

Abstract
A dissolution test for tablets containing 40 mg of olmesartan medoxomil (OLM) was developed and validated using both LC-UV and UV methods. After evaluation of the sink condition, dissolution medium, and stability of the drug, the method was validated using USP apparatus 2, 50 rpm rotation speed, and 900 ml of deaerated H2O + 0.5% sodium lauryl sulfate (w/v) at pH 6.8 (adjusted with 18% phosphoric acid) as the dissolution medium. The model-independent method using difference factor (f1) and similarity factor (f2), model-dependent method, and dissolution efficiency were employed to compare dissolution profiles. The kinetic parameters of drug release were also investigated. The obtained results provided adequate dissolution profiles. The developed dissolution test was validated according to international guidelines. Since there is no monograph for this drug in tablets, the dissolution method presented here can be used as a quality control test for OLM in this dosage form, especially in a batch to batch evaluation.
Key words: dissolution kinetic, dissolution test, LC-UV method, olmesartan medoxomil, UV method
INTRODUCTION
Dissolution testing has emerged in the pharmaceutical field as a very important tool to characterize drug product performance. The significance of this test is based on the fact that the rate and extent of drug absorption depend on its dissolution from the dosage form. Therefore, dissolution test is used not only for quality control of the final dosage form, but also to assess several stages of formulation. Moreover, when an in vitro/in vivo correlation is demonstrated, dissolution can be used as a surrogate test to predict the in vivo bioavailability of pharmaceutical formulations (1).
The interest in the in vitro dissolution of poorly soluble drugs has increased over the years due to the fact that this class of drugs is more likely to present a meaningful correlation between dissolution and absorption (2). Difficulties are usually encountered in selecting a dissolution medium of acceptable volume and composition that also presents a good discriminating power (3,4).
Olmesartan medoxomil (OLM; 5-methyl-(2-oxo-1,3-dioxol-4-yl)methyl-4-(1-hydroxy-1-methylethyl)-2-propyl-1-[2′(1H)-tetrazol-5yl)1,1′biphenyl-(4-yl)methyl]-1H-imidazole-5-carboxylate; Fig. 1) is a prodrug that is rapidly and completely hydrolyzed to the active metabolite olmesartan (OL; Fig. 1) by both arylesterase and albumin during gastrointestinal absorption (5). It is a selective angiotensin II receptor blocker which was approved by the US Food and Drug Administration (FDA) in 2002 for the treatment of hypertension, either alone or in combination with other drugs. OLM is also reported to be effective in animal models of atherosclerosis, liver disorders, and diabetic nephropathy (6). This drug is considered a weakly basic compound (pKa = 4.3), highly lipophilic, and with an oral bioavailability of about 29% (7), being classified as class IV, according to the Biopharmaceutical Classification System where its dissolution is the rate-limiting step for absorption (8). A dissolution method for OLM associated with hydrochlorothiazide in tablets was previously reported in the literature (6); however, the method described was not validated according to the parameters established by the official compendious.
Fig. 1.

Chemical structure of OLM and its active metabolite OL
In this context, the purpose of the present study is to develop and validate a dissolution test for OLM in tablets, based on its physicochemical characteristics. Additionally, the method was tested in two different formulations (Benicar® and Olmetec®) using both model-independent method and dissolution efficiency (%DE) approaches to characterize their dissolution profiles. The kinetics dissolution was determined using model-dependent methods. The parameters such as dissolution rate constant (k), time to promote the dissolution half-life (t50%), and time for release drug amount ≥80% (t80%) were also evaluated.
MATERIALS AND METHODS
Chemicals
OLM reference substance (assigned purity 99.3%) was purchased from Sequoia Research Products® (Oxford, UK). Benicar® (Sankyo Pharma, Brazil) and Olmetec® (Pfizer, Brazil) tablets containing 40 mg of OLM were obtained from commercial sources. Sodium lauryl sulfate (SLS) was obtained from Synth (São Paulo, Brazil). The excipients used to simulate those found in the dosage forms (cellulose microcrystalline, low-substitution hyprolose, lactose monohydrated, hyprolose, magnesium stearate, talc, titanium dioxide, hypromellose) were all of pharmaceutical grade and acquired from different distributors. LC-grade acetonitrile was obtained from Tedia® (Fairfield, USA). Purified water (Milli-Q Plus, Millipore®, MA, USA) was used throughout the analysis. All other reagents and solvents used for the preparation of buffer solutions were of analytical grade. The 0.1 and 0.01 M HCl and monobasic potassium phosphate USP buffers (pH 6.8 and 7.5) were prepared as described in USP 32 (11).
Instrumentation
The development and validation of the dissolution test was performed using a VANKEL® VK 8000 dissolution autosampling station consisting of a VK-type bidirectional peristaltic pump, VK 750D digitally controlled heater/circulator, VK 7010 multibath (n = 8) dissolution testing station with automated sampling manifold.
Chromatographic analysis was carried out using a Shimadzu LC system (Kyoto, Japan) which consisted of a LC-10AD pump, a SLA-10ADVP system controller, a SPD-10A (SPM-20A in the selectivity test) detector, a DGU-14A degasser, and a Rheodyne® 7725i manual injector with a 20-µl loop. Data integration was performed using the CLASS-VP software (version 6.1).
A UV–Vis spectrophotometer (UV-160A, Shimadzu) using 1.0 cm quartz cells and SPECTRA MANAGER software was used for all absorbance measurements.
An Ultrabasic potentiometer (Denver, Colorado, USA) was used to determine the pH of all solutions. A Thornton T50 ultrasonic bath (Metler-Toledo, Bedford, MA) was used for deaeration.
Three filters were evaluated for sample filtration: Millipore® (nylon membrane, 0.45 µm), Framex® (quantitative filter, 25 µm), and Vankel® (qualitative filter, 10 µm).
Analytical Conditions
A previously developed stability-indicating LC-UV method (9) was used to determine the percentage of OLM released. As described earlier, a Phenomenex® RP-18 column (250 × 4.6 mm, 5 µm) and UV detection at 257 nm were used. The mobile phase was composed of water/triethylamine/acetonitrile (60:0.3:40 v/v/v, pH adjusted to 6.3, with 18% phosphoric acid), previously filtered through a 0.45-μm membrane (Millipore®, Bedford, USA). The flow rate was 1.2 ml min−1 at room temperature (23 ± 2°C) and the injection volume was 20 µl. The run time was set to 10 min. For the specificity and stability studies, a SPD-M10ADVP photodiode array (PDA) detector was used.
The same samples analyzed by LC-UV were also analyzed by the UV method (λ = 257 nm) in order to compare the results using the dissolution medium as blank.
Tablets Analysis
The content analysis and dose uniformity test of Benicar® and Olmetec® tablets were carried out by the LC method previously validated (9) and described in the “Materials and Methods” section.
For content determination, 20 U of each formulation were ground and an amount equivalent to 10 mg of OLM was transferred to 20 ml volumetric flask containing 10 ml of acetonitrile. Then the flasks were sonicated for 15 min and the volume was completed with the same solvent. After filtration with qualitative filter, aliquots of 1 ml of the solutions were transferred to 25 ml volumetric flasks and diluted with the dissolution medium to obtain the final concentration of 20 µg ml−1. The analysis was performed in triplicate.
The dose uniformity test was evaluated after analysis of ten individual units of both formulations. Each tablet, previously ground, was transferred to 100 ml volumetric flask containing 50 ml of acetonitrile, which was kept in the ultrasonic bath for 30 min, and the volume was completed with the same solvent. After filtration with qualitative filter, aliquots of 1 ml of the solutions were transferred to 20 ml volumetric flasks and diluted with the dissolution medium to obtain the final concentration of 20 µg ml−1.
All solutions were filtered in a 0.45-µm nylon membrane before the chromatographic analysis.
Determination of Sink Conditions
OLM is a prodrug with pH-dependent solubility. In both acid and alkaline solutions, its solubility is high (pH 1.2 and 8.0) and it reaches its lower solubility in the pH range of 4.0 to 6.0 at 20°C (7). The sink conditions were determined in 0.1 M HCl, 0.01 M HCl, pH 6.8 and 7.5 USP buffered solutions. Different concentrations of SLS (0.5%, 1.0%, and 1.5% (w/v)) in the dissolution medium were also evaluated due to the low solubility of this drug. Vessels (n = 3) containing 250 ml of medium were preheated in a thermostatically controlled water bath at 37 ± 0.5°C, before adding an excess of OLM reference substance (approximately 40 mg). The suspensions were gently agitated. Aliquots of 10 ml were removed from each vessel after 1 and 2 h and filtered. An aliquot of 2.5 ml was transferred into 20 ml volumetric flask, diluted with the mobile phase, and analyzed by LC-UV methods.
Dissolution Test Conditions
Dissolution testing of tablets was performed in compliance with USP 32 (11) using a paddle (USP apparatus 2) and 900 ml of dissolution medium preheated at 37 ± 0.5°C. The rotation speed selection was done based on the recommended range (50–75 rpm) for apparatus 2 and the usual values for tablets (10,11). Samples were withdrawn at 15, 30, 45, 60, and 120 min for early validation work. After dissolution optimization, aliquots were withdrawn at 10, 15, 30, 45, 60, and 120 min. Automatic sampling was performed using 8 ml aliquots and these solutions were immediately filtered through 10 µm filters connected into the equipment. The percentage of drug dissolved was determined using both LC-UV and UV methods.
The reference substance solution was prepared using an amount of powder equivalent to 11.1 mg of OLM that was transferred to a 50-ml volumetric flask with acetonitrile (222.2 µg ml−1). An aliquot of 1.0 ml of this solution was transferred to a 10-ml volumetric flask and diluted with dissolution medium, obtaining the final concentration of 22.2 µg ml−1. The solution was filtered in a 0.45-μm membrane filter before analysis.
Dissolution Method Validation
In order to demonstrate that the method was adequate for dissolution test purposes, it was validated using both LC-UV and UV methods, evaluating stability, specificity, linearity, accuracy, precision, robustness, and filter suitability according to USP 32 (11) and ICH guidelines (10,12,13).
Standard and Sample Solution Stability
The stability of OLM in the dissolution medium was evaluated using reference substance and samples. The solutions were submitted to dissolution test in H2O containing 0.5% of SLS (w/v) at pH 6.8, corrected with 18% phosphoric acid, at 37 ± 0.5°C, 50 rpm, for 2 h. After this, both solutions were evaluated in 0 h at room temperature (23 ± 1°C), and after 15 and 24 h, at room and refrigerator (8 ± 2°C) temperatures. During this test, solutions were protected from light. The assay was performed in triplicate. All the results were within 98–102% of the initial value and no degradation product was observed.
Specificity
Specificity was evaluated by preparing a placebo based on the commercial tablets in their usual concentration. An amount equivalent to that contained in one tablet was transferred to vessels with 900 ml of medium, at 37 ± 0.5°C, and stirred for 1 h at 150 rpm, using USP apparatus 2. Aliquots of this solution were filtered prior to analysis. The concentrations of the excipients, listed in the “Materials and Methods” section, were based on the literature (14).
Linearity
Aliquots of a stock solution containing 100 µg ml−1 of OLM reference substance, prepared in acetonitrile, were transferred to 25 ml volumetric flasks and diluted with the dissolution medium to final concentrations of 5, 10, 15, 20, 25, and 30 µg ml−1, for LC-UV and UV analysis. The solutions were analyzed in triplicate every day, during three consecutive days. The linearity was evaluated by linear regression analysis, which was calculated by the least square regression method and analysis of variance (ANOVA).
Accuracy/Precision
Accuracy of the method was evaluated by the recovery test of known amounts of OLM reference substance added to the placebo. A stock solution containing 2 µg ml−1 was prepared in acetonitrile. Aliquots of 8, 10, and 12 ml of this solution were added to a vessel containing the medium for a final volume of 900 ml, preheated at 37 ± 0.5°C, and rotated for 1 h at 150 rpm. The final concentrations were 17.7, 22.2, and 26.6 µg ml−1, corresponding respectively to 80%, 100%, and 120% of the nominal assay concentration. The analyses were done in triplicate, in different days. Placebo samples were prepared in the same way described in the specificity test.
The same solutions used in the accuracy test were analyzed in order to evaluate the precision of the method. Repeatability (intraday) and intermediate precision (interday) were evaluated based on the relative standard deviation (RSD) of the results.
Robustness
Robustness was evaluated during development by making small, but deliberate, changes to the method's parameters. The release of OLM tablets in different pH values of the dissolution medium (6.4 and 6.8) was evaluated.
Filter Suitability
The filter evaluation is necessary to determine its adequacy and to verify that it does not adsorb the drug and that it removes insoluble excipients that may otherwise cause high background or turbidity (10). The reference substance and sample solutions were prepared in dissolution medium with a final concentration of 22.2 µg ml−1.
The sample solutions were prepared using a placebo, like that described in the specificity test, and transferred to the vessel that was gently rotated for 1 h at 37 ± 0.5°C. Four aliquots of 10 ml were withdrawn at the same point and submitted to one of the following procedures: centrifugation for 5 min at 3,000 rpm or filtration with a qualitative, quantitative, and nylon filters.
For a filter to be acceptable for use, the results must be within the 98–102% range (10,15).
Applied Methods to Compare Dissolution Profiles
The dissolution profiles were compared using a model-independent method and dissolution efficiency (%DE).
When comparing dissolution profiles, FDA guidance recommend approaches such as the model-independent method, based on the calculation of difference (f1) and similarity (f2) factors, which is currently applied (Table I). The f1 factor measures the percent error between two curves over all time points. The f2 factor is a logarithmic transformation of the sum-squared error of differences between the test and the reference products over all time points. Two dissolution profiles are considered to be similar if f1 has a value between 0 and 15 and if f2 has a value between 50 and 100 (16,17).
Table I.
Equation Descriptions for the Application of Dependent and Independent Models
| Model | Equation |
|---|---|
| Dependent | |
| Zero-order | Q t = Q 0 − k 0 × t |
| First-order | lnQ t = lnQ 0 − k 1 × t |
| Higuchi | Q 0 = k H × t 1/2 |
| Hixson–Crowell | Q 1/30 − Q t 1/3 = k HC × t |
| Korsmeyer–Peppas | Q t/Q α = k K × t n |
| Independent | |
| Difference factor |
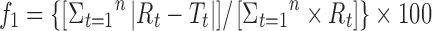
|
| Similarity factor |
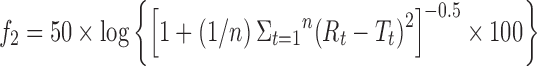
|
Q t amount of drug released in time t, Q α amount of drug released at infinite time t, Q 0 initial amount of drug in the tablet, k 0, k 1, k H, k HC, k K release rate constants, R dissolution measurements of reference, T dissolution measurements of test, n release exponent (indicative of drug release mechanism)
The dissolution efficiency (%DE) was calculated from the values obtained in the area under curve (AUC) of the dissolution profile, up to 120 min, applying the trapezoidal method (16). The %DE was determined considering the rectangle total area (AUCRT) defined by ordinate (100% of dissolution) and abscise (120 min).
To analyze the variance among the average values of %DE obtained from Benicar® and Olmetec®, both determined by LC-UV and UV methods, the ANOVA was applied.
Evaluation of Release Kinetic
To evaluate OLM release kinetics, five mathematical models were applied: zero-order, first-order, Higuchi, Hixon–Croweel, and Korsmeyers–Peppas equations, which are presented in Table I (16,17).
The suitability of models to experimental data was evaluated using the software MicroMath Scientist 3.0 (Micromath, EUA), based on the model selection criteria (MSC).
The curves were constructed by applying the kinetic models listed, considering only the ascendant points of drug released (0 to 30 min).
Other parameters were used to characterize the drug release profile such as dissolution half-life (t50%), which corresponds to the time necessary to release 50% of the drug from the pharmaceutical dosage form, and the sampling time for ≥80% of drug dissolution (t80%). Frequently, pharmacopeias use this parameter as an acceptance limit for the dissolution test (18).
RESULTS AND DISCUSSION
Tablets Analysis
The OLM content in tablets by the LC method for Benicar® and Olmetec® was 99.1 ± 1.1% and 98.9 ± 0.5%, respectively.
In relation to the uniformity test, the analyses for both Benicar® (100.7 ± 4.4%) and Olmetec® (95.7 ± 1.5%) were carried out using the general specifications of USP 32 (11).
According to these analyses, the tablets were considered appropriate for the dissolution tests.
Determination of Sink Conditions and Dissolution Conditions
For poorly soluble drugs, medium selection for dissolution tests is an important step in method validation due to the difficulty to achieve sink condition (19), which is defined as the volume of medium at least three times greater than that required to dissolve the dose of the drug being tested (10). Among the media tested, sink condition was observed only in H2O with 1.5% SLS (w/v) and pH 6.8 (Table II). However, media that fail to provide these conditions may be acceptable, if they prove to be more discriminating (10,11,20). The use of aqueous solutions containing a percentage of surfactant is suggested for poorly soluble compounds (10,20,21). The media tested were: 0.1 M HCl (pH 1.2); 0.01 M HCl (pH 1.8); USP pH 6.8 buffer; H2O + 0.5% SLS (w/v) pH 6.8; H2O + 1.0% SLS (w/v) pH 6.8; and H2O + 1.5% SLS (w/v) pH 6.8 (Fig. 2). USP pH 7.5 buffer medium was not used because preliminary tests showed the formation of degradation products. Dissolution tests were performed using USP apparatus 2 at a rotation speed of 50 rpm at 37 ± 0.5°C.
Table II.
Solubility of OLM Reference Substance at Different Dissolutions Media (40 mg OLM Bulk in 250 ml of Medium)
| Medium | Solubility (%) | |
|---|---|---|
| 1 h | 2 h | |
| 0.1 M HCl (pH 1.2) | 58.4 | 63.5 |
| 0.01 M HCl (pH 1.8) | 15.7 | 45.2 |
| USP pH 6.8 buffer | 34.7 | 38.5 |
| USP pH 7.5 buffer | 59.4 | 62.8 |
| H2O + 0.5% SLS pH 6.8 | 43.9 | 73.3 |
| H2O + 1.0% SLS pH 6.8 | 53.3 | 76.2 |
| H2O + 1.5% SLS pH 6.8 | 78.5 | 97.5 |
Fig. 2.

Dissolution profiles of Benicar® tablets (n = 3) using 0.1 M HCl, 0.01 M HCl, H2O + 0.5% SLS pH 6.8, H2O + 1.0% SLS pH 6.8, H2O + 1.5% SLS pH 6.8, and USP pH 6.8 buffer at 37 ± 0.5°C, USP apparatus 2 at 50 rpm, analyzed by LC-UV method
The use of surfactants in the dissolution medium composition for sparingly aqueous-soluble drugs products is physiologically relevant, because it can better simulate the environment of the gastrointestinal tract than a medium containing other solvents than water (4,8). Shah et al. recommend the use of the lowest amount of surfactant necessary to achieve 75–80% of drug release in a reasonable amount of time (60–90 min) (22).
Reference Substance and Sample Solution Stability
The stability test of the reference substance stock solution and samples demonstrated that OLM was stable in the test conditions for up to 15 h (considering the analysis time for routine quality control and dissolution profiles determination) at refrigerator temperature (8 ± 2°C). After 15 h at room temperature (23 ± 2°C), a decrease in OLM concentration above 2% was observed.
Specificity
The specificity of the dissolution test by the LC-UV method demonstrated no interference of excipients, using a PDA detector (Fig. 3). The same analysis was done using the UV method. The results obtained suggest that the UV method could also be used for the quantitation of OLM, once the formulation excipients had no interference at 257 nm (Fig. 4).
Fig. 3.

Specificity test for OLM before dissolution in H2O + 0.5% SLS pH 6.8 at 37 ± 0.5°C and apparatus 2 at 150 rpm, analyzed by the LC-UV method
Fig. 4.

UV spectrum of OLM before dissolution in H2O + 0.5% SLS pH 6.8 at 37 ± 0.5°C and apparatus 2 at 150 rpm
Linearity
Both LC-UV and UV methods showed good linearity at the concentration range of 5–30 µg ml−1. Correlation coefficients were closer to 1.0000 (>0.9999) for both methods. The slopes and intercepts obtained were 42,568 and 1,656 for LC-UV and 0.0434 and 0.0058 for UV, respectively. The analysis by ANOVA showed significant linear regression (p < 0.05) and no significant deviation from linearity (p > 0.05). These data indicate that both methods are linear for OLM.
Accuracy and Precision
The accuracy was demonstrated by the recovery of known amounts of OLM to the dissolution vessels. Percentage recoveries from 95% to 105% are recommended for this test (10,11). The accuracies of the methods were considered adequate (Table III). Interday and intraday precisions were evaluated at three different concentration levels (17.7, 22.2, and 26.6 µg ml−1) in 2 days. The low RSD (≤2%) demonstrate the good precision of both methods. The results are presented in Table IV.
Table III.
Accuracy Results for OLM (Percent Recovery) by LC-UV and UV Methods
| Sample | Concentration (%) | ||||||
|---|---|---|---|---|---|---|---|
| 17.7 µg ml−1 | 22.2 µg ml−1 | 26.6 µg ml−1 | |||||
| LC-UV | UV | LC-UV | UV | LC-UV | UV | ||
| Day 1 | 1 | 95.9 | 101.4 | 95.7 | 102.0 | 99.3 | 103.4 |
| 2 | 98.2 | 104.9 | 98.3 | 104.0 | 100.1 | 104.6 | |
| 3 | 97.1 | 102.7 | 98.6 | 104.4 | 99.0 | 103.7 | |
| Day 2 | 1 | 96.5 | 103.0 | 96.9 | 102.1 | 100.8 | 104.3 |
| 2 | 96.3 | 101.3 | 96.5 | 102.0 | 100.3 | 104.3 | |
| 3 | 96.0 | 100.4 | 97.0 | 103.0 | 99.4 | 103.7 | |
| Average (n = 6) | 96.7 | 102.3 | 97.1 | 102.9 | 99.8 | 104.0 | |
| Percent RSD | 0.9 | 1.5 | 1.1 | 1.0 | 0.7 | 0.4 | |
RSD relative standard deviation
Table IV.
Precision Results for OLM by LC-UV and UV Methods
| Day | Concentration (µg ml−1) | Intraday (µg ml−1) (±RSD) | Interday (µg ml−1) (±RSD) | ||
|---|---|---|---|---|---|
| LC-UV | UV | LC-UV | UV | ||
| 1 | 17.7 | 17.9 ± 1.2 | 18.2 ± 1.7 | 17.1 ± 0.9 | 18.1 ± 1.5 |
| 2 | 17.0 ± 0.2 | 17.9 ± 1.3 | |||
| 1 | 22.2 | 21.4 ± 1.6 | 22.9 ± 1.2 | 21.5 ± 1.1 | 22.8 ± 1.0 |
| 2 | 21.5 ± 0.3 | 22.7 ± 0.5 | |||
| 1 | 26.6 | 26.4 ± 0.6 | 27.6 ± 0.6 | 26.5 ± 0.7 | 27.6 ± 0.4 |
| 2 | 26.6 ± 0.7 | 27.6 ± 0.3 | |||
RSD relative standard deviation
Robustness
In the evaluation of method robustness, the influence of different pH values (6.4 and 6.8) was tested and the results (data not shown), analyzed by ANOVA, showed no significative difference among the OLM released from Benicar® by LC-UV (Fcalculated = 3.7 × 10−1 < Fcritical = 5.12; p > 0.05) and UV (Fcalculated = 6.1 × 10−1 < Fcritical = 5.12; p > 0.05) methods. These data demonstrated that the dissolution method is robust.
Filter Suitability
The results obtained from filtered and centrifuged solutions of reference substance and sample solutions by both methods showed average recoveries within 98–102%. These data demonstrate that the filters did not interfere in the results of analysis.
LC-UV Versus UV Quantitation and Dissolution Profiles Comparison
The in vitro dissolution profiles of Benicar® and Olmetec® are shown in Fig. 5. Each data point represents a mean of 12 measurements for each product. The RSD are in accordance with the literature, ≥20% for the first data point and then ≤10% (10,11).
Fig. 5.

Dissolution profiles of Benicar® and Olmetec® tablets (n = 12) using H2O + 0.5% SLS pH 6.8 at 37 ± 0.5°C, USP apparatus 2 at 50 rpm, analyzed by the LC-UV and UV methods
The percentage of drug dissolved, until 120 min, for both drug products were similar between them, as well as between the methods (Benicar®: LC-UV method, 93.1 ± 1.8% and UV method, 93.8 ± 4.4%; Olmetec®: LC-UV method, 95.6 ± 1.3% and UV method, 94.3 ± 2.2%).
The statistical investigation by ANOVA showed that both methods, LC-UV and UV, can be used to evaluate the percentage of drug released of OLM in tablets (Fcalculated = 8.0 × 10−4 < Fcritical = 4.84; p > 0.05).
Since Benicar® is the reference brand, the difference (f1) and similarity (f2) factors were calculated between Benicar® and Olmetec®. According to the FDA guidance (23), for the calculus of these parameters, only one point in the dissolution profile, after 85% of the drug is released, should be used. Therefore, this percentage for Benicar® was reached at 30 min (88.1% LC-UV and 85.3% UV) and for Olmetec® at 30 min (90.2% LC-UV) and 45 min (91.1% UV). The results of f1 and f2 were 5.4 and 62.6 for LC-UV and 5.1 and 93.7 for UV, respectively, showing that profiles were similar.
In order to evaluate the suitability of both methods with respect to each formulation, they were also analyzed using f1 and f2. For Benicar®, using LC-UV results as a reference, f1 and f2 were 1.3 and 97.4, respectively, and for Olmetec®, f1 and f2 were 5.9 and 93.7, respectively. These results demonstrate that both methods could be used to access the dissolution data for these formulations.
In accordance with the similarity results, the %DE data also showed no significant differences between the dissolution profiles of Benicar® (87.0 ± 1.2% LC-UV and 84.3 ± 3.2% UV) and Olmetec® (86.0 ± 1.3% LC-UV and 84.9 ± 1.7% UV; Fcalculated = 8.9 × 10−1 < Fcritical = 2.0; p > 0.05).
Kinetics of Drug Release
Dissolution profiles (LC-UV method) were used to evaluate the kinetics of drug release. The slope (k), determination coefficient (R2), and MSC are presented in Table V. Considering these values, obtained by mathematical modeling (MicroMath Scientist®), distinct release mechanisms were found for Benicar® (Hixson–Crowell) and Olmetec® (zero-order).
Table V.
Kinetic Parameters for Different Release Models of OLM in Tablets Analyzed by LC-UV Method
| Dissolution model | Benicar® | Olmetec® |
|---|---|---|
| Zero-order k 0 | 3.0832 | 3.0433 |
| R 2 | 0.9742 | 0.9927 |
| MSC | 3.1587 | 4.4273 |
| First-order k 1 | 0.0523 | 0.0493 |
| R 2 | 0.9529 | 0.9285 |
| MSC | 2.5573 | 2.1393 |
| Higuchi k H | 14.4125 | 14.1377 |
| R 2 | 0.9219 | 0.8894 |
| MSC | 2.0498 | 1.7023 |
| Hixson–Crowell k HC | 0.0696 | 0.0663 |
| R 2 | 0.9756 | 0.9592 |
| MSC | 3.2171 | 2.6998 |
| Korsmeyers–Peppas k K | 0.0501 | 0.0322 |
| n | 0.8466 | 0.9820 |
| R 2 | 0.9844 | 0.9928 |
| MSC | 3.1634 | 3.9442 |
| t 50% (min) | 15.1 | 15.7 |
| t 80% (min) | 25.9 | 26.3 |
k 0, k 1, k H, k HC, k K release rate constants, MSC model selection criteria, n release exponent (indicative of drug release mechanism), t 50% dissolution half-life (zero-order kinetics), t 80% time to release 80% of the drug (zero-order kinetics)
The Hixson–Crowell model assumes that the drug release mechanism from the dosage form is a function of the cube root of the unreleased drug fraction with time. So, the geometric tablet shape diminishes proportionally over time, assuming that the release rate is limited by the dissolution rate of drug particles (16). The zero-order model describes the systems where the drug release rate is independent of its concentration (16).
The zero-order model, which also described the data adequately, was used to calculate t50% and t80% (Table V). The results revealed that both formulations showed similar values for t50%, while t80% values confirmed that more than 80% of OLM in both formulations was released. Thus, these results are in accordance with the data obtained in the dissolution prolife for Benicar® and Olmetec® by both methods.
CONCLUSION
A dissolution test was developed and validated for OLM tablets, according to the USP 32 (11) and ICH guideline (10,12,13). Final conditions are 900 ml of H2O + 0.5% SLS (w/v) at pH 6.8 as dissolution medium, at 37 ± 0.5°C, and USP apparatus 2 at 50 rpm. The percentage of drug dissolved was determined by the LC-UV and UV methods, and the results showed no significant difference (p > 0.05), when applied to two drug products. Dissolution profiles were evaluated using model-independent approaches and dissolution efficiency (%DE). Difference (f1) and similarity (f2) factors, as well as %DE, showed similar profiles for Benicar® and Olmetec®, when analyzed by both methods. Kinetics of drug release was better described by the Hixson–Crowell model, for Benicar®, and the zero-order model, for Olmetec®. The kinetic parameters (t50% and t80%) estimated by the zero-order model revealed that both formulations showed similar performance. Considering this, the proposed methods could be used as a routine quality control test, since there is no fully validated available method for OLM in tablets.
Acknowledgements
We acknowledge the Conselho Nacional de Pesquisa, Brazil for the financial support.
References
- 1.Ansari M, Kazemipour M, Talebnia J. The development and validation of a dissolution method for clomipramine solid dosage forms. Dissol Technol. 2004;11:16–24. [Google Scholar]
- 2.Abdou HM. Remington: the science and practice of pharmacy. In: Gennaro AR, editors. Philadelphia: Lippincott Williams & Wilkins; 2000. p. 654–66
- 3.Löbenberg R, Krämer J, Shah MP, Amidon GL, Dressman JB. Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of glibenclamide. Pharm Res. 2000;17:439–444. doi: 10.1023/A:1007529020774. [DOI] [PubMed] [Google Scholar]
- 4.Shah VP, Konecny JJ, Everett RL, McCullough B, Noorizadeh AC, Skelly JP. In vitro dissolution profile of water-insoluble drug dosage forms in the presence of solubilizers. Pharm Res. 1989;6:612–618. doi: 10.1023/A:1015909716312. [DOI] [PubMed] [Google Scholar]
- 5.Yoshihara K, Gao Y, Shiga H, Wada R, Hisaoka M. Population pharmacokinetics of olmesartan following oral administration of it prodrug, olmesartan medoxomil in healthy volunteers and hypertensive patients. Clin Pharmacokinet. 2005;44:1324–1329. doi: 10.2165/00003088-200544120-00011. [DOI] [PubMed] [Google Scholar]
- 6.Sagirli O, Onal A, Toker SE, Sensoy D. Simultaneous HPLC analysis of olmesartan and hydrochlorothiazide in combined tablets and in vitro dissolution studies. Chromatographia. 2007;66:213–218. doi: 10.1365/s10337-007-0304-9. [DOI] [Google Scholar]
- 7.Koike H, Konse T, Sada T, Ikeda T, Hyogo A, Hinman D, et al. Olmesartan medoxomil, a novel potent angiotensin II blocker. Annu Rep Sankyo Res Lab. 2003;55:1–91. [Google Scholar]
- 8.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 9.Bajerski L, Rossi RC, Dias CL, Fröehlich PE, Bergold AM. Stability-indicating LC determination of a new antihypertensive, olmesartan medoxomil in tablets. Chromatographia. 2008;68:991–996. doi: 10.1365/s10337-008-0811-3. [DOI] [Google Scholar]
- 10.Pharmacopeial Forum. Pharmacopeial previews. 2004; 30:351–63.
- 11.Pharmacopeia US . USP 32 NF 27. Rockville: USP; 2009. [Google Scholar]
- 12.International Conference on Harmonisation (ICH). ICH harmonized tripartite guideline: validation of analytical procedure Q2A; 1994.
- 13.International Conference on Harmonisation (ICH). ICH harmonized tripartite guideline: validation of analytical procedure Q2B; 1994.
- 14.Rowe RC, Sheskey PJ, Welle PJ. Handbook of pharmaceutical excipients. In: Rowe RC, Sheskey PJ, Welle PJ, editors. Washington; 2000.
- 15.Marques MRC, Brown W. Desenvolvimento e validação de métodos de dissolução para formas farmacêuticas sólidas orais. Analytica. 2002;1:48–51. [Google Scholar]
- 16.Costa PJC. Avaliação in vitro da lioequivalência de formulações farmacêuticas. Braz J Pharm Sci. 2002;38:141–153. [Google Scholar]
- 17.Ferraz HG, Consiglieri VO, Storpirtis S. Avaliação da cinética de dissolução de ampicilina em comprimidos comercializados no Brasil. Braz J Pharm Sci. 1998;34:93–109. [Google Scholar]
- 18.Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 19.Khan MZI. Dissolution testing for sustained or controlled release oral dosage forms and correlation with in vivo data: challenges and opportunities. Int J Pharm. 1996;140:131–143. doi: 10.1016/0378-5173(96)04561-9. [DOI] [Google Scholar]
- 20.Brown CK, Chokshi HP, Nicherson B, Reed RA, Rohrs BR, Shah PA. Acceptable analytical practices of dissolution testing of poorly soluble compounds. Pharm Technol. 2004; 56–65.
- 21.Rohrs BR. Dissolution method development for poorly soluble compounds. Dissol Technol. 2001;8:1–5. [Google Scholar]
- 22.Swanepoel E, Liebenberg W, Devarakonda B, Villiers MM. Developing a discriminating dissolution test for three mebendazole polymorphs based on solubility differences. Pharmazie. 2003;58:117–121. [PubMed] [Google Scholar]
- 23.Food and Drugs Administration (FDA) FDA guidance for industry: dissolution testing of immediate release solid oral dosage forms. Rockville: FDA; 1997. [Google Scholar]