

Abstract
Atorvastatin calcium (ATC), an anti-lipid BCS class II drug, is marketed in crystalline and amorphous solid forms. The objective of this study was to perform solid state characterization of commercial crystalline and amorphous ATC drug samples available in the Indian market. Six samples each of crystalline and amorphous ATC were characterized using X-ray powder diffractometry (XRPD), differential scanning calorimetry (DSC), thermogravimetric analysis, Karl Fisher titrimetry, microscopy (hot stage microscopy, scanning electron microscopy), contact angle, and intrinsic dissolution rate (IDR). All crystalline ATC samples were found to be stable form I, however one sample possessed polymorphic impurity, evidenced in XRPD and DSC analysis. Amongst the amorphous ATC samples, XRPD demonstrated five samples to be amorphous ‘form 27’, while, one matched amorphous ‘form 23’. Thermal behavior of amorphous ATC samples was compared to amorphous ATC generated by melt quenching in DSC. ATC was found to be an excellent glass former with Tg/Tm of 0.95. Residual crystallinity was detected in two of the amorphous samples by complementary use of conventional and modulated DSC techniques. The wettability and IDR of all amorphous samples was found to be higher than the crystalline samples. In conclusion, commercial ATC samples exhibited diverse solid state behavior that can impact the performance and stability of the dosage forms.
Key words: atorvastatin calcium, physical characterization, thermal analysis, X-ray powder diffraction
INTRODUCTION
Majority of active pharmaceutical ingredients (APIs) can exist in different solid-state forms like polymorphs, solvates, and amorphous state (1). These solid forms can differ widely in their physicochemical, mechanical, and biopharmaceutical properties, and thus can influence the quality, safety, and efficacy of the drug product (1,2). Therefore, in addition to optimal solid form selection during the early drug development phase (3,4), a vital aspect gaining credence is the characterization and monitoring of solid state purity in commercial bulk drug manufacturing (5,6). This need is emphasized by stringent regulatory standards (2,7) and intellectual property related issues (8,9).
Apart from the solid state form, particle level properties of API can also critically influence the drug product performance. These properties include particle shape, size, particle size distribution, surface area, porosity, and surface morphology. A comprehensive understanding of these properties is essential to understand the behavior of API and drug product performance (4). This assumes higher importance in case of drugs that have solubility/dissolution rate limited oral bioavailability, such as the Biopharmaceutical Classification System (BCS) class II drugs (1).
Atorvastatin calcium (ATC), chemically designated as [R-(R*, R*)]-2-(4-fluorophenyl)-β, δ-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid, calcium salt (2:1) trihydrate ([C33H34FN2O5]2Ca.3H2O; MW 1209.42; Fig. 1), is a blockbuster synthetic lipid lowering agent and was first marketed by Pfizer (10). The drug belongs to BCS class II (11) and has an aqueous solubility of 0.1 mg/ml (12). Although, ATC is well-absorbed, it has an absolute bioavailability of 14% due to pre-systemic clearance in gastrointestinal mucosa and/or hepatic first pass metabolism (10).
Fig. 1.

Chemical structure of ATC
The theoretical biopharmaceutical parameters, based on BCS, were calculated for ATC using its highest dose of 80 mg (10), particle size of 100 µm (i.e., unmicronized), effective permeability (Peff) of 2x10−4 cm/s and small intestinal transit time (Tsi) of 199 min (13). ATC showed a dose number (D0) of 3.2, which was greater than the favorable value of 1 (12), a dissolution time (Tdisso) of 200 min leading to dissolution number (Dn) of 0.995 and an absorbable dose (Dabs) of 191 mg. These values suggest ATC to exhibit dissolution rate limited oral bioavailability. This was in agreement with the reported information that the extent of oral absorption for ATC increases in proportion to its dose (10). Thus, modification of solid form and particle characteristics can potentially be used to alter the fraction of drug absorbed.
Till date, ATC is reported to exist in about 41 crystalline (14) and two ‘amorphous’ forms (15). Numerous patents/patent applications have characterized these solid forms based on crystallographic and spectroscopic techniques. However, there is limited information on thermal characterization of ATC solid forms (16–18).
ATC is available in crystalline and amorphous forms in the Indian market. The objective of the present work was to comprehensively characterize commercial drug samples of crystalline and amorphous ATC for their molecular and particle level properties. The impact of these properties on API behavior was evaluated by wettability and intrinsic dissolution rate (IDR) determination. This study is expected to highlight the diversity in the solid state properties of ATC available in the Indian market and provide valuable information for developing drug substance specification for ATC physical form purity.
MATERIALS AND METHODS
Materials
Six commercial samples each of crystalline and amorphous ATC, produced by API manufacturers based in India were obtained as gift samples and coded as ATC-1 to ATC-6 (crystalline samples) and ATC-7 to ATC-12 (amorphous samples), respectively. The drug samples had chemical purity greater than 98% w/w (high performance liquid chromatography assay) according to the certificate of analysis of the manufacturers. Milli-Q water was used for the contact angle and IDR studies. All other chemicals used were of analytical grade.
Methods
Preparation of Amorphous ATC
Amorphous ATC was generated in situ by melt quenching the crystalline drug in the differential scanning calorimetry (DSC) instrument. Drug samples (3-5 mg) were taken in pin-holed aluminum pans and heated from 25°C to 171°C at a heating rate of 20°C/min, held isothermally for 30 s and immediately cooled to 25°C at the same rate. HPLC analysis confirmed 98% w/w assay of the treated sample, thereby ruling out significant degradation of the drug during melt quenching.
The HPLC analysis was performed using HPLC Shimadzu system (Shimadzu Corporation, Kyoto, Japan) which included a system controller (CBM-20A), a isocratic pump (LC-10AT), a degasser (DGU-20A5), a cooling autosampler (SIL-20AC), a column oven (CTO-10A), and a photodiode array detector (SPD-M20A) with Class-VP (Release 6.12) software. The HPLC analysis was performed on Lichrospher 100 RP-18e column (250 × 4 mm, 5 µm) maintained at temperature of 30°C. The mobile phase of 0.96% w/v citric acid anhydrous in water (pH 4.0 adjusted with ammonia solution)/acetonitrile/tetrahydrofuran (5.3:2.7:2 v/v) was pumped at a flow rate of 1.0 ml/min at 25°C. The injection volume was 20 µl and the UV detector was set at a wavelength of 244 nm.
X-ray Powder Diffraction
X-ray powder diffraction (XRPD) patterns of samples were recorded at room temperature on Bruker’s D8 Advance diffractometer (Karlsruhe, West Germany) with Cu Kα radiation (1.54 Å), at 40 kV, 40 mA passing through nickel filter with divergence slit (0.5°), antiscattering slit (0.5°), and receiving slit (0.1 mm). The diffractometer was equipped with a 2θ compensating slit, and was calibrated for accuracy of peak positions with corundum. XRPD scans were recorded in continuous mode with a step size of 0.01° and step time of 1 s for crystalline samples and step size of 0.02° and step time of 1 s for amorphous samples, over an angular range of 3-40° 2θ. Accurately weighed amount of powder (300 mg) was loaded in a 25 mm polymethyl methacrylate holder and pressed by a clean glass slide to ensure coplanarity of the powder surface with the surface of the holder. The sample holder was rotated in plane parallel to their surface at 30 rpm during the measurement. Obtained diffractograms were analyzed with DIFFRACplus EVA (version 9.0) diffraction software.
Differential Scanning Calorimetry
Conventional DSC (cDSC) studies were performed on Mettler Toledo 821e DSC (Mettler Toledo, Switzerland) operating with Stare software (version 9.0). Accurately weighed samples (3-5 mg) were analyzed in pin-holed aluminum pans. Heating rates of 10, 20, and 50°C/min under dry nitrogen purge of 40 ml/min were applied. Low heating rates helped to resolve the overlapping thermal events and fast heating rates allowed to maintain the original solid form of the sample (19). Glass transition temperatures (Tg) and endothermic transitions were reported as the onset temperatures. Melting onset was determined as the intersection point of the tangent of baseline before the transition and the inflection tangent. The instrument was calibrated for temperature and heat flow using high purity indium and zinc standard. A tau lag calibration of instrument for different heating rates was performed with indium. For heat-cool-heat cycle samples were heated from 25°C to 170°C, cooled to 100°C and reheated to 200°C at a scan rate of 20°C/min.
Modulated temperature DSC (MTDSC) studies were conducted using DSC, Model Q2000 (TA Instruments, Delaware, USA). Accurately weighed samples (3-5 mg) were analyzed in crimped pans in temperature range of 80 to 200°C under dry nitrogen purge of 50 ml/min. ‘Heat-cool’ MTDSC method using a heating rate of 2°C/min, modulation amplitude of 0.5°C and period of 40 s was employed. The instrument was calibrated for temperature and heat flow using high purity indium standard.
Thermogravimetric Analysis
Thermogravimetric analysis (TGA) was performed using Mettler Toledo 851e TGA/SDTA (Mettler Toledo, Switzerland) operating with Stare software version Solaris 2.5.1. Drug samples (5-7 mg) were weighed and analyzed under nitrogen purge (20 ml/min) in alumina crucibles at a heating rate of 20°C/min over a temperature range of 25-250°C.
Karl Fischer Analysis
Water content (% w/w) of the samples (200 mg) was determined by Karl Fischer titrimetry (716 DMS Titrino, Metrohm Limited, Switzerland). The instrument was calibrated using deionized water, before sample analysis.
Microscopy
Scanning Electron Microscopy
Powder sample morphology and particle size distribution was studied using scanning electron microscopy (Hitachi Electron Microscope, S-3400 N, Japan). Samples were mounted onto aluminum stubs using double sided adhesive tape and sputter coated with a thin layer of gold at 10 Pa vacuum before examination (Hitachi, Ion sputter, E1010, Japan). The specimens were scanned with an electron beam of acceleration potential of 25 kV. Particle size analysis was performed by measuring the length along the longest axis of individual particles. Particle size of about 100 particles was measured and mean diameter was determined.
Hot Stage Microscopy
Thermal transitions were observed under Leica DMLP polarized microscope (Leica Microsystems Wetzlar GmbH, Wetzlar, Germany) equipped with Linkam LTS 350 hot stage. Photomicrographs were acquired using JVS color video camera and analyzed using Linksys32 software. Samples were mounted in oil and heated from 25°C to 200°C at a heating rate of 20°C/min.
Sessile Drop Contact Angle Measurement
Contact angle of powder samples were measured by sessile drop method using Drop Shape Analyser instrument (FTA 1000, First Ten Angstrom, Virginia, USA). Compacts were prepared by compacting 100 mg of powder at 800 psi pressure in a hydraulic press (Hydraulic Unit Model 3912, Carver Inc., WI, USA), with a dwell time of 15 s, in 8 mm punch die set (surface area 0.5 cm2). Analysis of disc by XRPD confirmed that no solid-state transition occurred during compaction. Compacts were mounted on glass slide and drop of Milli-Q water saturated with ATC was dispensed on them. The video was captured by the FTA image analyser. Contact angle was calculated by the instrument by fitting mathematical expression to the shape of the drop and then calculating the slope of the tangent to the drop at the liquid-solid-vapor interface line. The contact angle made at 0.2 s from the time of contact of drop to the solid surface was taken for comparative assessment of powder samples. The surface tension of Milli-Q water and ATC saturated water were measured to be 73.1 ± 0.5 and 71.5 ± 0.4 mN/m at 25 ± 2°C, respectively. All measurements were performed in air under ambient conditions of 25 ± 2°C/55 ± 5 %RH, with the reported values being an average of six measurements.
Intrinsic Dissolution Rate
Intrinsic dissolution of crystalline and amorphous ATC samples was performed by the stationary disc method using USP 24 intrinsic dissolution apparatus. The compacts were prepared by method as described for contact angle measurement. Dissolution studies were performed in 500 ml of 50 mM sodium dihydrogen phosphate buffer (pH 6.8) containing 2% w/v sodium lauryl sulfate maintained at a temperature of 37 ± 0.5°C and rotational speed of 100 rpm. Aliquots were withdrawn at predefined time points, replaced with dissolution medium, and analyzed using a UV spectrophotometer at 242 nm. All measurements were performed in triplicates.
IDR is the rate of mass transfer from solid to liquid state when conditions such as surface area, pH, ionic strength and stirring speed are kept constant. IDR was determined using Eq. 1 (20)
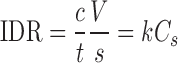 |
1 |
Where, C, is the drug concentration at time t, V is the volume of test solution, S is the surface area of compact, K is intrinsic dissolution rate constant, and Cs is the saturation solubility of drug.
RESULTS AND DISCUSSION
Particle Morphology
Morphology of ATC drug samples by SEM showed crystalline samples (ATC-2 to ATC-6) to consist primarily of rod shaped crystals, whereas ATC-1 showed irregular particles (Fig. 2). All amorphous samples (ATC-7 to ATC-12) comprised of irregular shaped particles. All the crystalline samples showed birefringence under plane polarized light microscope, while it was absent in the amorphous samples. Particle size analysis by SEM showed samples ATC-4, ATC-5 ATC-9 and ATC-10 had a mean diameter of about 5 µm, ATC-6, ATC-7 and ATC-12 had a mean diameter of about 3 µm and ATC-1, ATC-2, ATC-3, ATC-8 and ATC-11 exhibited a mean diameter of less than 1 µm.
Fig. 2.

Scanning electron microphotographs of crystalline samples: a ATC-1, b ATC-2, c ATC-3, d ATC-4, e ATC-5, and f ATC-6; and amorphous samples: g ATC-7, h ATC-8, i ATC-9, j ATC-10, k ATC-11, and l ATC-12
X-ray Powder Diffraction
Crystalline ATC
XRPD pattern of ATC-1 to ATC-6 revealed them to be of crystalline nature (Fig. 3). Samples ATC-2 to ATC-6 showed peaks at 2θ values of 9.07, 9.41, 10.18, 10.46, 11.76, 12.10, and 16.96° corresponding to crystalline ATC form I (21). No extra peaks were observed indicating the purity of the solid form. In contrast, ATC-1 showed numerous peaks additional to the ATC form I XRPD pattern. The peaks not corresponding to ATC form I was observed at 4.25, 4.54, 6.34, 6.84, 8.07, 8.61, 9.24, 10.71, 12.67, and 16.23° 2θ. Also, ATC-1 XRPD peaks were of low intensity and diffused. This indicated that ATC-1 had poor crystallinity and was a mixture of polymorphic forms. Additionally, Fourier transform infrared (FTIR) spectra of all crystalline samples except ATC-1, matched the reported crystalline ATC form-1 (17; data not shown).
Fig. 3.

X-ray powder diffractograms of crystalline ATC samples: a ATC-1, b ATC-2, c ATC-3, d ATC-4, e ATC-5, and f ATC-6. Samples ATC-2 to ATC-6 were form I while ATC-1 was a polymorphic mixture; arrows indicate peaks of ATC-1 not corresponding to form I
Amorphous ATC
All amorphous drug samples (ATC-7 to ATC-12) were found to be X-ray amorphous (Fig. 4). When compared to the XRPD patterns of the reported amorphous ATC forms 23 and 27 (15), halo pattern of ATC-7, ATC-8, ATC-9, ATC-10, and ATC-12 matched with form 27, while ATC-11 matched form 23. FTIR spectra for all amorphous ATC samples were found to be qualitatively similar (data not shown).
Fig. 4.

X-ray powder diffractograms of amorphous ATC samples: a ATC-7, b ATC-8, c ATC-9, d ATC-10, e ATC-11 and f ATC-12. Samples ATC-7, ATC-8, ATC-9, ATC-10, and ATC-12 exhibited XRPD pattern similar to that reported for amorphous ATC form 27, while XRPD pattern of ATC-11 was similar to amorphous ATC form 23
Thermal Characterization
Crystalline ATC
TGA analysis of ATC samples at heating rate of 20°C/min (Fig. 5) was studied to elucidate the weight loss corresponding to the thermal transitions. The TGA scan of ATC-2 to ATC-6 showed three steps weight loss in the temperature region of 40-60°C, 70-120°C, and 120-158°C, respectively. The first, relatively smaller, step change was most likely due to the loss of loosely bound water. Subsequent, second and third step changes were due to the loss of water of hydration. Fall in baseline above temperature of 207°C was due to its degradation. The Karl Fischer titration analysis of crystalline samples showed water content in the range of 4-6% w/w, which matched the total weight loss observed in the TGA. Theoretical water loss for ATC trihydrate has been reported as 4.46% w/w (17). ATC-1 exhibited a total weight loss of 4.39% w/w; however, the contribution of loosely bound water (1.55% w/w) was considerably higher in comparison to all other crystalline samples.
Fig. 5.

TGA overlay of crystalline ATC samples: a ATC-1, b ATC-2, c ATC-3, d ATC-4, e ATC-5, and f ATC-6, at heating rate of 20°C/min. Values in parentheses indicate the total weight loss (% w/w)
DSC analysis, at a heating rate of 20°C/min, for samples ATC-2 to ATC-6 showed broad endotherm of water loss in the temperature range of 80-125°C, followed by a second endotherm for water loss and melting with an onset temperature in the range of 141-148°C (Fig. 6). Thus, the melting peak included partial loss of hydrate water (from TGA data) contributing to asymmetry, with shouldering being observed in some cases (ATC-3 and ATC-4). The enthalpy of fusion (ΔHm) ranged from 81.4 to 87.1 J/g and matched with the earlier reported values (18). Samples ATC-2 to ATC-6 showed similar behavior in DSC and were confirmed to be ATC polymorphic form I.
Fig. 6.

DSC thermograms at heating rate of 20°C/min of crystalline ATC samples: a ATC-1, b ATC-2, c ATC-3, d ATC-4, e ATC-5, and f ATC-6; dashed boxes depict water loss and melting endotherms, and the arrow shows the zoomed DSC scan of ATC-1
ATC-1 exhibited a DSC plot different from all other crystalline samples. It showed an endotherm of water loss in the temperature range of 91-120°C. The second endotherm had an onset of 128°C and a shoulder peak in the tailing end at temperature of 156°C with a significantly lower ΔHm of 14.03 J/g. This was followed by third endotherm at temperature of 184°C of enthalpy 18.39 J/g. The multiple endothermic peaks were probably due to polymorphic impurity. The MTDSC scan of ATC-1 showed an absence of Tg in the reversible signal (data not shown), which negated the possibility of amorphous solid form impurity.
Samples were also evaluated at lower (10°C/min) and higher (50°C/min) heating rates in DSC. At scan rate of 10°C/min, the melting endotherms had an early onset, and the peaks became broad and showed prominent shouldering (Fig. 7i). This can be ascribed to the molecular reorganization occurring at lower heating rates due to longer time of analysis (22). Further, at scan rate of 50°C/min the endothermic transitions merged to give single sharp melting peak with a shift in onset temperature to the range of 150-160°C (Fig. 7ii). This apparent increase in melting temperature occurred due to delayed loss of water of hydration. The thermal transitions were visually examined by hot stage microscopy (HSM) at a heating rate of 20°C/min. Water loss was observed as generation of tiny droplets around the solid particles followed by melting in the temperature regions as seen in DSC profiles. ATC-1 showed two separate melting transitions which correspond with the two different melting endotherms observed in the DSC profile.
Fig. 7.

DSC thermograms at heating rate of (i) 10 and (ii) 50°C/min of crystalline ATC samples: a ATC-1, b ATC-2, c ATC-3, d ATC-4, e ATC-5, and f ATC-6; dashed boxes depict water loss and melting endotherms; melting endotherms show shouldering
Amorphous ATC
Amorphous ATC was generated in DSC by in situ method and used as a standard for comparison of the commercial samples. Its DSC profile showed a glass transition onset at temperature of 140°C, followed by degradation as a fall in baseline at temperature of 190°C. No recrystallization was observed in the heating run. ATC exhibited a Tg/Tm of 0.95. The ratio of Tg/Tm greater than 0.7 indicates excellent glass forming ability (23), and thus ATC could be considered to be an excellent glass former.
In comparison to the crystalline samples, the TGA scan of amorphous ATC samples (Fig. 8) showed only one broad step weight change, in the temperature range of 40-120°C, followed by degradation at temperature of about 207°C. The Karl Fischer titration analysis of amorphous samples showed water content in the range of 1-4% w/w.
Fig. 8.

TGA overlay of amorphous ATC samples: a ATC-7, b ATC-8, c ATC-9, d ATC-10, e ATC-11, and f ATC-12, at heating rate of 20°C/min. Values in parentheses indicate the total weight loss (% w/w)
All amorphous samples at a heating rate of 20°C/min showed a broad endotherm for water loss in the temperature range of 40-120°C, followed by glass transition onset in the temperature range of 145-149°C (Fig. 9ii). The use of pierced pan in DSC study ensured removal of water before the onset of Tg, and hence, the glass transition behavior is expected to remain unaffected by water. However, determination of Tg in the first heating run can have interference from enthalpy relaxation, which primarily depends on the storage history of the sample. All the amorphous samples showed different extent of enthalpy relaxation accompanying the glass transition event. DSC profile of ATC-11, when compared to other samples, was characterized to have a much sharper endothermic event at Tg. This, coupled with its XRPD pattern indicated a greater ‘molecular order’ in amorphous form 23. Further, in case of ATC-11 and ATC-12, glass transition was followed by an additional endotherm at temperature of 171 and 164°C, respectively. HSM study of amorphous ATC samples at 20°C/min showed water loss followed by liquification in the temperature range of 145-170°C.
Fig. 9.

DSC thermograms at heating rate of (i) 10, (ii) 20, and (iii) 50°C/min of amorphous ATC samples: a ATC-7, b ATC-8, c ATC-9, d ATC-10, e ATC-11, and f ATC-12; dashed boxes depict water loss and glass transitions and arrows indicate post T g endotherm
Samples were also analyzed at heating rates of 10 and 50°C/min (Fig. 9). Glass transition onset shifted to temperature range of 140-147°C at 10°C/min and to temperature range of 148-153°C at a higher heating rate of 50°C/min. Enthalpy relaxation were observed at all heating rates. However, post Tg endotherm was observed for ATC-7, ATC-9, ATC-11, and ATC-12 at heating rate of 10°C/min, and for only ATC-11 and ATC-12 at higher heating rates (20 and 50°C/min). The appearance of endotherm at even high heating rate in case of ATC-11 and ATC-12 indicated towards presence of impurity in the ‘ab initio’ samples. The impurity could be due to presence of multiple ‘amorphous’ forms or residual crystalline content. To confirm this possibility, samples ATC-11 and ATC-12 were further investigated by heat-cool-heat cycle and MTDSC studies.
Heat-cool-heat Cycle
ATC-12 was assessed by heat-cool-heat treatment at scan rate of 20°C/min in DSC to understand the post Tg endotherm (Fig. 10). In the first heating cycle, Tg was observed at temperature of 145°C followed by an endotherm at temperature of 164°C, while during the cooling phase, no transition was visible. In the second heating cycle, only Tg (at 143°C) was observed. Similar results were observed with ATC-11 (data not shown). Heating the sample beyond the melting temperature and cooling back resulted in generation of a single amorphous phase and subsequent heating run showed a single Tg. This highlighted the kinetic nature of the post Tg endotherm, which can be correlated with the fact that the post Tg endotherms were observed in four amorphous samples at lower (10°C/min) heating rate in the DSC, but were only visible for ATC-11 and 12 at higher (20 and 50°C/min) heating rates. ATC-11 and ATC-12 samples were further assessed by MTDSC technique.
Fig. 10.

Heat-cool-heat DSC thermogram for ATC-12 at scan rate of 20°C/min; dashed lines indicate heating and cooling phase(s) and the arrow indicates post T g endotherm; insets depict zoomed version of T g and post T g endotherms
MTDSC
Second heating run in heat-cool-heat cycle resulted in disappearance of the post Tg endothermic events. However, it could not reveal whether the second endothermic event was because of crystallinity or due to multiple amorphous states. MTDSC scan of ATC-11 and ATC-12 showed the post Tg endotherm in the non-reversing heat flow signal (Fig. 11). Absence of a baseline shift in the reversing heat flow ruled out the presence of multiple amorphous states. A MTDSC scan of crystalline ATC showed melting endotherm in the non-reversing signal (data not shown). Therefore, the events in ATC-11 and 12 could be attributed to the melting of crystalline content.
Fig. 11.

MTDSC scans of amorphous ATV samples: a ATC-11 and b ATC-12; baseline shift in the reversing heat flow curve is due to glass transition; dashed boxes depict the enthalpy relaxation and arrows indicate post T g endotherm in the non-reversing heat flow signal
Wettability Analysis
Surface wetting precedes dissolution and can critically affect the in vitro and biological performance of the drug (24). Wettability is a surface property and is dependent on the molecular and particle level properties of the material. The sessile drop contact angle measurement is performed on compacted disc of pharmaceutical powders (25). This method has limitations, as compaction can influence the surface properties. However, the technique is widely used for qualitative ranking of materials (26).
The contact angle was found to be significantly different for crystalline and amorphous ATC samples (Table I). However, samples within a group showed similar values. Crystalline samples showed higher contact angle (90-94°) than the amorphous samples (72-80°), indicating their lower wetting tendency with water. The amorphous form, due to its lack of long range order, is expected to posses more random surface chemistry, which results in a net greater number of energetic sites, as compared to the crystalline counterpart. This high surface polarity of amorphous form improves its wettability with water and can facilitate dissolution (27,28).
Table I.
Advancing Contact Angle Measured with Saturated ATC Aqueous Solution and Intrinsic Dissolution Rate in 50 mM Sodium Dihydrogen Phosphate Buffer (pH 6.8) Containing 2% w/v Sodium Lauryl Sulfate for ATC samples (mean ± SD)
| Crystalline ATC | Amorphous ATC | ||||
|---|---|---|---|---|---|
| Sample | Advancing contact anglea (°) | Intrinsic dissolution ratea (mg/min/cm2) | Sample | Advancing contact angle (°) | Intrinsic dissolution rate (mg/min/cm2) |
| ATC-1 | 93.6 ± 2.7 | 0.112 ± 0.008 | ATC-7 | 79.1 ± 2.2 | 0.239 ± 0.010 |
| ATC-2 | 93.1 ± 1.9 | 0.119 ± 0.005 | ATC-8 | 72.8 ± 3.2 | 0.233 ± 0.008 |
| ATC-3 | 90.7 ± 2.3 | 0.112 ± 0.010 | ATC-9 | -b | -b |
| ATC-4 | -b | -b | ATC-10 | 78.8 ± 3.1 | 0.224 ± 0.011 |
| ATC-5 | 90.7 ± 2.4 | 0.124 ± 0.006 | ATC-11 | 73.7 ± 1.7 | 0.252 ± 0.015 |
| ATC-6 | 93.6 ± 3.1 | 0.114 ± 0.004 | ATC-12 | 73.6 ± 2.5 | 0.183 ± 0.012 |
aFor advancing contact angle n = 6 and for dissolution rate n = 3
bValue not determined due to limited sample amount
Intrinsic Dissolution Rate
Intrinsic dissolution profiles for crystalline and amorphous ATC are provided in Fig. 12a and b, respectively. IDR was calculated from slope of each curve for time period of 2-30 min. Crystalline ATC samples showed IDR values in the close range of 0.112-0.124 mg/min/cm2 (Table I). Amongst the crystalline samples, all samples had shown similar solid state behavior, except for ATC-1. Although, ATC-1 was a polymorphic mixture, presence of polymorphic impurity did not impact the IDR significantly. Amorphous samples showed IDR in the range of 0.183-0.252 mg/min/cm2, which was about twofold higher than the crystalline samples. Amorphous samples ATC-7, ATC-8, and ATC-10 had a similar solid state behavior and gave similar IDR. ATC-12 showed a reduced IDR value, which could be associated with the residual crystalline content as observed in the DSC analysis. Despite the presence of crystalline impurity, ATC-11 exhibited highest IDR. This sample had XRPD pattern similar to the amorphous form 23, which was different from all other amorphous forms. Hence, the cause for relatively high IDR obtained for ATC-11 sample could not be conclusively identified. Crystalline and amorphous samples, within each group, had shown similar wettability behavior with water and hence wettability was less expected be a contributor to the differences in IDR.
Fig. 12.

Intrinsic dissolution patterns for a crystalline ATC samples and b amorphous ATC samples (n = 3, mean ± SD)
CONCLUSION
The study emphasizes on importance of physical form characterization for bulk drug substances. Solid state characterization of six commercial samples each of crystalline and amorphous ATC, available in the Indian market, revealed variations in the solid form purity. The study also unveiled critical information on the thermal characterization of ATC samples. All crystalline samples were found to be stable form I. However, one of the sample possessed polymorphic impurity, which was evident in the XRPD (presence of additional peaks) and DSC (fall in heat of fusion) analysis. The DSC profile of ATC trihydrate crystalline form I varied with different heating rates, indicating the kinetic nature of dehydration. At lower heating rate, endothermic peak for loss of water of hydration and collapse of crystal lattice appeared as a doublet; whereas at higher heating rates the transitions merged to give a sharp endotherm.
ATC was found to be an excellent glass former with Tg/Tm ratio of 0.95. Amorphous ATC has a Tg onset at temperature of about 141°C, which is close to the melting onset of stable crystalline form I. This may necessitate the use of sound characterization tools to prevent erroneous conclusions. All amorphous drug samples were found to be X-ray amorphous. However, one of the samples showed a characteristically different halo pattern, which was indicative of a different amorphous phase. Thermal analysis of amorphous samples showed varying levels of enthalpy relaxation indicating different aging conditions. Post Tg endotherms found in two amorphous samples were successfully characterized to be due to residual crystalline content. Particle wetting was higher for the metastable amorphous form as compared to the crystalline counterpart, with no significant variations observed within the groups. The IDR of amorphous form was found to be about twofold higher than the crystalline form. Thus, the study provides insights into the solid state behavior of crystalline and amorphous ATC that can aid in setting polymorph specification and product development.
Acknowledgments
Vibha Puri acknowledges the financial assistance provided by Indian Council of Medical Research (ICMR, New Delhi, India) as senior research fellowship (45/12/2006/BMS/PHA). Lokesh Kumar would also like to acknowledge the financial support from the Department of Science and Technology, Government of India, as junior research fellowship (SR/SO/HS-23/2006).
References
- 1.Grant DJW. Theory and origin of polymorphism. In: Brittain HG, editor. Polymorphism in pharmaceutical solids. New York: Marcel Dekker; 1999. pp. 1–33. [Google Scholar]
- 2.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry ANDAs: Pharmaceutical Solid Polymorphism; 2007; Rockville, MD.
- 3.Aaltonen J, AllesØ M, Mirza S, Koradia V, Gordon KC, Rantanen J. Solid form screening—a review. Eur J Pharm Biopharm. 2009;71:23–37. doi: 10.1016/j.ejpb.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Brittain HG, Bogdanowich SJ, Bugay DE, DeVincentis J, Lewen G, Newman AW. Physical characterization of pharmaceutical solids. Pharm Res. 1991;8:963–73. doi: 10.1023/A:1015888520352. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal S, Ashokraj Y, Bharatam PV, Pillai O, Panchagnula R. Solid-state characterization of rifampicin samples and its biopharmaceutic relevance. Eur J Pharm Sci. 2004;22:127–44. doi: 10.1016/j.ejps.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Carini JP, Pavei C, Silva APC, Machado G, Mexias AS, Pereira VP, et al. Solid state evaluation of some thalidomide raw materials. Int J Pharm. 2009;372:17–23. doi: 10.1016/j.ijpharm.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 7.Byrn S, Pfeiffer R, Ganey M, Hoiberg C, Poochikian G. Pharmaceutical solids: a strategic approach to regulatory considerations. Pharm Res. 1995;12:945–54. doi: 10.1023/A:1016241927429. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein J. Polymorphism and patents from a chemist's point of view. In: Hilfiker R, editor. Polymorphism in the Pharmaceutical Industry. 1. Weinheim: Wiley-VCH; 2006. pp. 365–84. [Google Scholar]
- 9.Gardner CR, Walsh CT, Almarsson Ö. Drugs as materials: valuing physical form in drug discovery. Nat Rev. 2004;3:926–34. doi: 10.1038/nrd1550. [DOI] [PubMed] [Google Scholar]
- 10.Lipitor. Package Insert, Pfizer Ireland Pharmaceuticals, Dublin, Ireland. Parke Davis, Division of Pfizer Inc, New York; 2009.
- 11.Lau YY, Okochi H, Huang Y, Benet LZ. Pharmacokinetics of atorvastatin and its hydroxy metabolites in rats and the effects of concomitant rifampicin single doses: relevance of first-pass effect from hepatic uptake transporters, and intestinal and hepatic metabolism. Drug Metab Dispos. 2006;34:1175–81. doi: 10.1124/dmd.105.009076. [DOI] [PubMed] [Google Scholar]
- 12.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennerna H, Hussain AS, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharmaceutics. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 13.Yu LX. An integrated model for determining causes of poor oral drug absorption. Pharm Res. 1999;16:1883–7. doi: 10.1023/A:1018911728161. [DOI] [PubMed] [Google Scholar]
- 14.Sonje VM, Kumar L, Meena CL, Kohli G, Puri V, Jain R, et al. Atorvastatin calcium. Profiles of drug substances, excipients and related methodology: Elsevier Inc.; 2010. doi:10.1016/S1075-6280(10)35001-4. [DOI] [PubMed]
- 15.Pfizer Citizen Petition. Docket no. 2005P-0452. FDA's decision concerning approvals of generic versions of atorvastatin. 2005.
- 16.Kim JS, Kim MS, Park HJ, Jin SJ, Lee S, Hwang SJ. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spray-drying and SAS process. Int J Pharm. 2008;359:211–9. doi: 10.1016/j.ijpharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 17.Kim MS, Jin SJ, Kim JS, Park HJ, Song HS, Neubert RH, et al. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69:454–65. doi: 10.1016/j.ejpb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H, Wang J, Zhang Z, Le Y, Shen Z, Chen J. Micronization of atorvastatin calcium by antisolvent precipitation process. Int J Pharm. 2009;374:106–13. doi: 10.1016/j.ijpharm.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Thomas LC. TA Instruments application notes—use of multiple heating rate DSC and modulated temperature DSC to detect and analyze temperature-time-dependent transitions in materials. 2001.
- 20.Nogami H, Nagai T, Suzuki A. Studies on powdered preparations. XVII. Dissolution rate of sulfonamides by rotating disk method. Chem Pharm Bull. 1966;14:329–38. doi: 10.1248/cpb.14.329. [DOI] [PubMed] [Google Scholar]
- 21.Briggs CA, Wade JR, Harasawa K, Ichikawa S, Minohara K, Nakagawa S, Inventors; Crystalline [R- (R*, R*)]-2-(4-difluorophenyl)3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin) US Patent 5969156. 1999.
- 22.Schawe JEK, Strobl GR. Superheating effect during the melting of crystallites of syndiotactic polypropyleene analysed by temperature-modulated differential scanning calorimetry. Polymer. 1998;39:3745–51. doi: 10.1016/S0032-3861(97)10330-5. [DOI] [Google Scholar]
- 23.Zallen R. The formation of amorphous solids. In: The Physics of amorphous solids. New York: Wiley; 1983. p. 1–22.
- 24.Buckton G. Wetting, solubility, dissolution and absorption. In: Buckton G, editor. Interfacial phenomena in drug delivery and targeting. Switzerland: Harwood Academic Publishers; 1995. pp. 197–206. [Google Scholar]
- 25.Prestidge CA, George T. Wettability studies of morphine sulfate powders. Int J Pharm. 2000;198:201–12. doi: 10.1016/S0378-5173(00)00341-0. [DOI] [PubMed] [Google Scholar]
- 26.Buckton G. Surface characterization: understanding sources of variability in the production and use of pharmaceuticals. J Pharm Pharmacol. 1995;47:265–75. doi: 10.1111/j.2042-7158.1995.tb05794.x. [DOI] [PubMed] [Google Scholar]
- 27.Buckton G. Characterisation of small changes in the physical properties of powders of significance for dry powder inhaler formulations. Adv Drug Delivery Rev. 1997;26:17–27. doi: 10.1016/S0169-409X(97)00507-3. [DOI] [PubMed] [Google Scholar]
- 28.Newell HE, Buckton G, Butler DA, Thielmann F, Williams DR. The use of inverse gas chromatography to measure the surface energy of crystalline, amorphous, and recently milled lactose. Pharm Res. 2001;18:662–666. doi: 10.1023/A:1011089511959. [DOI] [PubMed] [Google Scholar]