

Abstract
Dissolution studies cannot distinguish phenomena occurring inside the dosage forms when studying formulation with similar dissolution profiles—such formulations can behave differently when considering their physical changes. The application of flow-through dissolution apparatus integrated with magnetic resonance imaging (MRI) system for discriminative evaluation of controlled release dosage forms with similar dissolution profiles was presented. Hydrodynamically balanced systems (HBS) containing l-dopa and various grades hydroxypropyl methylcelluloses were prepared. The dissolution studies of l-dopa were performed at high field (4.7 T) MR system with MR-compatible flow-through cell. MRI was done with 0.14 × 0.14 × 1-mm spatial resolution and temporal resolution of 10 min to record changes of HBS parameters during dissolution in 0.1 M HCl. Structural and geometrical changes were evaluated using the following parameters: total area of HBS cross-section, its Feret’s diameter, perimeter and circularity, area of hydrogel layer, and “dry core” area. While the dissolution profiles of l-dopa were similar, the image analysis revealed differences in the structural and geometrical changes of the HBS. The mechanism of drug release from polymeric matrices is a result of synergy of several different phenomena occurring during dissolution and may differ between formulations, yet giving similar dissolution profiles. A multivariate analysis was performed to create a model taking into account dissolution data, data from MRI, information about chemical structure, and polymer viscosity. It provided a single model for all the formulations which was confirmed to be competent. The presented method has merit as a potential Process Analytical Technology tool.
Key words: dissolution testing, flow-through cell, magnetic resonance imaging, MRI, PAT
INTRODUCTION
The drug dissolution test is often considered as an indicator of potential drug release characteristics of a product in vivo. In certain cases, the dissolution test is even used as a surrogate marker for bioavailability. Therefore, one of the important goals of dissolution testing is to provide some predictive estimates of the drug release in respect to the in vivo performance of a drug product. Although, in the literature data, studies concerning formulations with similar dissolution profiles and variable biological characteristics can be found (1,2), combining the dissolution studies with other analytical methods to improve the specificity and sensitivity of dosage form evaluation is still in its infancy. Imaging of the processes occurring during drug dissolution may bring valuable additional information about the nature of the dissolution phenomena (3,4). However, application of the integrated methods of imaging and drug dissolution is limited; thus, the need of such studies is pointed out in monograph 1092 of USP 30: “Visual observations and recordings of product dissolution and disintegration behavior are very useful, because dissolution and disintegration patterns can be indicative of variables in the formulation or manufacturing process.”
For the last decade, the application of imaging techniques in pharmaceutical technology has been of increasing interest to researchers (5,6). One of the most promising techniques is magnetic resonance imaging (MRI). MRI gives opportunity for non-destructive recording of spatial distribution of certain nuclei, e.g., protons, in the sample. Proton MRI (1H MRI) is most often applied in imaging of the pharmaceutical formulations (7); however, the examples of imaging of other nuclei, e.g., 19F are also presented in the literature (8). The 1H MRI allows for the measurement of water signal in aqueous solutions interacting with the dosage form. It allows visualizing the dosage form since MRI signal intensity is proportional to water content. MRI is noninvasive and does not influence or alter the processes of diffusion, swelling, and erosion of the dosage form. Because of the nature of MRI technique, it seems to be predestined for combination with the drug dissolution methods. The only limitations of such experiments are special requirements for the materials used in strong magnetic fields—paramagnetic or ferromagnetic elements of the apparatus must be substituted by non-magnetic materials. The literature data concerning simultaneous MR imaging and dissolution studies are still limited. Fyfe and Blazek-Welsh (9) described the system for performing MRI experiments within USP apparatus 4. The system was applied to record the physical changes of the small tablets simultaneously with the dissolution study. Recently, commercial, small benchtop MRI systems equipped with low field solid magnet dedicated for pharmaceutical studies were introduced into the market. The results of the application of such systems were already presented in few papers; however, it must be pointed out that the dissolution studies in these works were carried out using paddle or basket method and the samples were placed in the MRI unit for imaging purpose only (10–12).
In the previous works (13,14), we described the application of modified flow-through cell apparatus for simultaneous dissolution studies and MR imaging of the hydrodynamically balanced systems (HBS) under flow conditions. Since, in some cases, we did not observe any significant differences in the drug dissolution between various formulations, we decided to apply quantitative MR imaging to discriminate their physicochemical properties.
Hydroxypropyl methylcellulose (HPMC) derivatives were chosen as model excipients. HPMC is one of the most frequently used polymers in controlled drug delivery systems (15). The polymer is available in several grades with different viscosities and various degrees of substitution of the hydroxypropyl and methoxyl groups. Because all of these parameters influence the drug release rate, there is an opportunity to obtain a specific drug release profile by changing the type of the polymer (16). However, in many cases, despite differences in dosage form composition, the drug dissolution profiles may be similar or even overlapping. The question remains how to distinguish and parameterize the behavior of such formulations.
We chose HBS formulations as a model drug delivery system. It is an illustrative example of sustained release, floating dosage forms with drug release localized in stomach (17–19). The simplicity of the HBS formulations allows for observations of interactions between drug and polymer, not disturbed by the presence of additional excipients, e.g., lubricants, fillers, and binders. The capsular form of HBS ensures the avoidance of additional technological manipulations, e.g., granulation or tableting, which can alter properties of the formulations.
The aims of the study were:
▪to assess the usefulness of the system for simultaneous dissolution studies and MRI of sustained release, polymer-based dosage forms for differentiation of the formulations with similar dissolution profiles and
▪to obtain quantitative information on the macroscopic physicochemical and geometrical changes of the systems from MR images to differentiate HBS formulations with similar dissolution profiles.
MATERIALS AND METHODS
Materials
Four samples of HPMC (Shin-Etsu) with different viscosity and substitution types were used in the study (Table I). l-dopa (LD, Sigma) and hard gelatin capsules Coni-Snap® (Capsugel) were used for the preparation of HBS. All other materials were of analytical grade.
Table I.
Properties of Metoloses Used in the Experiment
| Metolose type | Viscosity (cP) | Substitution type according to USP classification | Methoxyl groups (%) | Hydroxypropyl groups (%) |
|---|---|---|---|---|
| 60SH | 10,000 | 2910 | 28–30 | 7–12 |
| 65SH | 4,000 | 2906 | 27–30 | 4–7.5 |
| 65SH | 400 | 2906 | 27–30 | 4–7.5 |
| 90SH | 400 | 2208 | 19–24 | 4–12 |
The mixtures of l-dopa and Metoloses in 1:1 ratio were prepared in the mortar. Capsules were filled manually with non-compressed powders with the amount corresponding to 250 mg of l-dopa.
Methods
MRI Studies
The system for simultaneous dissolution and MRI studies, described in details elsewhere (14), was used in the experiment. The system was based on 4.7 T (1H Larmor frequency of 200 MHz) superconducting magnet (Bruker, Germany) and digital MR console MARAN DRX (Resonance Instruments, Great Britain). A special non-magnetic flow-through cell designed specifically for use in high magnetic fields (i.e., inside the magnet) combined with MR resonator was used to perform MR imaging during the dissolution study. The flow of the solution was forced with USP compliant piston pump (HKP 60 Erweka GmbH, Germany).
The apparatus enables performing dissolution study and simultaneously tracking overall swelling of the system and evolution of the hydrogel layer, partially hydrated polymer layer, and “dry core.” It supports wide pH range of the dissolution media, is equipped with temperature control, and provides pulsatile flow according to USP apparatus 4. Flow-compensated, spin-echo MR sequence was used to obtain images of the dosage form inside the flow-through cell at continuous flow condition. Field of view 35 × 35 mm covers swelling “real-size” systems—for example capsule size 0 or tablets 12–15 mm in diameter.
Imaging sequence parameters were as follows: echo time of 19 ms, repetition time of 625 ms, and number of excitations of 4. Matrix size of 256 × 256 for the 3.5-cm field of view was used and the slice thickness was 1 mm, resulting in spatial resolution of 0.14 × 0.14 × 1 mm and temporal resolution of 15 minutes (total scan time of 10 min).
Dissolution Study and MR Imaging
The experiments were carried out using 0.1 M HCl solution. The capsule was inserted into the cell and the solution circulation was started. The total volume of the dissolution medium was 1,000 mL circulated in the closed loop, the circulation rate was 40 mL/min, and the temperature was maintained at 37°C using thermostatic water bath. The experiment was carried out for 5 h.
For dissolution study, the samples of solution were withdrawn every 15 min during the experiment. The volume of each sample was 5 mL. Equal amounts of dissolution media were replaced immediately after withdrawal of each sample. Released LD amounts were determined by measuring the UV absorption at 280 nm and calculated using calibration curve of the drug. Dissolution experiments were performed in triplicate.
Dissolution Data Treatment
The drug dissolution profiles were compared using similarity factor f2 introduced by Moore and Flanner (20) and defined as follows:
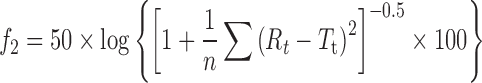 |
1 |
where Rt is the percentage of dissolved product for a reference batch at time point t, Tt is the percentage of dissolved product for the test batch, and n is the number of time points. The calculations were made on the mean values for three independent studies for each formulation.
To perform the modeling of dissolution data, “Kinet_DS” software was applied. It is the own-written software for integrated analysis of dissolution data. It enables simultaneous analysis of many dissolution profiles and application of several kinetic and empirical models (Korsmeyer–Peppas, Hixson–Crowell, Higuchi, zero, first-, second-, and third-order kinetic, etc.).
After initial suitability analysis of several equations used for the identification and description of mechanisms of drug release, Korsmeyer–Peppas model was chosen as the most suitable and universal approach. The model is described by Eq. 2.
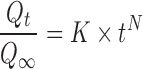 |
2 |
where 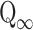 is the amount of drug released in the infinity (%), N is the release exponent, and K is the release constant Korsmeyer–Peppas model which is expected to be valid only up to approximately 60% cumulative drug released (21). Acquired dissolution data satisfied the condition. Coefficient of determination (R2 statistics) was used for the evaluation of model fit accuracy.
is the amount of drug released in the infinity (%), N is the release exponent, and K is the release constant Korsmeyer–Peppas model which is expected to be valid only up to approximately 60% cumulative drug released (21). Acquired dissolution data satisfied the condition. Coefficient of determination (R2 statistics) was used for the evaluation of model fit accuracy.
Flotation Force Measurements
For in vitro monitoring of the total vertical force acting on an immersed HBS, an experimental setup using an apparatus described by Timmermans and Moës (22) was applied. A detailed description of the apparatus for the flotation force measurements was presented earlier (23).
HBS was placed in the beaker filled with 1,000 mL of hydrochloric acid solution (0.1 M) maintained in thermostatic water bath (37°C). Floating HBS was positioned by cylindrical basket connected with lever. For calibration, 5-g weight was used. The results were shown on the display of the balance. The flotation force may be used to quantify and further to characterize the floating behavior of the HBS.
Image Analysis
MR images were reconstructed from multidimensional raw data (RImage files native MARAN DRX format) with two-dimensional digital Fourier transform. Pixel intensity in the images obtained with spin echo sequence was proportional to proton concentration weighted by relaxation times T1 and T2. Black and dark gray colors (values near zero level) characterized “dry” regions of the HBS. Higher values corresponded to increasing water concentration in partially and fully hydrated parts of the dosage forms.
Image analysis was performed using ImageJ 1.39u software for Mac Os X (NIH, Bethesda, MD, USA). The first images with a cross-section of the central region of the HBS were chosen. Then, the regions of interest (ROI) containing HBS were selected. In the next step, the histograms of signal intensities in ROIs were made. Based on the information from histograms, the thresholds were determined (14). The thresholds were used to separate the areas in the image with specific intensity characteristics. After thresholding-based segmentation, the areas of segmented regions were measured, i.e., total area of HBS cross-section, dry core of HBS, and hydrogel layer. Boundaries of the regions were detected using Sobel edge detector filter.
The geometry of the systems was complicated and time-dependent; therefore, these changes cannot be neglected. Hence, formulation size and shape were measured and quantified using Feret’s diameter, perimeter, and the circularity. Feret’s diameter was evaluated as the longest distance between any two points along the selection boundary. Perimeter was the length of the outside boundary of the selection. The circularity was calculated using the following equation:
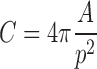 |
3 |
where C is the circularity, A is the total HBS area, and p is the perimeter.
A C value of 1.0 indicates a perfect circle. As it approaches 0, it indicates an increasingly elongated polygon. Additionally, the mean thickness of the hydrogel layer was calculated. The measurements of hydrogel thickness were carried out in ten points distributed regularly around the core of HBS. Image analysis was performed three times independently.
Modeling and Statistical Analysis
Multiple linear regression was used for modeling the drug release relationships from the formulation parameters. Data linearization was achieved by logarithmic transformation and arithmetic product of available parameters. The final model was the result of empirical experiments with inclusion/exclusion of the parameters to the equation. Coefficient of determination (R2), root mean squared error (RMSE), and relative root mean squared error (REL RMSE) were the model fit criteria. REL RMSE was computed as a percentage value of RMSE over the output variable minimum and maximum values range. OpenCalc 3.2 was used for computations.
Non-parametric Kruskal–Wallis test was performed in order to estimate the statistical significance of differences between time profiles measured for different formulations. It is regarded as an alternative for one-way ANOVA for the comparison of more than two groups where it could not be assumed that population distributions are normal (24). For statistical analysis, SPSS 16.0 (SPSS Inc., USA) was applied.
RESULTS
Dissolution Studies
Dissolution profiles for different formulations are presented in Fig. 1. Despite the fact that various types of HPMC were used, the dissolution profiles of l-dopa were similar. After 5 h, 41% to 46% of the drug was dissolved. The value of f2 was 69.5 for utmost curves, i.e., formulation with Metolose 65SH400 was compared with the formulation containing Metolose 90SH400.
Fig. 1.

Release of l-dopa from HBS formulations. Bars represent the standard deviation
The application of Korsmeyer–Peppas model (Eq. 2) in the analysis of dissolution data showed that the values of exponent “N” were in the range 0.8799 for the formulation containing Metolose 60SH10000 to 0.8984 for HBS with Metolose 90SH400. The magnitude of the release exponent “N” indicates the release mechanism (i.e., Fickian diffusion, case II transport, or anomalous transport). According to the cylindrical geometry of the HBS formulations, in the present study, the limits considered were N = 0.45 (indicates a classical Fickian diffusion-controlled drug release) and N = 0.89 (indicates a case II relaxation release transport; non-Fickian, zero-order release) (25). The values of N between 0.45 and 0.89 can be regarded as an indicator of both phenomena (drug diffusion in the hydrated matrix and the polymer relaxation) commonly called anomalous transport. The values of release exponents in the Korsmeyer–Peppas model indicated that l-dopa was released according to the anomalous transport mechanism (i.e., non-Fickian). In the case of HBS containing Metolose 90SH400, case II transport was also involved in the mechanism of drug release (Table II).
Table II.
Parameters of Korsmeyer–Peppas Equation
| Formulation | Equation parameters | ||||
|---|---|---|---|---|---|
| K | N | SE (N) | REL SE (N, %) | R 2 | |
| 60SH10000 | 0.2944 | 0.8799 | 0.0133 | 1.50 | 0.9957 |
| 65SH400 | 0.3235 | 0.8842 | 0.0099 | 1.12 | 0.9976 |
| 65SH4000 | 0.2688 | 0.8817 | 0.0205 | 2.33 | 0.9898 |
| 90SH400 | 0.2438 | 0.8984 | 0.0466 | 5.19 | 0.9513 |
SE (N) standard error of parameter N estimation, REL SE relative standard error of N estimation in percentages of actual N value, R 2 coefficient of determination
Imaging of the HBS and Image Analysis
Mean Hydrogel Thickness and Areas of Cross-sections of HBS
Under experimental conditions (0.1 M HCl at 37°C), the capsule shell dissolved completely and gelatin was conveyed away from the flow-through cell by the flowing solution. The hydrogel layer was formed simultaneously with the capsule dissolution. The structure of the surface layer was not uniform and the concentrations of water and polymer were changing toward the core, which affected its physical properties. Some authors distinguish particular sub-layers in the surface layer, e.g., the infiltration layer, the swollen solid layer, the gel layer, and the liquid layer (26). In pharmaceutical publications, the term “moving front” was introduced (27–29). It denotes the part of the matrix where the physical conditions sharply change and may be addressed to boundaries of identified layers. Usually, the swelling and erosion fronts were identified. Swelling front denotes a position which separates dry polymer in glassy state and moistened polymer in rubbery state. Erosion front separates hydrogel layer from the solvent. The hydrogel located between swelling and erosion fronts is called the diffusion layer.
In Fig. 2, examples of MR images recorded after 30 and 300 min are presented. Edge detection was carried out for better visualization of particular areas in the image. The outer white line encircles the hydrogel and indicates the position of the erosion front, while the inner line encircles the dry core and indicates the position of the swelling front.
Fig. 2.

Sagittal MR images of HBS formulations containing various types of HPMC at 30 and 300 min; white lines show positions of swelling front and erosion front: a 65SH400, 30 min, b 65SH400, 300 min, c 90SH400, 30 min, d 90SH400, 300 min, e 65SH4000, 30 min, f 65SH4000, 300 min, g 60SH10000, 30 min, h 60SH10000, 300 min
The images recorded at 30 min show the formulations after dissolution of gelatin capsule where the dry core was covered by a thin layer of hydrogel (Fig. 3a, c, e, g). The hydration of the polymers was most effective for formulations containing Metoloses with low viscosities (90SH 400 cP and 65SH 400 cP). At 30 min, the mean thickness of the surface layers was 1.05 and 0.84 mm, respectively. In other cases, the thicknesses of hydrogel layers were 0.67 mm (65SH 4,000 cP) and 0.5 mm (60SH 10,000 cP).
Fig. 3.

Time evolution of total area of a HBS, b hydrogel area, and c dry core area. Bars represent the standard deviation
At 5 h, the dry core was maintained in all investigated formulations (Fig. 3b, d, f, h). However, the differences between HBS containing various types of HPMC were visible. The thickest hydrogel layer (2.75 mm) was observed for the HBS with Metolose 60SH10000. In this case, the hydrogel was regularly distributed around the core of the system (Fig. 3h). In other cases, the mean thickness of hydrogel was in the range 2.29–2.46 mm.
The total areas of the HBS, hydrogel, and dry core regions, as previously defined, were measured with temporal resolution of 15 min. The results of these measurements are presented in Fig. 3. The overview of changes in particular regions of HBS is presented in Table III. Initially, total areas of HBS cross-sections were in the range 184–206 mm2. For the HBS containing Metolose 60SH10000, the total area of the system increased linearly from 184 mm2 at 30 min to 251 mm2 at 165 min (R2 = 0.987), with a rate of 0.50 mm2/min. The maximum, 262 mm2, was reached at 210 min. Subsequently, the total area of the system slowly decreased down to 257 mm2. Simultaneously, the linear increase of hydrogel area with a rate of 0.33 mm2/min (R2 = 0.984) was observed. The area of the dry core of the system increased initially from 137 mm2 at 15 min to 145 mm2 at 105 min. This phenomenon was caused by initial stretching of the system induced by swelling polymeric particles. When the hydrogel reached appropriate thickness and became resistant enough to balance the stretching of the dosage form, the increase of dry core area was stopped. From 105 min up to the end of the experiment, the decrease of “dry core” was observed. Finally, it reached 108 mm2. Linear growth of hydrogel was accompanied with the linear decrease of dry core (R2 = 0.981). Decrease rate of the dry core was 0.19 mm2/min. For formulations with Metolose 65SH4000 and Metolose 65SH400, the total area of the systems increased slightly throughout the experiment. Initial area of the system with Metolose 65SH400 was 185 mm2 and reached 211 mm2 at 5 h. In the case of formulation containing Metolose 65SH4000, initial area was 206 mm2 and reached 227 mm2 at 5 h.
Table III.
Changes of Particular Areas of HBS Formulations
| Formulation l-dopa + Metolose | Measured area | Time (min) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 | 30 | 45 | 60 | 75 | 90 | 105 | 120 | 135 | 150 | 165 | 180 | 195 | 210 | 225 | 240 | 255 | 270 | 285 | 300 | ||
| Metolose 65SH 400 | Total area | ➚Increase (nonlinear) | |||||||||||||||||||
| Hydrogel area | ➚Increase (linear), rate 0.28 mm2/min | ||||||||||||||||||||
| Dry core area | ➘ Decrease (linear), rate 0.23 mm2/min | ||||||||||||||||||||
| Metolose 90SH400 | Total area | ➚Increase (nonlinear) | ➘Decrease (nonlinear) | ||||||||||||||||||
| Hydrogel area | ➚Increase (linear), rate 0.29 mm2/min | ||||||||||||||||||||
| Dry core area | ➘Decrease (nonlinear) | ||||||||||||||||||||
| Metolose 65SH4000 | Total area | ➚Increase (nonlinear) | |||||||||||||||||||
| Hydrogel area | ➚Increase (linear), rate 0.24 mm2/min | ||||||||||||||||||||
| Dry core area | ➘Decrease (linear), rate 0.15 mm2/min | ||||||||||||||||||||
| Metolose 60SH10000 | Total area | ➚Increase (linear), rate 0.50 mm2/min | ➘Decrease (nonlinear) | ||||||||||||||||||
| Hydrogel area | ➚Increase (linear), rate 0.33 mm2/min | ||||||||||||||||||||
| Dry core area | ➚Increase (nonlinear) | ➘Decrease (linear), rate 0.19 mm2/min | |||||||||||||||||||
For HBS with Metolose 90SH 400, the time evolution of total area was complicated. For the first 75 min of experiment, the total area of HBS was almost constant. It ranged from 188 to 206 mm2. Afterwards, the increase of the total area to 233 mm2 at 165 min was observed. Subsequently, it decreased to 216 mm2 at 5 h.
The time evolution of hydrogel areas had nearly linear nature unlike the time evolution of total areas. For the systems with Metoloses 65SH4000, 65SH400, and 90SH400, the rates of the hydrogel areas formation were 0.24 mm2/min (R2 = 0.955), 0.29 mm2/min (R2 = 0.968), and 0.28 mm2/min (R2 = 0.958), respectively.
In the case of HBS containing Metolose 65SH4000 and 65SH400, the dry core areas decreased linearly with rates 0.15 mm2/min (R2 = 0.974) and 0.23 mm2/min (R2 = 0.961), respectively. Such a decrease was not observed for the formulation with Metolose 90SH400.
During the experiment, the geometrical changes of the HBS were observed. They were assessed using perimeter, Feret’s diameter, and circularity. The temporal changes (profiles) of the parameters were obtained. In Table IV, initial and final values are presented. In the beginning, the perimeters of HBS ranged between 54.1 and 57.5 mm. The swelling of the systems effected the increase of their parameters. At 5 h, they were in the range from 59.8 to 65.0 mm.
Table IV.
Summary of the Results of Image Analysis
| Formulation | Metolose 90SH400 | Metolose 65SH400 | Metolose 65SH4000 | Metolose 60SH10000 | ||||
|---|---|---|---|---|---|---|---|---|
| Time (min) | 30 | 300 | 30 | 300 | 30 | 300 | 30 | 300 |
| Total area (mm2) | 147.4 | 184.1 | 143.7 | 165.4 | 156.8 | 170.8 | 159.9 | 200.8 |
| Perimeter (mm) | 54.8 | 65.0 | 54.1 | 59.8 | 57.5 | 63.1 | 55.4 | 62.2 |
| Feret’s diameter (mm) | 22.7 | 28.1 | 22.6 | 25.6 | 24.5 | 28.1 | 23.5 | 26.1 |
| Circularity | 0.616 | 0.547 | 0.617 | 0.582 | 0.595 | 0.539 | 0.653 | 0.651 |
Perimeter—the length of the outside boundary of the selection
Feret’s diameter—the longest distance between any two points along the selection boundary
Circularity—a value of 1.0 indicates a perfect circle; as the value approaches 0.0, it indicates an increasingly elongated polygon
The increase of Feret’s diameters and decrease of circularity of the systems during the experiment indicate that the swelling of the HBS were not uniform in all directions. The increase of Feret’s diameters suggests that the most preferred swelling direction was parallel to the horizontal axis of the systems.
Floating Properties
All developed formulations floated for 5 h (Fig. 4). According to identical proportions of HPMC and LD and size of investigated formulations, the initial floating force of HBS was in the range 4.40–4.57 mN. In most cases, floating force profiles could be divided into two parts: initial expansion of the systems and final erosion of the swollen polymer. For formulations with Metoloses 60SH 10,000 and 90SH 400, the maxima of floating force (5.48 and 5.39 mN, respectively) were observed at 2 h. The maximum floating force, 5.40 mN, of the formulation with Metolose 65SH4000 was achieved after 3 h. The floating capacity of HBS with Metolose 65SH400 decreased gradually throughout the experiment. Finally, the floating force of this system was 2.83 mN, while for the other formulations, the final values of floating force were in the range 3.66–4.45 mN.
Fig. 4.

Floating properties of HBS formulations containing various types of HPMC. Bars represent the standard deviation
Preliminary Data Modeling
In the case of hydrogel-based formulations like HBS, a relationship between drug dissolution rate and viscosity could be expected. However, in this study, it was impossible to correlate drug dissolution pattern with the standard 2% polymer solution viscosity. In order to provide a relationship between drug dissolution and formulation parameters, an empirical formula was created (Eq. 4)
 |
4 |
where CORE is the dry core area, HDG is the hydrogel area, HYP is the amount of hydroxypropyl groups (%), METOX is the amount of methoxyl groups (%), VISC is the viscosity (cP), and a,b,c are parameters of the equation.
Parameters of the empirical equation were summarized in Table V. Based on the R2 and RMSE values, it could be concluded that the model has predictive performance suitable for further analysis and reasoning (Fig. 5).
Table V.
Parameters of Eq. 4
| Equations parameters | Goodness of fit | ||||
|---|---|---|---|---|---|
| a | b | c | R 2 | RMSE | REL RMSE (%) |
| −23.44 | 23.35 | −45.18 | 0.92371 | 1.77 | 3.88 |
Fig. 5.

Predicted vs. observed values for the model described by Eq. 4
DISCUSSION
Formulations with similar dissolution profiles and different bioavailability were previously described. In the study presented by Sawada et al. (1), three compression-coated tablet formulations with similar in vitro drug release profiles but different core erosion characteristics were administered to the fasted dogs. The pharmacokinetic parameters obtained after administration of the tablets were different: AUC (µg h/mL) ranged between 0.4 and 1.4, and Cmax (µg/mL) ranged between 0.07 and 0.4. The correlation between a greater core erosion ratio and larger AUC and Cmax values after oral administration was observed. Sako et al. (2) showed that changes in dissolution conditions are not sufficient for discriminatory evaluation of tablets containing HPMC, while the bioavailability studies have demonstrated significant differences between the formulations.
The presented MRI study showed the differences in the physical properties and behavior of the HBS formulations with similar dissolution characteristics. For areas of dry core, hydrogel, and total area, statistically significant differences between time profiles were demonstrated (p value between 10−5 and 0.02), whereas dissolution profiles were not significantly different between each other (p = 0.76). These results supported the claim that dissolution test does not reveal all the differences between formulations. It means also that the mechanism of drug release from polymeric matrices is a result of synergy of several different phenomena occurring during dissolution and may differ between formulations. It may be expected that differences in physical properties of the systems during dissolution influence the biological performance of particular formulations. An attempt to conclude mechanism of drug dissolution from the dissolution profile parameterization only leads to oversimplification of the problem—the choice between Fickian and “anomalous” diffusion. The term “anomalous diffusion” denotes several interacting dissolution mechanisms, including Fickian diffusion, swelling, and erosion of the system—there is no possibility to obtain quantitative information concerning contribution of particular mechanism. That is the reason for the development of methods appropriate for parameterization of physicochemical properties behavior of the dosage forms during dissolution studies, particularly in the context of works by Sako et al. and Sawada et al. (1,2). These properties may influence the behavior of formulation in vivo. This is important because the results of dissolution studies are commonly used as prerequisite in bioavailability and bioequivalence studies (30,31).
Analysis of correlation between dissolution parameters and data from image analysis showed the linear relationships between some parameters. For example, the linear correlation was observed between the released amounts of l-dopa and mean hydrogel layer thickness. The correlation coefficients (R2) were in the range 0.92–0.98. These results indicate that the dissolution of l-dopa was connected with the formation of hydrogel and its thickness. It shows indirectly that in the case of l-dopa, which is freely soluble in water, the diffusional phenomena play an important role in dissolution mechanism. Interestingly, the results of total area measurements were strongly correlated linearly with dissolution results only in the case of HBS containing Metolose 60SH10000 (R2 = 0.94). In the remaining cases, the correlation coefficients were much lower (0.53–0.82). It suggests that in the case of formulations containing Metoloses with lower viscosity, the mechanisms of drug dissolution from polymeric matrices are complex and based on various phenomena occurring in the dosage form, which finally may result with similar dissolution profiles. For Metoloses 400 cP-based formulations, overall swelling of the system (total area of the system) is less efficient; hydrogel layer is thinner than in the case of the formulation with Metolose 10,000, but erosion of the system is more effective. These formulations are more “erosion-driven” than formulations containing Metoloses with higher viscosity grade. For all formulations, at a specific point of time, total area of the system reaches steady state. From that moment, hydrogel layer increases at “dry core” area expense only. Generally, evolutions of hydrogel layer and “dry core” are in opposite phase, i.e., “dry core” area is decreasing while hydrogel layer area is increasing.
When comparing “dry core” area profiles, it can be seen that they are more sensitive to functional group substitution than to viscosity. It was shown by Nokhodchi (32) that functional group substitution is of great significance at the initial phase of polymer hydration (i.e., have an impact on glassy to elastic state transition).
Simple relationships between drug release pattern and viscosity of the polymer solution were impossible to find. The universal model (Eq. 4) integrating all the formulations required inclusion of viscosity and substitution type of the polymers, together with dry core and hydrogel area evolution. Without any of these parameters, the model had no such predictive performance like that demonstrated in Table V. Therefore, multivariate approach was confirmed as necessary for appropriate quantification of analyzed relationships. It is the indirect proof that multiple processes are occurring simultaneously and compensate themselves, giving similar dissolution profiles. This also suggests the suitability of MRI technique to provide appropriate information about formulation behavior related to the drug dissolution pattern.
The literature data indicate that during drug dissolution from polymeric matrices, several physicochemical phenomena occur simultaneously (33). In contact of the polymer with water, the interface is formed and swelling of the polymeric particles occurs. Water acts as a plasticizer, and its presence causes a decrease of the polymer glass transition temperature (Tg) (34,35). When the concentration of water in swelling area is sufficient to lower Tg to the temperature of dissolution medium, phase transition from glassy to rubbery state occurs and hydrogel is formed (34–36). In the swelling boundary, the particles of the drug are initially moistened and subsequently dissolved. Hydrogel near the swelling boundary contains dissolved drug which diffuses toward the dissolution medium. The process of water penetration into the matrix is faster than the hydrogel formation. It was investigated by FTIR imaging applied during the drug dissolution from tablets (37). In the contact with small amounts of water, the grains of the polymer change their dimensions according to relaxation of polymer chains. It induces the swelling force, causing stretching of the system.
The stretching of the hydrogel layer induces the mechanical translocation of the dry particles in the core of the system toward the swelling boundary. The result is exposition of new particles of drug and polymer to the solvent.
CONCLUSIONS
Original findings from the study are detailed shortly below:
An integrated dissolution and MRI study performed in the flow-through cell on the specially selected samples demonstrated that the dosage forms with similar dissolution profiles may have different temporal changes of physical and geometrical properties during drug release studies in vitro.
Viscosity of 2% polymer solutions was not confirmed to be the release controlling factor as a single variable.
The mechanism of drug release from polymeric matrices is a result of synergy of several different phenomena occurring during dissolution and may differ between formulations—in the presented study, the factors influencing l-dopa dissolution (e.g., hydrogel layer volume, overall swelling, erosion of the system, and others) compensate themselves to give similar dissolution profiles.
Consequently, it is necessary to perform quantification of the physical changes of the dosage form because dissolution study analysis cannot supply complete information for analysis and understanding drug dissolution mechanisms.
Following complementary data sets, temporal changes of the following parameters were obtained: total area of HBS cross-section, its Feret’s diameter, perimeter and circularity, area of hydrogel layer, “dry core” area, and floating force changes—they reflect the physical changes of the systems.
A multivariate analysis was performed to create a model taking into account dissolution profiles, data from MRI, information about chemical structure, and viscosity of the polymer—it provided a single model for all the formulations, which was confirmed to be competent.
The method has merit as a potential Process Analytical Technology tool.
The presented study constitutes a good starting point for discussion about more precise modeling and parameterization of dissolution mechanisms in polymer-based controlled release dosage forms.
Acknowledgments
We are thankful to Prof. G. Stanisz from Sunnybrook Health Sciences Centre, Toronto Canada for critical reading of the manuscript. The work was supported by The Polish Ministry of Science and Higher Education grant NN518 40 74 38.
References
- 1.Sawada T, Sako K, Fukuji M, Yokohama S, Hayashi M. A new index, the core erosion ratio, of compression-coated timed-release tablets predicts the bioavailability of acetaminophen. Int J Pharm. 2003;265:55–63. doi: 10.1016/S0378-5173(03)00405-8. [DOI] [PubMed] [Google Scholar]
- 2.Sako K, Sawada T, Nakashima H, Shigeharu Y, Takashi S. Influence of water soluble fillers in hydroxypropylmethylcellulose matrices on in vitro and in vivo drug release. J Control Release. 2002;81:165–72. doi: 10.1016/S0168-3659(02)00067-6. [DOI] [PubMed] [Google Scholar]
- 3.Cao Q-R, Cho H-G, Kim D-C, Lee B-J. Release behavior and photo-image of nifedipine tablet coated with high viscosity grade hydroxypropylmethylcellulose: effect of coating conditions. Int J Pharm. 2004;274:107–17. doi: 10.1016/j.ijpharm.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Kazarian SG, van der Weerd J. Simultaneous FTIR spectroscopic imaging and visible photography to monitor tablet dissolution and drug release. Pharm Res. 2008;25:853–60. doi: 10.1007/s11095-007-9375-4. [DOI] [PubMed] [Google Scholar]
- 5.Zeitler JA, Gladden LF. In vitro tomography and non-destructive imaging at depth of pharmaceutical solid dosage forms. Eur J Pharm Biopharm. 2008;71:2–22. doi: 10.1016/j.ejpb.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Richardson JC, Bowtell RW, Mäder K, Melia CD. Pharmaceutical applications of magnetic resonance imaging (MRI) Adv Drug Del Rev. 2005;57:1191–209. doi: 10.1016/j.addr.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 7.Baumgartner S, Lahajnar G, Sepe A, Kristl J. Quantitative evaluation of polymer concentration profile during swelling of hydrophilic matrix tablets using 1H NMR and MRI methods. Eur J Pharm Biopharm. 2004;59:299–306. doi: 10.1016/j.ejpb.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Fyfe CA, Grondey H, Blazek-Welsh AI, Chopra SK, Fahie BJ. NMR imaging investigations of drug delivery devices using a flow-through USP dissolution apparatus. J Control Release. 2000;68:73–83. doi: 10.1016/S0168-3659(00)00237-6. [DOI] [PubMed] [Google Scholar]
- 9.Fyfe CA, Blazek-Welsh AI. Quantitative NMR imaging study of the mechanism of drug release from swelling hydroxypropylmethylcellulose tablets. J Control Release. 2000;68:313–33. doi: 10.1016/S0168-3659(00)00245-5. [DOI] [PubMed] [Google Scholar]
- 10.Strübing S, Abboud T, Vidor Contri R, Metz H, Mäder K. New insights on poly(vinyl acetate)-based coated floating tablets: characterisation of hydration and CO2 generation by benchtop MRI and its relation to drug release and floating strength. Eur J Pharm Biopharm. 2008;69:708–17. doi: 10.1016/j.ejpb.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Strübing S, Metz H, Mäder K. Characterization of poly(vinyl acetate) based floating matrix tablets. J Control Release. 2008;126:149–55. doi: 10.1016/j.jconrel.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Malaterre V, Metz H, Ogorka J, Gurny R, Loggia N, Mäder K. Benchtop-magnetic resonance imaging (BT-MRI) characterization of push–pull osmotic controlled release systems. J Control Release. 2009;133:31–6. doi: 10.1016/j.jconrel.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Dorożyński P, Kulinowski P, Jachowicz R, Jasiński A. Development of the system for simultaneous dissolution studies and magnetic resonance imaging of water transport in hydrodynamically balanced systems—a technical note. AAPS PharmSciTech. 2007;8:Article 15. [DOI] [PubMed]
- 14.Kulinowski P, Dorożyński P, Jachowicz R, Węglarz W. Integrated system for dissolution studies and magnetic resonance imaging of the hydrodynamically balanced systems—a tool for quantitative assessment of hydrogel formation process. J Pharm Biomed Anal. 2008;48:685–93. doi: 10.1016/j.jpba.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 15.Shah NH, Railkar AS, Phauapradit W, Zeng F, Chen A, Infeld MH, et al. Effect of processing techniques in controlling the release rate and mechanical strength of hydroxypropylmethylcellulose based hydrogel matrices. Eur J Pharm Biopharm. 1996;42:183–7. [Google Scholar]
- 16.Nellore RV, Rekhi GS, Hussain AS, Tillman LG, Augsburger LL. Developement of metoprolol tartrate extended-release matrix tablet formulations for regulatory policy consideration. J Control Release. 1998;50:247–56. doi: 10.1016/S0168-3659(97)00141-7. [DOI] [PubMed] [Google Scholar]
- 17.Ali J, Arora S, Ahuja A, Babbar AK, Sharma RK, Khar RK, et al. Formulation and development of hydrodynamically balanced system for metformin: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2007;67:196–201. doi: 10.1016/j.ejpb.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 18.Bardonnet PL, Faivre V, Pugh WJ, Piffaretti JC, Falson F. Gastroretentive dosage forms: overview and special case of Helicobacter pylori. J Control Release. 2006;111:1–18. doi: 10.1016/j.jconrel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 19.Singh BN, Kim KH. Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release. 2000;63:235–59. doi: 10.1016/S0168-3659(99)00204-7. [DOI] [PubMed] [Google Scholar]
- 20.Moore JW, Flanner HH. Mathematical comparison of curves with an emphasis on in vitro dissolution profiles. Pharm Technol. 1996;6:64–74. [Google Scholar]
- 21.Ritger PL, Peppas NA. A simple equation for description of solute release. II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5:37–42. doi: 10.1016/0168-3659(87)90035-6. [DOI] [PubMed] [Google Scholar]
- 22.Timmermans J, Moës AJ. How well do floating dosage forms float? Int J Pharm. 1990;62:207–16. doi: 10.1016/0378-5173(90)90234-U. [DOI] [Google Scholar]
- 23.Dorożyński P, Jachowicz R, Kulinowski P, Kwieciński S, Szybiński K, Skórka T, et al. The macromolecular polymers for the preparation of hydrodynamically balanced systems—methods of evaluation. Drug Dev Ind Pharm. 2004;30:947–57. doi: 10.1081/DDC-200037179. [DOI] [PubMed] [Google Scholar]
- 24.Negro S, Herrero-Vanrell R, Barcia E, Villegas S. Comparative study of the dissolution profiles of a commercial theophylline product after storage. Arch Pharm Res. 2001;24:568–71. doi: 10.1007/BF02975167. [DOI] [PubMed] [Google Scholar]
- 25.Costa P, Sousa Lobo JM. Modelling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–33. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 26.Miller-Chou BA, Koenig JL. A review of polymer dissolution. Prog Polym Sci. 2003;28:1223–70. doi: 10.1016/S0079-6700(03)00045-5. [DOI] [Google Scholar]
- 27.Colombo P, Bettini R, Massimo G, Catellani PL, Santi P, Peppas NA. Drug diffusion front movement is important in drug release control from swellable matrix tablets. J Pharm Sci. 1995;84:991–7. doi: 10.1002/jps.2600840816. [DOI] [PubMed] [Google Scholar]
- 28.Colombo P, Bettini R, Catellani PL, Santi P, Peppas NA. Drug volume fraction profile in the gel phase and drug release kinetics in hydroxypropylmethyl cellulose matrices containing a soluble drug. Eur J Pharm Sci. 1999;9:33–40. doi: 10.1016/S0928-0987(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 29.Bettini R, Catellani PL, Santi P, Massimo G, Peppas NA, Colombo P. Translocation of drug particles in HPMC matrix gel layer: effect of drug solubility and influence on release rate. J Control Release. 2001;70:383–91. doi: 10.1016/S0168-3659(00)00366-7. [DOI] [PubMed] [Google Scholar]
- 30.CPMP/EWP/QWP/1401/98 Rev 1: Guideline on Investigation of Bioequivalence (Draft), London, 24 July 2008.
- 31.US Department of Health and Human Services FDA CDER: Guidance for industry—dissolution testing of immediate release solid oral dosage forms. August 1997.
- 32.Nokhodchi A, Ford JL, Rubinstein MH. Studies on the interaction between water and (hydroxypropyl)methylcellulose. J Pharm Sci. 1997;86:608–15. doi: 10.1021/js960279a. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Hardy RJ, Gu X. Effect of drug solubility on polymer hydration and drug dissolution from polyethylene oxide (PEO) matrix tablets. AAPS PharmSciTech. 2008;9:437–43. doi: 10.1208/s12249-008-9060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouwstra JA, Salomons-de Vries MA, van Miltenburg JC. The thermal behavior of water in hydrogels. Thermochim Acta. 1995;248:319–27. doi: 10.1016/0040-6031(94)01948-G. [DOI] [Google Scholar]
- 35.Ford JL. Thermal analysis of hydroxypropylmethylcellulose and methylcellulose: powders, gels and matrix tablets. Int J Pharm. 1999;179:209–28. doi: 10.1016/S0378-5173(98)00339-1. [DOI] [PubMed] [Google Scholar]
- 36.Gómez-Carracedo A, Alvarez-Lorenzo C, Gómez-Amoza JL, Concheiro A. Glass transitions and viscoelastic properties of Carbopol® and Noveon® compacts. Int J Pharm. 2004;274:233–43. doi: 10.1016/j.ijpharm.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 37.van der Weerd J, Kazarian SG. Combined approach of FTIR imaging and conventional dissolution tests applied to drug release. J Control Release. 2004;98:295–305. doi: 10.1016/j.jconrel.2004.05.007. [DOI] [PubMed] [Google Scholar]