.08 | 101.4 | 2.82 | 2.01 |
| BUSP11 | 1 | −1 | 0 | 4,301.4 ± 263.5 | 47.8 ± 0.35 | 0.56 ± 0.11 | 99.5 | 2.45 | 4.80 |
| BUSP12 | 0 | 1 | 1 | 5,272.5 ± 425.0 | 55.0 ± 0.27 | 0.40 ± 0.10 | 100.7 | 2.08 | 5.46 |
| BUSP13 | −1 | 0 | 1 | 5,525.3 ± 334.1 | 59.3 ± 0.58 | 0.27 ± 0.08 | 98.8 | 1.34 | 6.00 |
Table II.
Variables of Box–Behnken Design for Buspirone Transdermal Therapeutic Systems
| Independent variables | Levels used, actual (coded) | ||
|---|---|---|---|
| Low (−1) | Medium (0) | High (+1) | |
| X 1 = HPMC K 4 M (g) | 0.25 | 1.13 | 2 |
| X 2 = d-limonene (mL) | 4 | 8 | 12 |
| X 3 = Propylene glycol (mL) | 5 | 10 | 15 |
Rheological Measurements
The rheological measurements were performed using a controlled stress rheometer with the cone (24 mm) and plate geometry (Brookfield Programmable DVIII+Digital Rheometer, Brookfield Engineering Laboratories Inc., MA, USA). The viscosity was determined by torque sweep from 10% to 110%. All the measurements were performed in triplicate at 25°C. The equilibrium time before every measurement was 5 min and sample quantity used was approximately 0.5 gm. Calculation of rheological properties were performed using Rheocalc 32 software (Brookfield Engineering Laboratories Inc., USA).
Determination of Drug Content
Weighed quantity of about 1.0 g of reservoir system was placed in 100 mL of distilled water, sonicated for 10 min using bath sonicator and filtered through 0.45 μm membrane filter. The filtrate was suitably diluted, and the drug content in the sample was determined using high-pressure liquid chromatography (HPLC) (9). The experiments were conducted in triplicate.
Preparation of Rat Abdominal Skin
The animal study was conducted in accordance with the approval of the animal ethical committee, Kakatiya University, India. Albino rats weighing 150–200 gm were sacrificed using anaesthetic ether. The hair of test animals was carefully trimmed with electrical clippers and the full thickness skin was removed from abdominal region. The epidermis was prepared surgically by heat separation technique (10), which involved soaking the entire abdominal skin in water at 60°C for 45 s, followed by careful removal of the epidermis. The epidermis was washed with water and used for ex vivo permeability studies.
Ex vivo Permeation Studies
The ex vivo permeation of BUSP from reservoir system containing 10 mg of BUSP in 2 gm, was studied using Franz diffusion cell with a surface area of 3.8 cm2. The rat skin was mounted between donor and receptor compartments of the diffusion cell with stratum corneum facing the donor compartment. The reservoir system was placed in pre-activated dialysis membrane (1.4 × 2.8 mm, Himedia 70KD) and was further placed in donor compartment. Phosphate buffer saline (13 mL, PBS; pH 7.4) was placed in receptor compartment. The whole assembly was kept on a multi magnetic stirrer (Cintex, Mumbai, India) and the contents of receptor compartment were agitated at 400 rpm. The study was conducted at 37°C and samples of 1 mL were collected at predetermined time points and replenished with PBS (pH 7.4). The cumulative amount of BUSP permeated was determined using HPLC (9) and concentration was corrected for sampling effects according to Eq. 1 (11)
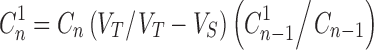 |
1 |
Where 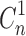 and Cn are the corrected and measured concentration of BUSP respectively in nth sample.
and Cn are the corrected and measured concentration of BUSP respectively in nth sample. 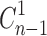 and Cn-1 are corrected and measured concentration of BUSP, respectively in (n-1)th sample. VT and Vs are the total volume of the receiver fluid and volume of sample drawn, respectively.
and Cn-1 are corrected and measured concentration of BUSP, respectively in (n-1)th sample. VT and Vs are the total volume of the receiver fluid and volume of sample drawn, respectively.
The steady state flux was calculated from slope of the steady state portion of line in the plot of drug amount permeated Vs time. Permeability coefficient (Kp) was calculated by dividing the flux with concentration of drug in TTS. The lag time was calculated from intercept on time axis in the plot of cumulative amount permeated Vs time. The target flux was calculated using Eq. 2.
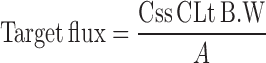 |
2 |
Css, the BUSP concentration at therapeutic level (2.5 μg L−1) and CLt the total body clearance, 1,700 mL h−1 (1), BW the standard human body weight of 60 kg, A represents surface area of diffusion cell (i.e., 3.8 cm2). The calculated target flux value for BUSP was 67.1 μg cm−2 h−1.
Determination of BUSP in Skin Layers
Drug content in the skin layers and donor compartment was determined for BUSP optimized (BUSP-OPT) formulation. After 24 h of study, the skin was homogenized using tissue homogenizer (Remi, Mumbai, India) and the drug was extracted into dichloromethane solvent system, evaporated to dryness and the drug content was estimated using HPLC (9). The experiment was performed in triplicate.
Check Point Analysis and Optimization Model Validation
Statistical validation of the polynomial equations generated by Design Expert was established on the basis of ANOVA provision in the software. The models were evaluated in terms of statistically significant coefficients and r2 values. Various feasibility and grid searches were performed to find the optimum parameters and seven optimum check point formulations were selected to validate the experimental model and polynomial equations. The optimized check point formulation factors were evaluated for various responses. The resultant experimental values of responses were quantitatively compared with predicted values to calculate the percentage prediction error.
Stability Studies
The stability study was conducted for BUSP-OPT. Sufficient samples were placed in clear glass containers and were further placed at 40 ± 2°C/75 ± 5% R.H. (Skylab Instruments and Engineering Pvt Ltd., Thane, India) for 3 months. Samples were withdrawn at time intervals of 1, 2, and 3 months. The ex vivo permeation study was conducted according to the procedure described earlier and drug content in the formulations was estimated using HPLC (9)
Skin Irritation Studies
The skin irritation study was performed on six rabbits. The hair of rabbits on dorsal side was shaved with electrical shaver and BUSP-OPT (2 gm) formulation in activated dialysis membrane was directly applied. The development of erythema was monitored for 7 days.
In vivo Bioavailability Study in Rabbits
The animal study protocol was reviewed and approved by the institutional animal ethical committee, University College of Pharmaceutical Sciences, Kakatiya University, India. White New Zealand rabbits weighing 2.1 ± 0.13 Kg were selected for study. The bioavailability of BUSP (10 mg in 2 g of reservoir system) from BUSP-OPT was compared with an oral solution (4 mL of 0.25% w/v BUSP in distilled water). They were allowed free access to food and water, until night prior to dosing and were fasted for 10 h. Latin square crossover design was followed; the animals were divided into two groups each consisting of three rabbits. The rabbits to be used for application of BUSP-OPT were shaved carefully with the help of electrical shaver before application of BUSP-OPT followed by cleaning with water. To one group, oral solution (2.5 mg mL−1) was administered through feeding tube followed by rinsing with 10 mL of water and BUSP-OPT to another group in first phase. In second phase, vice versa was followed and was conducted after 15 days of wash out period. BUSP-OPT was applied on dorsal side and was covered with a water impermeable backing membrane and was further adhered with the help of adhesive tape USP. Blood samples (1.5 mL) from marginal ear vein were collected at preset intervals of 0, 0.5, 1, 2, 4, 8, 12, 24, 36, 48, and 72 h; 0, 1, 2, 4, 6, 8, 12, 18, 24, 36, 48, and 72 h, respectively, after administration of oral solution and application of BUSP-OPT. All blood samples were allowed to clot and centrifuged for 10 min at 4,000 rpm. The serum was separated and transferred into clean micro centrifuge tubes and stored at −20°C until HPLC analysis. The amount of BUSP in the samples was estimated using HPLC (9).
Pharmacokinetic Analysis
Pharmacokinetic parameters of BUSP after administration of BUSP-OPT and oral solution were estimated for each rabbit using a computer program, KINETICA 2000 (Version 3.0, Innaphase corporation, Philadelphia, USA). Non-compartmental analysis was used to calculate the pharmacokinetic parameters, CMax, TMax and area under the curve (AUC). CMax (ng mL−1) and TMax (h) were the observed maximal drug concentration and its time, respectively. The relative bioavailability for transdermal drug delivery system was calculated using the Eq. 3
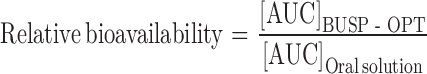 |
3 |
Ex vivo–In vivo Correlation
The cumulative amount of BUSP permeated across rat abdominal skin ex vivo from BUSP-OPT was compared against extent of absorption, i.e., cumulative AUC values for a possible ex vivo–in vivo correlation.
Statistical Analysis
Statistical comparisons were made using Student's t test using Sigmastat software (Jandel Corp., CA, USA). Results were considered significant at 95% confidence interval (p < 0.05) and results were expressed as mean ± SD.
RESULTS
Rheological Measurements
Spindle (CP-52) was used for viscometric characterization of BUSP reservoir systems. The decrease in viscosity of reservoir system was observed with an increasing shear rates, can be described well by an exponential function and hence the obtained data were analyzed using “Power Law” (12) as expressed by the Eq. 4
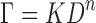 |
4 |
where τ is shear stress; K is gel index (GI) or consistency index; D is shear rate; and n is flow index. ‘Rheocalc 32’ software was used to automatically apply the model to generated data, and the value of GI was recorded. The GI value for different formulations is presented in Table I. The gel index was found to be ranging from 0.73 to 2.82. BUSP permeation was found to be decreased with increasing gel index.
Drug Content
The results of drug content showed in Table I. The assay ranged from 98.3 in BUSP3 to 102.3 in BUSP7. The results indicate that consistency in drug content.
Ex vivo Skin Permeation Experiments
The permeation profiles of BUSP-TTS through rat skin are shown in Fig. 1 and permeation parameters, Q24, flux and lag time in Table I. The Q24 ranged from 2,344.2 to 8,921.6 µg, flux ranged from 20.4 to 108.4 µg cm−2 h−1 and lag time ranged from 0.18 to 0.64 h. Formulation BUSP9 showed highest amount of BUSP permeated (8,921.6 µg) with a flux of 108.4 µg cm−2 h−1 and a lag time of 0.45 h. BUSP9-C (control) (DLM and PG free) showed a cumulative amount of 2,322.8 µg permeated with a flux of 26.3 µg cm−2 h−1 and a lag time of 0.48 h. The enhancement ratio of BUSP9 compared with BUSP9-C was 4.12-fold higher than the BUSP9-C.
Fig. 1.

a–b Ex vivo permeation profiles of BUSP (Buspirone) from reservoir-based TTS (Transdermal Therapeutic Systems), BUSP-OPT, and BUSP9-C (control), values represented are mean ± SD (n = 3)
The results of retained drug in skin layers for BUSP-OPT were shown in Table III. The drug content in skin layers and donor compartment were found to be 115.8 and 1,135 µg, respectively.
Table III.
Drug Content in Donor Compartment and Skin Layers after ex vivo Permeation of BUSP-OPT (Optimized Formulation)
| Drug content (µg) | Franz diffusion cell | Mean | SD | ||
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| Cumulative amount permeated | 8,825.1 | 8,434.5 | 8,916.2 | 8,725.3 | 255.9 |
| Donor compartment | 1,037.1 | 1,431.3 | 936.6 | 1,135.0 | 261.5 |
| Drug in skin layers | 98.5 | 136.4 | 112.5 | 115.8 | 19.2 |
| Sum | 9,960.7 | 10,002.2 | 9,965.3 | 9,976.1 | 22.7 |
Formulation Optimization by Experimental Design
The independent variables and responses for all 13 experiments are given in Tables I and II. The contour plots and 3D response surface plots drawn using Design Expert software are shown in Fig. 2. Design Expert software was used to optimize the formulation and develop the quadratic Eqs. 5, 6, and 7)
Fig. 2.

Contour plot showing effect of a HPMC (Hydroxypropyl methylcellulose) (X 1) and DLM (X 2); b HPMC (Hydroxypropyl methylcellulose) (X 1) and PG (X 3); c DLM (d-limonene) (X 2) and PG (propylene glycol) (X 3) on response Y 1 (Q 24); corresponding response surface plots (d–f)
The responses, Y1 and Y2 were found to be significantly higher (Y1, 7,191.8–8,921.6 µg; Y2, 84.3 to 108.4 µg cm−2 h−1) only when HPMC and DLM were used at 0.25 w/w and 4% or 8% v/w concentration level, respectively. The lag time (Y3) was found to be ranging from 0.64–0.18 h at low to medium levels of DLM. The ranges of other responses, Y1 and Y2 were 2,344.2–8,921.6 µg and 20.4–108.4 µg cm−2 h−1, respectively.
The responses of formulations ranged from low drug penetration of 2,344.2 µg (BUSP10, high level of HPMC and DLM and medium level of PG) to a higher penetration of 8,921.6 µg (BUSP9, low level of HPMC, DLM, medium level of PG). For estimation of quantitative effects of different combination of factors and their levels on Y1, Y2, and Y3, the response surface models were calculated. The model described could be represented as:
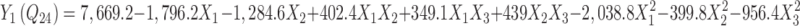 |
5 |
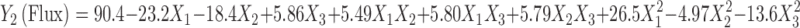 |
6 |
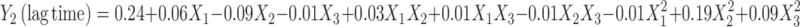 |
7 |
Stability Studies
The BUSP content in stability samples was found to be 99.2, 99.5 and 98.4% respectively after 1, 2, and 3 months. The gel strength was found to be 0.81, 0.78, and 0.75 cP after 1, 2, and 3 months of study. The results reveal that BUSP was stable during study. The ex vivo permeation profiles are shown in Fig. 1. Formulation BUSP-OPT-S showed 8,587.6 ± 263.9 µg of BUSP permeated in 24 h with a flux of 101 µg cm−2 h−1. The results suggested that the formulations did not show significant difference (p > 0.05) in permeation profiles compared with initial permeation profiles indicating the formulation is stable.
Skin Irritation Study
The skin irritation studies could not find any irritation, erythyma indicating that BUSP-OPT is non irritant.
In vivo Bioavailability Studies
The CMax and TMax were found to be 34 and 37.2 ng mL−1; 1.7 and 11.7 h, respectively, after administration of oral solution and BUSP-OPT. The AUC0-n and 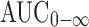 were found to be respectively 366.3 and 969.8 ng h mL−1; 394.6 and 1,078 ng h mL−1 after administration of oral solution and BUSP-OPT.
were found to be respectively 366.3 and 969.8 ng h mL−1; 394.6 and 1,078 ng h mL−1 after administration of oral solution and BUSP-OPT.
DISCUSSION
Ex vivo Skin Permeation Experiments
In our preliminary study, DLM showed a potential enhancement effect on BUSP permeation through rat abdominal skin. DLM had low solubility in aqueous systems; a co-solvent is required to improve its solubility. Additionally, some reports (13–15) have indicated that specific combinations of vehicles and enhancers such as DLM in ethanol, and PG had shown an increased in drug penetration. Therefore, in this study, the combination of 20% v/v ethanol and PG was used in the preparation of reservoir-based TTS. DLM in ethanol and PG was used to produce the synergistic enhancement effect on penetration rate of BUSP and to decrease the used amount of enhancers. Ethanol was used to solubilize DLM, and it also possesses penetration enhancement properties. The permeation enhancement effect by limonene suggests that there are possible multiple mechanisms that could have resulted in a more permeable intercellular pathway for BUSP. They include partitioning of BUSP into stratum corneum (SC) lipids, partial extraction of SC lipids (16), phase separation within the SC lipid lamellae (17) and limonene-PG synergy (18). Formulation BUSP9 showed maximum Q24 and flux among the formulations and was also showed statistically significant (p < 0.05) difference compared with that of Q24 and flux of BUSP9C. All formulations were found to follow zero-order kinetics as it was evidenced from correlation coefficients (r > 0.981). The results indicating that the permeation parameters of BUSP from reservoir-based TTS were markedly influenced by the composition. The drug permeation was found to be decreased with increasing concentration (low to high) of HPMC. As the concentration of HPMC increases the viscosity of TTS was increased it further increases the diffusion path to traverse the drug. The drug permeation was also found to be decreased with increasing concentrations of DLM. This was due to more affinity of BUSP (log P, 1.6) towards DLM (log P, 4.58) could decrease the release and thus permeation.
The mass balance was matching for BUSP (Table III) after ex vivo permeation study of optimized formulation. The results suggesting that BUSP was released from TTS and permeated across skin.
Fitting of Data to the Model
Formulation BUSP9 showed a significantly higher value of Y1 and Y2 among formulations. The responses observed for 13 formulations prepared were simultaneously fit to first order, second order, and quadratic models using Design Expert 7.1.5. It was observed that the best fit model was quadratic model and the comparative values of r2, standard deviation and percentage coefficient of variation are given in Table IV. A positive value represents an effect that favors the optimization, while a negative value indicates an inverse relationship between factor and response. It is evident that the independent variable X1 is having positive effect on the response Y3.
Table IV.
Summary of Results of Regression Analysis for Responses Y 1, Y 2, and Y 3 for Fitting to Quadratic Model
| Response | r 2 | Adjusted r 2 | Adequate Precision | SD | % CV |
|---|---|---|---|---|---|
| Y 1 | 0.995 | 0.982 | 24.3 | 289.12 | 5.11 |
| Y 2 | 0.996 | 0.985 | 26.7 | 3.51 | 5.58 |
| Y 3 | 0.939 | 0.756 | 6.87 | 0.07 | 17.34 |
SD standard deviation, CV coefficient of variation
The 3D response surface plots (Fig. 2d–f) were drawn to estimate the effects of independent variables on response and to select optimal formulation. The required flux to reach therapeutic concentration calculated was found to be about 67.1 µg cm−2 h−1. Hence, the penetration rate of optimal formulations in the optimization process was set at above 67.1 µg cm−2 h−1. Formulation, BUSP9 showed maximum flux of 108.4 µg cm−2 h−1 and could meet the target flux indicating that the concentrations may be enough to elicit the pharmacological effect.
Data Analysis
Formulations BUSP1, BUSP3, BUSP5, BUSP8, and BUSP9 had the highest Q24 and flux (Table I). The Q24 and flux obtained at various levels of the three independent variables was subjected to multiple regression analysis to yield a second-order polynomial equation. The value of the correlation coefficient (r2) of Eq. 5 was found to be 0.995, indicating good fit (Table IV). “Adeq Precision” measures the signal to noise ratio. A ratio greater than 4 is desirable, the ratio of 24.3 (Table IV) indicates an adequate signal. The Q24 values measured for different formulations showed wide variation (i.e., values ranged from a minimum of 2,344.2 µg in BUSP10 to a maximum of 8,921.6 µg in BUSP9). The results clearly indicate that Q24 value is strongly affected by the variables selected for the study. The main effects of X1, X2, and X3 represent the average result of changing one variable at a time from its low level to its high level. The interaction terms 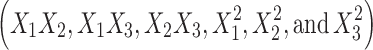 show how the Q24 changes when two variables are simultaneously changed. The negative coefficients indicate an unfavorable effect on Q24, while the positive coefficients for interactions between two variables indicate a favorable effect on Q24. Among the three independent variables, the lowest coefficient value is for X1 (–1,796.2), indicating that this variable is insignificant in prediction of Q24.
show how the Q24 changes when two variables are simultaneously changed. The negative coefficients indicate an unfavorable effect on Q24, while the positive coefficients for interactions between two variables indicate a favorable effect on Q24. Among the three independent variables, the lowest coefficient value is for X1 (–1,796.2), indicating that this variable is insignificant in prediction of Q24.
The value of r2 of Eq. 6 was found to be 0.996, indicating good fit (Table IV). The “Adeq Precision” was found to be 26.7, indicating an adequate signal. The flux values of BUSP1, BUSP3, BUSP5, BUSP8, and BUSP9 were found to be more among formulations. The flux values were found to be increased from medium to low levels of X1 and X2 and high to low levels of X3. The flux values measured for the different formulations showed wide variation (i.e., values ranged from a minimum of 20.4 µg cm−2 h−1 in BUSP10 to a maximum of 108.4 µg cm−2 h−1 in BUSP9). The interaction terms 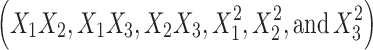 show how the flux changes when two variables are simultaneously changed. The positive coefficients (X1X2, X1X3 and X2X3) for the interactions between two variables indicate a favorable effect on flux. Among the three independent variables, the lowest coefficient value is for X1 (−23.2), indicating that this variable is insignificant in prediction of flux.
show how the flux changes when two variables are simultaneously changed. The positive coefficients (X1X2, X1X3 and X2X3) for the interactions between two variables indicate a favorable effect on flux. Among the three independent variables, the lowest coefficient value is for X1 (−23.2), indicating that this variable is insignificant in prediction of flux.
The lag time values of BUSP1, BUSP3, BUSP4, BUSP5, and BUSP13 were found to be less among the formulations. The lag time values were found to be increased from low to high levels of X1; high to low levels of X2 and X3. The results attributed to that the deposition of drug from reservoir within the layers of stratum corneum, might increase the lag time. The interaction terms 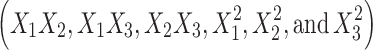 show how the lag time changes when two variables are simultaneously changed. The positive coefficients
show how the lag time changes when two variables are simultaneously changed. The positive coefficients 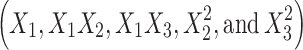 for the interactions between two variables indicate a favorable effect on lag time. Among the three independent variables, the lowest coefficient value is for X3 (−0.09), indicating that this variable is insignificant in prediction of lag time.
for the interactions between two variables indicate a favorable effect on lag time. Among the three independent variables, the lowest coefficient value is for X3 (−0.09), indicating that this variable is insignificant in prediction of lag time.
Contour Plots and Response Surface Analysis
Two-dimensional contour plots and 3D response surface plots are shown in Fig. 2, which are useful to study the interaction effects of the factors on responses at one time. In all the presented figures, the third factor was kept at a constant level. All the relationships among the three variables are non-linear (Fig. 2). Factors X2 and X3 have curvilinear relationship at all levels of the two variables on response Y2. Response surface plots show the relationship between these factors even more clearly. The Q24 and flux were found to have increased with decreasing concentrations of either HPMC or DLM (from medium to low level).
Optimization
The optimum formulation was selected based on criteria of attaining maximum value of Q24, flux and low value of lag time by applying constraints on Y1 (7,500 ≤ Y ≤ 9,500 µg), Y2 (65 ≤ Y≤110 µg cm−2 h−1) and Y3 (0.15 ≤ Y ≤ 0.50 h). Upon trading of various responses and exhaustive grid search, the formulation composition with HPMC concentration of 0.5%, DLM, 5.85% and PG 8.3% was found to fulfill the maximum requisite of an optimum formulation because of maximum Q24 (8,725.3 µg), flux (104.6 µg cm−2 h−1) and low lag time (0.31 h) values. The flux of reservoir TTS was found to meet the target flux (67.1 µg cm−2 h−1). The gel index of BUSP-OPT was found to be 0.81 cP.
Validation of Response Surface Methodology
Seven checkpoint formulations were obtained from response surface methodology (RSM), the composition and predicted responses were shown in Table V. To confirm the validity of calculated optimal parameters and predicted responses, the optimum formulations were prepared according to the values of factors (Table V) and subjected to ex vivo permeation studies. The predicted error was below 15%, indicating that the observed responses were very close to the predicted values (Table V). Percentage prediction error is helpful in establishing the validity of generated equations and to describe the domain of applicability of RSM model. Linear correlation plots between experimental and predicted responses were shown in Fig. 3. The linear correlation plots drawn between experimental and predicted values demonstrated high values of r2 (Q24, 0.9782; flux, 0.9632; lag time, 0.9507) indicating goodness of fit.
Table V.
Composition of Checkpoint Formulations, the Predicted and Experimental Values of Response Variables and Percentage Prediction Error
| Optimized formulation composition (X 1/X 2/X 3) | Response variable | Experimental value | Predicted value | Percentage prediction error |
|---|---|---|---|---|
| 0.251:6.05:8.2 | Y 1 | 8,275.1 | 8,409.5 | −1.62 |
| Y 2 | 98.4 | 101.4 | −3.06 | |
| Y 3 | 0.27 | 0.29 | −7.25 | |
| 0.751:8.85:7.6 | Y 1 | 7,840.3 | 7,736.1 | 1.33 |
| Y 2 | 92.1 | 91.2 | 0.97 | |
| Y 3 | 0.25 | 0.22 | 9.94 | |
| 0.73:8.23:7.4 | Y 1 | 7,794.6 | 7,965.9 | −2.20 |
| Y 2 | 92.8 | 94.4 | −1.79 | |
| Y 3 | 0.21 | 0.24 | −12.35 | |
| 0.79:5.75:11.1 | Y 1 | 8,436.1 | 8,516.9 | −0.96 |
| Y 2 | 100.5 | 102.1 | −1.57 | |
| Y 3 | 0.36 | 0.33 | 7.23 | |
| 0.93:5.3:13.4 | Y 1 | 7,720.8 | 7,747.1 | −0.34 |
| Y 2 | 92.4 | 91.3 | 1.17 | |
| Y 3 | 0.42 | 0.41 | 2.34 | |
| 0.5:5.85:8.3 | Y 1 | 8,725.3 | 8,836.6 | −1.28 |
| Y 2 | 104.6 | 106.9 | −2.29 | |
| Y 3 | 0.31 | 0.32 | −4.72 | |
| 0.88:7.1:9.8 | Y 1 | 8,310.5 | 8,280.6 | 0.36 |
| Y 2 | 97.1 | 98.7 | −1.63 | |
| Y 3 | 0.23 | 0.25 | −9.17 |
Fig. 3.

a–c Linear correlation plots between actual and predicted values
In vivo Bioavailability Studies
The results of bioavailability study (Table VI, Fig. 4) reveal that BUSP is released and permeated well from reservoir system (BUSP-OPT) by transdermal route, as compared with oral solution. The CMax, TMax, and AUC profiles were compared. In four rabbits, CMax was higher for transdermal route than oral route and in the remaining rabbits CMax for oral route was higher than the transdermal route. Greater CMax values (in four rabbits) are attributed due to avoidance of first pass hepatic metabolism after transdermal administration. In the remaining two rabbits, slightly higher CMax values (38.4 and 34.3 ng mL−1; 36.3 and 32.9 ng mL−1 after administration of oral solution and TTS, respectively) were obtained compared with transdermal route. The TMax values in all rabbits were higher for transdermal administration than oral administration, and the difference was statistically significant (p < 0.05). This difference was because of stratum corneum that could delay the permeation of BUSP from reservoir system in contrast, solution administered by oral route is an immediate release dosage form. The overall mean value of AUC0−72 by transdermal route was 2.65 times higher than that of oral route, and the difference was found to be statistically significant (p < 0.05) demonstrating improved bioavailability of BUSP from reservoir TTS. This could be due to avoidance of first pass hepatic metabolism by transdermal route. The reported oral bioavailability of BUSP was 4% (1), because of first pass metabolism. In the present study the bioavailability of BUSP by transdermal route was found to be 11%. Therefore for the effective management of anxiety disorders, BUSP in the form of reservoir TTS could provide an effective treatment.
Table VI.
Pharmacokinetic Parameters of BUSP (Buspirone) in Rabbits after Administration of Oral Solution and BUSP-OPT (Optimized Formulation) Each Containing 10 mg of BUSP, Values Represented are mean ± SD (n = 6)
| Formulation | C Max (ng mL−1) | T Max (h) | AUC0–t (ng h mL−1) | AUC0-∞ (ng h mL−1) | T1/2 (h) |
|---|---|---|---|---|---|
| Oral solution | 34.0 ± 7.57 | 1.7 ± 0.52 | 366.3 ± 85.97 | 394.6 ± 93.69 | 5.4 ± 1.68 |
| BUSP-OPT | 37.2 ± 11.7 | 11.7 ± 5.85 | 969.8 ± 269.64 | 1,078.0 ± 329.05 | 18.7 ± 3.56 |
Fig. 4.

Mean serum profiles of BUSP (Buspirone) in rabbits, after administration of oral solution and reservoir-based TTS (Transdermal Therapeutic System), each containing 10 mg of BUSP, values represented are mean ± SD (n = 6)
Ex vivo–In vivo Correlation
Ex vivo–in vivo correlation between cumulative amount of drug permeated across rat abdominal skin and AUC showed a biphasic curve pattern (Fig. 5), which can be distinguished into two regions for BUSP-OPT. Good linear correlation was observed with correlation coefficients, r2 = 0.979 during lag phase and r2 = 0.998 during absorption phase. Point to point correlation of ex vivo permeation of drug to in vivo performance was observed, indicating that it follows type A correlation (19). The slow permeation of BUSP through skin in initial stages is explained as follows; in first phase BUSP was released and permeated through skin and deposition of BUSP took place in skin layers and concentration build up was maintained. Permeation and concentration buildup at skin is the lag phase observed in first region. Concentration built up resulted in flux establishment and AUC increased at a rapid rate in the second phase. This indicates that initially drug permeated into stratum corneum rapidly but it takes some time for permeation and absorption. Once the necessary flux is established, absorption was rapid as large amount of drug is deposited in the layers of skin.
Fig. 5.

Ex vivo–in vivo correlation of cumulative amount permeated ex vivo Vs AUC (Area Under the Curve)
CONCLUSIONS
In this work, the optimization of the buspirone transdermal therapeutic system was satisfactory and performed by Box–Behnken design. The response surface methodology and contour plot allowed finding the optimum conditions for TTS preparation. The results demonstrated that the formulation was nonirritating and did not cause any erythyma upon transdermal administration. Results of bioavailability study showed improved permeation of drug from reservoir TTS compared with oral solution. An improvement of bioavailability by TTS to the extent of 2.65 times over oral solution was obtained. Good ex vivo–in vivo correlation was obtained with correlation coefficients of 0.979 and 0.998 during lag and permeation phase, respectively.
Acknowledgements
One of the authors (Ramesh Gannu) thank AICTE, New Delhi, India for providing financial assistance in the form of National Doctoral Fellowship (NDF). The authors also acknowledge the liberal help of Dr. Reddys Laboratories, Hyderabad, India for providing Buspirone as gift sample.
References
- 1.Iftekhar M, Chandra S. Clinical pharmacokinetics and pharmacodynamics of buspirone an anxiolytic drug. Clin Pharmacokinet. 1999;36(4):277–287. doi: 10.2165/00003088-199936040-00003. [DOI] [PubMed] [Google Scholar]
- 2.Dollery C. Therapeutic Drugs. Edinburgh: Churchill Livingstone; 1999. [Google Scholar]
- 3.Clarke's analysis of drugs and poisons (2007). Available at www.medicinescomplete.com.
- 4.Victor MM, Mohammad AK, Michniak BB. Enhanced iontophoretic delivery of buspirone hydrochloride across human skin using chemical enhancers. Int J Pharm. 2003;264(1–2):73–83. doi: 10.1016/s0378-5173(03)00390-9. [DOI] [PubMed] [Google Scholar]
- 5.Vaddi H, Wang P, Chan S. Effect of some enhancers on the permeation of haloperidol through rat skin in vitro. Int J Pharm. 2001;212(2):247–255. doi: 10.1016/S0378-5173(00)00616-5. [DOI] [PubMed] [Google Scholar]
- 6.Mohammad AK, Victor MM, Michniak BB. Iontophoretic transdermal delivery of buspirone hydrochloride in hairless mouse skin. AAPS PharmSci. 2003;5(2):61–71. doi: 10.1208/ps050214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamed E, Sakr A. Application of multiple response optimization technique to extended release formulations design. J Control Rel. 2001;73(2–3):329–338. doi: 10.1016/S0168-3659(01)00356-X. [DOI] [PubMed] [Google Scholar]
- 8.Box GEP, Behnken DW. Some new three level designs for the study of quantitative variables. Technometrics. 1960;2(4):455–75. doi: 10.2307/1266454. [DOI] [Google Scholar]
- 9.Ramesh G, Shravan KY, Chinna RP, Vamshi VY, Harshini K, Madhusudan RY. Development of high performance liquid chromatography method for buspirone in rabbit serum: application to pharmacokinetic study. Anal Chim Acta. 2009;647(2):226–230. doi: 10.1016/j.aca.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Levang AK, Zhao K, Singh J. Effect of ethanol/propylene glycol on the in vitro percutaneous absorption of aspirin, biophysical changes and macroscopic barrier properties of the skin. Int J Pharm. 1999;181(2):255–263. doi: 10.1016/S0378-5173(99)00055-1. [DOI] [PubMed] [Google Scholar]
- 11.Hayton WL, Chen T. Correction of perfusate concentration for sample removal. J Pharm Sci. 1982;71(7):820–821. doi: 10.1002/jps.2600710726. [DOI] [PubMed] [Google Scholar]
- 12.Bonacucina G, Martelli S, Palmieri GF. Rheological, mucoadhesive and release properties of Carbopol gels in hydrophilic cosolvents. Int J Pharm. 2004;282(1–2):115–130. doi: 10.1016/j.ijpharm.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Perry FCL, Xiang YL, Lifeng K, Paul CLH, Yew WC, Sui YC. Limonene GP1/PG organogel as a vehicle in transdermal delivery of haloperidol. Int J Pharm. 2006;311(1–2):157–164. doi: 10.1016/j.ijpharm.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 14.Osamu S, Akira O, Shinobu T, Sueaki I, Takashi S, Kozo T, Tsuneji N. Synergistic effect of d-limonene and ethanol on the transdermal permeation of NB-818. Drug Dev Ind Pharm. 1995;21(4):411–425. doi: 10.3109/03639049509026632. [DOI] [Google Scholar]
- 15.Kozo T, Tsuneji N. Limonene and related compounds as potential skin penetration promoters. Drug Dev Ind Pharm. 1994;20(4):677–684. doi: 10.3109/03639049409038325. [DOI] [Google Scholar]
- 16.Krishnaiah YSR, Satyanarayana V, Bhaskar P. Effect of limonene on the in vitro permeation of nicardipine hydrochloride across the excised rat abdominal skin. Pharmazie. 2002;57:842–847. [PubMed] [Google Scholar]
- 17.Moghimi H, Williams AC, Barry BW. A lamellar matrix model for stratum corneum intercellular lipids. V. Effects of terpene penetration enhancers on the structure and thermal behaviour of the matrix. Int J Pharm. 1997;146:41–54. doi: 10.1016/S0378-5173(96)04766-7. [DOI] [Google Scholar]
- 18.Barry BW. Lipid–protein-partitioning theory of skin penetration enhancement. J Control Rel. 1991;15:237–248. doi: 10.1016/0168-3659(91)90115-T. [DOI] [PubMed] [Google Scholar]
- 19.Emami J. In vitro–in vivo correlations: from theory to applications. J Pharm Pharm Sci. 2006;9(2):31–51. [PubMed] [Google Scholar]