

Abstract
The purpose of this present study was to evaluate the antiangiogenic activity of sterically stabilized liposomes containing paclitaxel (SSL-PTX). The SSL-PTX was prepared by the thin-film method. The release of paclitaxel from SSL-PTX was analyzed using a dialysis method. The effect of SSL-PTX on endothelial cell proliferation and migration was investigated in vitro. The antitumor and antiangiogenic activity of SSL-PTX was evaluated in MDA-MB-231 tumor xenograft growth in BALB/c nude mice. The release of paclitaxel from SSL-PTX was 22% within 24 h. Our in vitro results indicated that SSL-PTX could effectively inhibit the endothelial cell proliferation and migration at a concentration-dependent manner. We also observed that metronomic SSL-PTX induced marked tumor growth inhibition in MDA-MB-231 xenograft model via the antiangiogenic mechanism, unlike that in paclitaxel injection (Taxol) formulated in Cremophor EL (CrEL). Overall, our results suggested that metronomic chemotherapy with low-dose, CrEL-free SSL-PTX should be feasible and effective.
Key words: antiangiogenic activity, migration, paclitaxel, proliferation, sterically stabilized liposomes
INTRODUCTION
Maximum tolerated dose (MTD) chemotherapy is a common strategy which directly kills tumor cells by cytotoxic drugs (1). However, this regimen shows toxic side effect by resting for a long time to recover before next administration (2). Recently, metronomic chemotherapy has been reported as an alternative strategy to MTD protocol (3,4). Metronomic delivery or frequent administration of drugs at doses much lower than MTD have antiangiogenic properties and may be more effective (5). This metronomic regimen could maximize the growth-limiting effects on the tumor vasculature. The advantage of metronomic strategy is lower toxicity and risk of emergence of drug-resistant tumor cells compared with MTD administration (6).
Paclitaxel is a potent cytotoxic agent that is widely used against various refractory and metastatic malignancies. It appears to be a strong candidate for metronomic chemotherapy given its ability to inhibit endothelial cell functions relevant to angiogenesis in vitro at extraordinarily low concentrations (7). Unfortunately, clinically relevant concentrations of the formulation vehicle Cremophor EL (CrEL) in Taxol (commercial paclitaxel injection product, Squibb Co., Princeton, NJ, USA) were previously reported to nullify the antiangiogenic activity of paclitaxel (8).
There are a variety of nanoparticle carrier systems currently being explored for cancer therapeutics. The types of them currently used in research for cancer therapeutic applications include liposomes, polymeric nanoparticles, micelles, protein nanoparticles, viral nanoparticles, and metallic nanoparticles (9). Although many CrEL-free paclitaxel nanoparticle carrier systems have been developed, efforts were only focused on removing the use of CrEL to decrease the hypersensitive side effect or to improve the pharmacokinetic behavior of paclitaxel. Their antiangiogenic efficacy in metronomic administration has not been considered.
Among the aforementioned carrier systems, liposome has been used as one of the promising approaches since the association of drugs with lipid carrier results in a dramatic improvement of the pharmacokinetics of the drug, resulting in reduced toxicities and improved therapeutic efficacies (10). It has been reported that liposomal paclitaxel improved its solubility and showed similar in vitro cytotoxicity against a variety of tumor cell lines compared to that of Taxol (11). However, a major drawback of conventional liposome is its rapid uptake and accumulation by phagocytic cells of the mononuclear phagocyte system (reticuloendothelial system or RES) after systemic administration (12). Grafting of the liposome with the inert and biocompatible polymer polyethylene glycol (PEG) leads to the formation of a protective, hydrophilic layer on the surface of the liposome. This modification prevents the recognition of liposome by opsonins and therefore reduces their clearance by cells of the RES (13). The PEGylated liposome is therefore often referred to as sterically stabilized liposome (SSL). Using PEGylated phospholipids, the apparent terminal half-life of such long-circulating liposome can be extended in humans from a timescale of minutes to days (14). It has been reported that SSL could spontaneously accumulate in solid tumors via the enhanced permeability and retention (EPR) effect through the passive targeting mechanism (15). Now, sterically stabilized liposomes containing paclitaxel (SSL-PTX) has been reported (16–18). The pharmacokinetic and antitumor efficiency of this SSL-PTX has been evaluated. However, the antiangiogenic efficacy of SSL-PTX in metronomic administration was still unclear.
Albumin-bound paclitaxel ABI-007 (Abraxane®; Abraxis BioScience and AstraZeneca) is a novel, albumin-bound, 130-nm particle formulation of paclitaxel, free from any kind of solvent (19). This formulation is prepared by high-pressure homogenization of paclitaxel in the presence of serum albumin, resulting in a nanoparticle colloidal suspension. The albumin concentration is 3–4%, which is similar to the albumin concentration in the blood (20). Preclinical studies have demonstrated that ABI-007 has a higher penetration into tumor cells with an increased antitumor activity, compared with an equal dose of standard paclitaxel (21). ABI-007 has been approved by the US Food and Drug Administration for pretreated metastatic breast cancer patients. It is of interest that the antiangiogenic activity of ABI-007 has been confirmed in metronomic chemotherapy (22).
Considering the results of antiangiogenic activity obtained from ABI-007, we hypothesize that the CrEL-free SSL-PTX may also render paclitaxel-based antiangiogenic activity in metronomic chemotherapy. To test this hypothesis, the antiangiogenic activity of SSL-PTX was evaluated in vitro and in vivo.
MATERIALS AND METHODS
Materials
Paclitaxel was purchased from Yunnan Hande Bio-Tech Co., Ltd. (Kunming, Yunnan, China). Taxol was commercially available from the local hospital of Beijing (Bristol-Myers Squibb Co., Princeton, NJ, USA), and the formulation of Taxol contains 30 mg paclitaxel in 5 ml of 50% Cremophor EL (v/v) and 50% ethanol (v/v). Soybean phosphatidylcholine (S100PC) was provided by Lipoid GmbH (Ludwigshafen, Germany). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine [methoxy (polyethylene glycol)-2000] (mPEG2000-DSPE) was supplied by NOF Co. (Tokyo, Japan). Cholesterol, sulforhodamine B (SRB), and tris base were obtained from Sigma-Aldrich (St, Louis, MO, USA).
Cell Line
Human breast cancer cells, MDA-MB-231 (Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Beijing, China), were cultured in L-15 media (M&C Gene Tech Co., Ltd, Beijing, China) with 10% fetal bovine serum (Sijiqing Biological Engineering Materials Co. Ltd, Beijing, China) and maintained in a humidified atmosphere containing 5% CO2 at 37°C.
Human umbilical vein endothelial cells (HUVEC) were isolated from umbilical cords (Haidian Maternal and Child Health Hospital, Beijing, China) as previously reported (23). Briefly, culture medium consisted of M 199, containing 20% (v/v) newborn calf serum and 5% (v/v) pooled human serum with 2 mM l-glutamine. Cells were plated onto plates precoated with 0.02% (w/v) gelatin with a medium change after 24 h and every 2 days thereafter until confluent. Primary cultures of HUVEC were harvested by incubation with 0.05% trypsin and 0.02% EDTA, and the cells were collected.
Animals
Female BALB/c nude mice were purchased from the Academy of Military Medical Sciences (Beijing, China) at 6 weeks of age (initially weighing 20–25 g). All of the animal experiments adhered to the principles of care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of Peking University.
Preparation of SSL-PTX
The SSL-PTX, composed of S100PC, cholesterol, and mPEG2000-DSPE in the molar ratio of 90:10:5, was prepared by a thin-film hydration method, as described previously (16–18). Briefly, the mixture of paclitaxel, S100PC, cholesterol, and mPEG2000-DSPE was dissolved in chloroform and dried by an RE52 rotary evaporator (Shanghai Yarong Biochemistry Instrument Company, China) in a round-bottom flask at 40°C. The lipid film was flushed with nitrogen gas for 5 min and maintained overnight in a desiccator to remove traces of chloroform. The resulting thin-film was then hydrated in phosphate buffer saline (PBS, pH 7.4) by vortex and sonicated using a bath type sonicator at 50°C for 30 min to form the liposome suspensions. The liposome suspensions were purified by centrifuging at 1,000 rpm for 10 min and then passing through 0.2-µm pore size membrane to remove unentrapped paclitaxel. Then, the final SSL-PTX was obtained.
Characterization of SSL-PTX
The particle sizes and zeta potential of SSL-PTX were measured by photon correlation spectroscopy using Malvern Zeta sizer 3000 HS (Malvern, UK) at 25°C.
In Vitro Release of Paclitaxel from SSL-PTX
The release of paclitaxel from SSL-PTX was investigated using a dialysis method. Briefly, a volume of 0.2 ml of each SSL-PTX or Taxol (1 mg/ml) was placed into a dialysis tube (MWCO 7000) and was tightly sealed, respectively. Then, the dialysis tube was immersed in 200 ml of release medium (PBS (pH 7.4) containing 0.1% (v/v) Tween 80) and incubated in an orbital shaker for 24 h at room temperature. Samples (0.5 ml) were taken at predetermined time intervals from the release medium for 24 h, which was refilled with the same volume of fresh medium. The concentration of paclitaxel was determined by high-performance liquid chromatography (HPLC) after appropriate dilution with acetonitrile without further treatment.
Endothelial Cell Proliferation
The effect of SSL-PTX on endothelial cell proliferation was analyzed with the SRB assay as described previously (24). Briefly, HUCEC were seeded into 96-well plates at 5 × 103 cells per well and allowed to attach for 48 h. Then, HUVEC were exposed to various concentrations of paclitaxel in SSL-PTX (0.5–10 nM). After incubation for 48 h at 37°C, cells were fixed with trichloroacetic acid, washed, and stained with SRB. After shaking, the absorbance at 540 nm was recorded using a 96-well plate reader (Bio-Rad, 680, America). The survival percentages of HUVEC were calculated using the following formula: 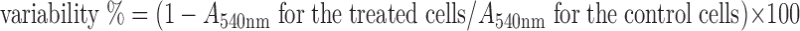 , where A540nm is the absorbance value. Each assay was repeated with a minimum of three times; there were quadruplicate determinations per dose level.
, where A540nm is the absorbance value. Each assay was repeated with a minimum of three times; there were quadruplicate determinations per dose level.
Migration Assay
The migration assay used was a monolayer denudation assay as described by Marcel et al. (25). Briefly, confluent HUVEC in 24-well plates (Costar, America) were mechanically “wounded” by scraping away a swath of cells with a pipette tip and denuding a strip of the monolayer 300 μm wide. Variation in the wound width within experiments was approximately 5%. Endothelial monolayers were washed twice with D-hanks solution to remove wound-derived loose and dislodged cells and incubated in media supplemented with SSL-PTX (0.01–10 nM). Control HUVEC cultures only received media alone. The extent of wound closure was observed and photographed after 24-h incubation. The effect of SSL-PTX on the progression of endothelial cell migration was represented by the wound width which was measured in six areas and compared with the untreated wound width at 0-h time point.
In Vivo Antitumor Efficacy
MDA-MB-231 cells were resuspended in serum-free cell culture medium. Approximately 4 × 106 MDA-MB-231 cells (100 μl) were subcutaneously injected into the armpits of the female BALB/c nude mice. Once tumor masses in xenografts reached to 150 to 200 mm3 in volume, mice were randomly assigned to five groups (five to six animals for each group). Each group was treated with physiological saline, MTD Taxol (15 mg/kg, i.v., qd × 4), MTD SSL-PTX (15 mg/kg, i.v., qd × 4), metronomic Taxol (6 mg/kg, i.v., five consecutive days and repeated second time with 6-day interval), and metronomic SSL-PTX (6 mg/kg, i.v., five consecutive days and repeated second time with 6-day interval), respectively. For the administration at each time, formulations were given to mice via tail vein in each group. The total dose of paclitaxel in all treatment groups was 60 mg/kg. Throughout the study, mice were weighed, and tumors were measured with a caliper every 2 days. Tumor volumes were calculated by the formula: 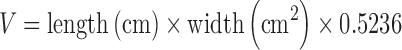 . Once animals in any group seemed moribund, all animals in the experiment were sacrificed, and then tumors were harvested. The harvested tumors were fixed in a formalin solution and paraffin-embedded for immunohistochemical analysis.
. Once animals in any group seemed moribund, all animals in the experiment were sacrificed, and then tumors were harvested. The harvested tumors were fixed in a formalin solution and paraffin-embedded for immunohistochemical analysis.
Assay of In Vivo Angiogenesis
CD31 staining was used to identify the microvessel density (MVD) in the tumor tissues by immunohistochemical method. Briefly, after paraffin-embedded tissue sections (5 µm thick) were deparaffinized in xylene and rehydrated in alcohol, sections were incubated in 0.3% H2O2 to block endogenous peroxidase activity. Each slide was incubated with normal goat serum for 20 min at room temperature and then incubated in the primary antibody at 4°C overnight. After incubation with the secondary antibody, biotinylated for 30 min at 37°C, each slide was rinsed in PBS and was incubated in the avidin–biotin peroxidase complex for 30 min at 37°C. The peroxidase was visualized with 3-3′-diamino-benzidinetetrahydrochloride solution and then counterstained with hematoxylin. MVD was assessed according to the international consensus report. Immunostained slides were scanned at ×100 magnification to identify the areas with the highest number of vessels (so called “hot spot”). Counts were performed on ten fields in the hot spot by two independent pathologists at ×400 magnification, and the mean was analyzed.
HPLC Analysis of Paclitaxel
The concentration of paclitaxel in SSL-PTX was determined by an HPLC method (26) by Waters HPLC system consisting of a 1525 pump and a 2487 ultraviolet detector (Waters Co. Inc., Westerville, OH, USA). Mobile phase consisted of methanol–water–tetrahydrofuran (67.5:30:2.5, v/v/v) and was delivered at a flow rate of 1 ml/min. Chromatographic separation was performed on a Phenomenex ODS3 column (250 × 4.6 mm, 5 μm, Torrance, CA, USA). Wavelength was set at 230 nm.
Statistical Analysis
Data are presented as the mean ± standard deviation (SD). One-way analysis of variance was used to determine significance among groups, after which post hoc tests with the Bonferroni correction were used for comparison between individual groups. Statistical significance was established at p < 0.05.
RESULTS
Characterization of SSL-PTX
The particle size of SSL-PTX was approximately 93.70 nm with the polydispersity of 0.23. The value of zeta potential of SSL-PTX showed a slightly negative charge (−0.73 mV). The entrapment efficiency of SSL-PTX was 90.68 ± 2.36% (n = 3).
In Vitro Release of Paclitaxel from SSL-PTX
In the in vitro release study, paclitaxel in Taxol released rapidly and was almost completed within 24 h. However, the SSL-PTX released 22% of paclitaxel within 24 h of dialysis at room temperature (Fig. 1).
Fig. 1.

The release of paclitaxel from SSL-PTX or Taxol at room temperature. The release medium was PBS (pH 7.4) containing 0.1% (v/v) of Tween 80. Each data represent the mean ± standard deviation (n = 3)
Endothelial Cell Proliferation
To determine the effects of SSL-PTX on endothelial cell proliferation, HUVEC were treated with SSL-PTX for 48 h. HUVEC proliferation was determined by the SRB method. The results indicated that HUVEC proliferation was significantly inhibited by SSL-PTX (p < 0.01), as shown in Fig. 2. The values of antiproliferative ratio (100% − variability%) at 5 and 10 nM were 32% and 26%, respectively. In addition, blank SSL did not affect HUVEC proliferation (data not shown).
Fig. 2.

Effects of SSL-PTX on HUVEC proliferation. The HUVEC were incubated in the absence or in the presence of SSL-PTX at different concentrations and harvested 48 h later. The proliferation of HUVEC was assessed by SRB method to calculate the proliferation variability rate (%). Values present the mean ± SD (n = 6). ** p < 0.01, vs vehicle as control
Endothelial Cell Migration
Migration of HUVECs was analyzed in the wound assay. The wounded HUVEC monolayers were incubated with SSL-PTX. The degree of wound closure was assessed after 24-h incubation, as shown in Fig. 3a. Untreated HUVEC migrated into the denuded areas, filling the wound by 84.2 ± 4.3%. In SSL-PTX treatment group, few migrated HUVEC were observed in the denuded areas (Fig. 3a). In this model, SSL-PTX significantly inhibited migration of HUVEC into the wounded area at a concentration-dependent manner (p < 0.01), as shown in Fig. 3b. Wound closure was inhibited by 54.8 ± 8.1% at the concentration at 0.01 nM. At the highest concentration (10 nM), the value of inhibited ratio was 20.8 ± 3.9% compared with untreated HUVEC group as a percentage of control wound width (17.5% vs 84.2%).
Fig. 3.

Migration assay. Wound assay was done to determine whether SSL-PTX inhibits HUVEC migration. After treatment with various concentrations of SSL-PTX, HUVEC were allowed to migrate into the denuded area for 24 h. HUVEC migration was visualized by light microscopy. a Typical photomicrographs (final magnification, ×25) were shown in unwounded, untreated in 0 hour, untreated in 24 h, and SSL-PTX treated in 24 h at the concentration of 10 nM. b Percentage of wound width which is measured in denuded areas at 24 h compared with the untreated wound width at 0-h time point versus SSL-PTX concentration. **, p < 0.01, vs untreated as control
In Vivo Antitumor Activity
Figure 4 describes the therapeutic efficacy of paclitaxel formulations against MDA-MB-231 human cells in vivo. Compared to the control group administered as physiological saline, the tumor growth was significantly inhibited in all treatment groups (p < 0.01), but the exerted effect varied. For MTD treatment, there were no significant differences in tumor growth inhibition between the Taxol group and the SSL-PTX group. For metronomic treatment, tumor growth inhibition in SSL-PTX group was significantly evident compared with that in Taxol group (p < 0.01). In fact, tumor growth inhibition in metronomic Taxol group was much lower than that in other three treatment groups (p < 0.01). When comparing the tumor growth inhibition among the MTD Taxol, MTD SSL-PTX, and metronomic PTX-SSL groups, there were no significant differences observed. In addition, there was no significant weight loss observed between treatment groups and control group (data not shown).
Fig. 4.

Tumor growth inhibition with MTD and metronomic paclitaxel formulations. MDA-MB-231 cells were implanted in the nude mice on the zeroth day, and the treatment was started on the 11th day when the tumor volume reached 150–200 mm3. The regimen of MTD treatment was administering Taxol or SSL-PTX (15 mg/kg) on the 11th, 15th, 19th, and 23 rd day, respectively. For metronomic treatment, Taxol or SSL-PTX (6 mg/kg) was administered from the 11th to 15th days and from the 22nd to 26th days, respectively. On the 32nd day, one of five mice in the control group began to seem moribund; thus, all nude mice were sacrificed, and the tumor was harvested. Data (change ratio for tumor volume (%)) are presented as the mean ± SD per group measured at indicated days after treatment (n = 5). **, p < 0.01, vs control treatment group at the 32nd day; ††, p < 0.01, vs metronomic Taxol treatment group at the 32nd day
In Vivo Angiogenesis
To evaluate whether reduced angiogenesis accounts for the suppressed in vivo growth of MDA-MB-231 cells, MVD was assessed by immunohistochemistry. As shown in Fig. 5a, microvessels were easily observed by CD31 staining. There were few microvessels observed in the metronomic SSL-PTX treatment group. Statistics analysis showed significantly less MVD present in the MTD treatment group of Taxol (15.2 ± 3.3) and SSL-PTX (11.6 ± 2.1) compared to the control group (24.2 ± 3.9; p < 0.01), as shown in Fig. 5b. The value of MVD in metronomic Taxol treatment group (19.8 ± 1.8) was slightly lower than that in control group with no significant difference (p > 0.05). However, significantly less MVD was shown in the metronomic SSL-PTX treatment group (5.2 ± 1.3) compared to the control group (p < 0.01). In addition, metronomic SSL-PTX group reduced MVD more remarkably compared with other treatment groups (p < 0.05 or p < 0.01).
Fig. 5.

Effect of SSL-PTX on MVD in xenograft MDA-MB-231 tumors. a Representative micrographs of immunohistochemical detection of CD31+ microvessel in xenograft MDA-MB-231 tumors in control group, MTD treatment groups of Taxol or SSL-PTX, and metronomic treatment groups of Taxol or SSL-PTX (final magnification, ×400). b Mean CD31+ microvessel count in xenograft MDA-MB-231 tumors in control group, MTD treatment groups of Taxol or SSL-PTX, and metronomic treatment groups of Taxol or SSL-PTX. Data are presented as the means ± SD (n = 5). **, p < 0.01, vs control treatment group; †, p < 0.05, vs MTD SSL-PTX treatment group. ††, p < 0.01, vs MTD Taxol and metronomic Taxol treatment group
DISCUSSION
Our results show that metronomic chemotherapy with SSL-PTX exhibits potent antiangiogenic activity in vivo. It has been reported that paclitaxel in DMSO solution suppressed rat aortic angiogenesis and HUVEC proliferation (8). Similar results were also confirmed in CrEL-free ABI-007 metronomic chemotherapy (22). In the present study, SSL-PTX induced responses similar to that in paclitaxel DMSO solution and ABI-007, indicating that the antiangiogenic property of paclitaxel was effectively delivered by CrEL-free SSL-PTX. For metronomic Taxol treatment group, tumor growth inhibition was significantly lower than that of other three treatment groups (p < 0.01). Considering the previous reports, this may be due to CrEL results nullifying the antiangiogenic effect (8,22,27). Therefore, our results confirm that the formulation vehicle of liposome in SSL-PTX could not affect the antiangiogenic activity of paclitaxel.
The results of immunohistochemistry indicated the antiangiogenic effect of metronomic SSL-PTX in vivo. We also observed the antiangiogenic effects in MTD treatment group, but this effect was much lower than that in metronomic SSL-PTX treatment group (p < 0.05), as shown in microvessel density assessment. These results indicate that frequent administration of SSL-PTX, at doses lower than MTD, has antiangiogenic properties that block the blood supply and may be more effective in suppressing tumor growth in vivo. However, for metronomic Taxol treatment group, there were no significant differences compared to control group, indicating that CrEL might nullify its antiangiogenic activity, as described in previous reports.
Angiogenesis is an important factor in the progression and enlargement of solid neoplasms and has a close relation to invasion and metastases. The proliferation of endothelial cell plays a key role in tumor angiogenesis. For this reason, many studies have focused on antiangiogenesis or target endothelial cell receptors, such as paclitaxel encapsulated in cationic liposomes (28–33). Paclitaxel encapsulated in cationic liposomes significantly inhibits tumor growth by a significant reduction of functional tumor microcirculation and induction of endothelial cell apoptosis. In addition, the antiangiogenic activity of CrEL-free ABI-007 has been also confirmed in metronomic therapy. Our results indicate that this antiangiogenic activity can also be obtained by simply administrating CrEL-free SSL-PTX in metronomic therapy.
Several studies have demonstrated that paclitaxel can efficaciously affect endothelial cells at ultralow concentrations. Paclitaxel can disturb most of the endothelial cell functions involved in angiogenesis, since they can inhibit endothelial cell proliferation, motility, and migration (8). Our in vitro results indicate that SSL-PTX could effectively inhibit the endothelial cell proliferation and migration at a concentration-dependent manner. These results are similar with that obtained from ABI-007 (22).
Because paclitaxel is extremely insoluble in water as well as in other vehicles commonly used in parenteral dosage forms, the current Taxol formulation consists of paclitaxel solubilized in 50:50 (v/v) CrEL and dehydrated alcohol. CrEL provokes formation of micelles, which entrap paclitaxel and carry it in the systemic circulation, thereby reducing the free drug fraction available for cellular partitioning (34,35). We suggest that this is a possible reason for nullifying the antiangiogenic activity of paclitaxel. Our in vitro release results indicate that the paclitaxel in Taxol released rapidly and completely (Fig. 1). However, we suggest that this paclitaxel was still entrapped within CrEL micelles, not the free drug. On the contrary, paclitaxel released from SSL-PTX was in free drug form, according to the previous literature (36).
Liposome represents a mature, versatile technology with considerable potential for entrapment of both lipophilic and hydrophilic drugs (37). In addition, liposome is relatively nonimmunogenic and easy to prepare (and employ) for large-scale manufacturing (38). There are several commercially available liposomal products for treatment of a number of neoplastic and infectious diseases (39). However, one of the major drawbacks of the liposomal formulation is its rapid clearance from blood by the RES. This rapid elimination can limit its overall effectiveness (38). Fortunately, when the surface of the liposome was modified with a flexible hydrophilic polymer such as PEG, the uptake by RES could be retarded. This results in the increase of the biological half-life and the spontaneous accumulation of liposome in solid tumor via the “EPR” effect through the passive targeting mechanism. It has been reported that SSL could accumulate in the tumor perivascular space, near the tumor vessels, but not directly associate with the vessel wall following intravenous administration (40). Therefore, we suggest that the paclitaxel which is released from SSL-PTX, like paclitaxel in solution, may render paclitaxel-based antiangiogenic activity.
CONCLUSION
In summary, we have shown that the CrEL-free SSL-PTX is a viable and effective drug carrier system for metronomic chemotherapy. Our in vitro results indicated that SSL-PTX could effectively inhibit the endothelial cell proliferation and migration at a concentration-dependent manner. We also observed that metronomic SSL-PTX inhibited tumor growth in MDA-MB-231 xenograft model via the antiangiogenic effect. Overall, our results suggest that metronomic chemotherapy with low-dose, CrEL-free SSL-PTX should be feasible and effective.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (no. 30873170) and the National Basic Research Program of China (973 Program 2007CB935800 and 2009CB930300).
References
- 1.Frei E, 3rd, Elias A, Wheeler C, Richardson P, Hryniuk W. The relationship between high-dose treatment and combination chemotherapy: the concept of summation dose intensity. Clin Cancer Res. 1998;4:2027–2037. [PubMed] [Google Scholar]
- 2.Nieto Y. The verdict is not in yet. Analysis of the randomized trials of high-dose chemotherapy for breast cancer. Haematologica. 2003;88:201–211. [PubMed] [Google Scholar]
- 3.Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4:423–436. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- 4.Laquente B, Viñals F, Germà JR. Metronomic chemotherapy: an antiangiogenic scheduling. Clin Transl Oncol. 2007;9:93–98. doi: 10.1007/s12094-007-0018-3. [DOI] [PubMed] [Google Scholar]
- 5.Browder T, Butterfield CE, Kräling BM, Shi B, Marshall B, O’Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- 6.Kamat AA, Kim TJ, Landen CN, Jr, Lu C, Han LY, Lin YG, et al. Metronomic chemotherapy enhances the efficacy of antivascular therapy in ovarian cancer. Cancer Res. 2007;67:281–288. doi: 10.1158/0008-5472.CAN-06-3282. [DOI] [PubMed] [Google Scholar]
- 7.Belotti D, Vergani V, Drudis T, Borsotti P, Pitelli MR, Viale G, et al. The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin Cancer Res. 1996;2:1843–1849. [PubMed] [Google Scholar]
- 8.Ng SS, Figg WD, Sparreboom A. Taxane-mediated antiangiogenesis in vitro: influence of formulation vehicles and binding proteins. Cancer Res. 2004;64:821–824. doi: 10.1158/0008-5472.CAN-03-3391. [DOI] [PubMed] [Google Scholar]
- 9.Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Straubinger RM, Arnold RD, Zhou R, Mazurchuk R, Slack JE. Antivascular and antitumor activities of liposome-associated drugs. Anticancer Res. 2004;24:397–404. [PubMed] [Google Scholar]
- 11.Sharma A, Sharma US, Straubinger RM. Paclitaxel-liposomes for intracavitary therapy of intraperitoneal P388 leukemia. Cancer Lett. 1996;107:265–272. doi: 10.1016/0304-3835(96)04380-7. [DOI] [PubMed] [Google Scholar]
- 12.Huwyler J, Drewe J, Krähenbuhl S. Tumor targeting using liposomal antineoplastic drugs. Int J Nanomedicine. 2008;3:21–29. doi: 10.2217/17435889.3.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moghimi SM, Patel HM. Opsonophagocytosis of liposomes by peritoneal macrophages and bone marrow reticuloendothelial cells. Biochim Biophys Acta. 1992;1135:269–274. doi: 10.1016/0167-4889(92)90230-9. [DOI] [PubMed] [Google Scholar]
- 14.Lasic DD. Doxorubicin in sterically stabilized liposomes. Nature. 1996;11(380):561–562. doi: 10.1038/380561a0. [DOI] [PubMed] [Google Scholar]
- 15.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 16.Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, et al. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm. 2007;338:317–326. doi: 10.1016/j.ijpharm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Yang T, Choi MK, Cui FD, Lee SJ, Chung SJ, Shim CK, et al. Antitumor effect of paclitaxel-loaded PEGylated immunoliposomes against human breast cancer cells. Pharm Res. 2007;24:2402–2411. doi: 10.1007/s11095-007-9425-y. [DOI] [PubMed] [Google Scholar]
- 18.Crosasso P, Ceruti M, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J Control Release. 2000;63:19–30. doi: 10.1016/S0168-3659(99)00166-2. [DOI] [PubMed] [Google Scholar]
- 19.Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stinchcombe TE. Nanoparticle albumin-bound paclitaxel: a novel Cremophor-EL free formulation of paclitaxel. Nanomedicine (Lond) 2007;2:415–423. doi: 10.2217/17435889.2.4.415. [DOI] [PubMed] [Google Scholar]
- 21.Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of Cremophor-free, albumin-bound paclitaxel, ABI-007, compared with Cremophor-based paclitaxel. Clin Cancer Res. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 22.Ng SS, Sparreboom A, Shaked Y, Lee C, Man S, Desai N, et al. Influence of formulation vehicle on metronomic taxane chemotherapy: albumin-bound versus Cremophor EL-based paclitaxel. Clin Cancer Res. 2006;12:4331–4338. doi: 10.1158/1078-0432.CCR-05-2762. [DOI] [PubMed] [Google Scholar]
- 23.Ashton AW, Yokota R, John G, Zhao S, Suadicani SO, Spray DC, et al. Inhibition of endothelial cell migration, intercellular communication, and vascular tube formation by thromboxane A(2) J Biol Chem. 1999;274:35562–35570. doi: 10.1074/jbc.274.50.35562. [DOI] [PubMed] [Google Scholar]
- 24.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 25.Bijman MN, Van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006;5:2348–2357. doi: 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]
- 26.Liu XR, Wu KC, Huang Y, Sun JB, Ke XY, Wang JC, et al. In vitro and in vivo studies on plasma-to-blood ratio of paclitaxel in human, rabbit and rat blood fractions. Biol Pharm Bull. 2008;31:1215–1220. doi: 10.1248/bpb.31.1215. [DOI] [PubMed] [Google Scholar]
- 27.Aapro MS, Von Minckwitzb G. Molecular basis for the development of novel taxanes in the treatment of metastatic breast cancer. EJC Supplements. 2008;6:3–11. [Google Scholar]
- 28.Kunstfeld R, Wickenhauser G, Michaelis U, Teifel M, Umek W, Naujoks K, et al. Paclitaxel encapsulated in cationic liposomes diminishes tumor angiogenesis and melanoma growth in a “humanized” SCID mouse model. J Invest Dermatol. 2003;120:476–482. doi: 10.1046/j.1523-1747.2003.12057.x. [DOI] [PubMed] [Google Scholar]
- 29.Schmitt-Sody M, Strieth S, Krasnici S, Sauer B, Schulze B, Teifel M, et al. Neovascular targeting therapy: paclitaxel encapsulated in cationic liposomes improves antitumoral efficacy. Clin Cancer Res. 2003;9:2335–2341. [PubMed] [Google Scholar]
- 30.Strieth S, Eichhorn ME, Sauer B, Schulze B, Teifel M, Michaelis U, et al. Neovascular targeting chemotherapy: encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int J Cancer. 2004;110:117–124. doi: 10.1002/ijc.20083. [DOI] [PubMed] [Google Scholar]
- 31.Strieth S, Nussbaum CF, Eichhorn ME, Fuhrmann M, Teifel M, Michaelis U, et al. Tumor-selective vessel occlusions by platelets after vascular targeting chemotherapy using paclitaxel encapsulated in cationic liposomes. Int J Cancer. 2008;122:452–460. doi: 10.1002/ijc.23088. [DOI] [PubMed] [Google Scholar]
- 32.Strieth S, Eichhorn ME, Werner A, Sauer B, Teifel M, Michaelis U, et al. Paclitaxel encapsulated in cationic liposomes increases tumor microvessel leakiness and improves therapeutic efficacy in combination with Cisplatin. Clin Cancer Res. 2008;14:4603–4611. doi: 10.1158/1078-0432.CCR-07-4738. [DOI] [PubMed] [Google Scholar]
- 33.Bode C, Trojan L, Weiss C, Kraenzlin B, Michaelis U, Teifel M, et al. Paclitaxel encapsulated in cationic liposomes: a new option for neovascular targeting for the treatment of prostate cancer. Oncol Rep. 2009;22:321–326. [PubMed] [Google Scholar]
- 34.Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res. 1999;59:1454–1457. [PubMed] [Google Scholar]
- 35.Ellis AG, Webster LK. Inhibition of paclitaxel elimination in the isolated perfused rat liver by Cremophor EL. Cancer Chemother Pharmacol. 1999;43:13–18. doi: 10.1007/s002800050857. [DOI] [PubMed] [Google Scholar]
- 36.Marcel Musteata F, Pawliszyn J. Determination of free concentration of paclitaxel in liposome formulation. J Pharm Pharm Sci. 2006;9:231–237. [PubMed] [Google Scholar]
- 37.Fielding RM. Liposomal drug delivery. Advantages and limitations from a clinical pharmacokinetic and therapeutic perspective. Clin Pharmacokinet. 1991;21:155–164. doi: 10.2165/00003088-199121030-00001. [DOI] [PubMed] [Google Scholar]
- 38.Campbell RB, Ying B, Kuesters GM, Hemphill R. Fighting cancer: from the bench to bedside using second generation cationic liposomal therapeutics. J Pharm Sci. 2009;98:411–429. doi: 10.1002/jps.21458. [DOI] [PubMed] [Google Scholar]
- 39.Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 40.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]