

Abstract
In this study, hot melt extrusion (HME) and KinetiSol® Dispersing (KSD) were utilized to prepare dissolution-enhanced solid dispersions of Roche Research Compound A (ROA), a BCS class II drug. Preformulation characterization studies showed that ROA was chemically unstable at elevated temperatures and acidic pH values. Eudragit® L100-55 and AQOAT® LF (HPMCAS) were evaluated as carrier polymers. Dispersions were characterized for ROA recovery, crystallinity, homogeneity, and non-sink dissolution. Eudragit® L100-55 dispersions prepared by HME required the use of micronized ROA and reduced residence times in order to become substantially amorphous. Compositions containing HPMCAS were also prepared by HME, but an amorphous dispersion could not be obtained. All HME compositions contained ROA-related impurities. KSD was investigated as a method to reduce the decomposition of ROA while rendering compositions amorphous. Substantially amorphous, plasticizer free compositions were processed successfully by KSD with significantly higher ROA recovery values and amorphous character than those achieved by HME. A near-infrared chemical imaging analysis was conducted on the solid dispersions as a measure of homogeneity. A statistical analysis showed similar levels of homogeneity in compositions containing Eudragit® L100-55, while differences were observed in those containing HMPCAS. Non-sink dissolution analysis of all compositions showed rapid supersaturation after pH adjustment to approximately two to three times the equilibrium solubility of ROA, which was maintained for at least 24 h. The results of the study demonstrated that KSD is an effective method of forming dissolution-enhanced amorphous solid solutions in cases where HME is not a feasible technique.
Key words: acid labile, hot melt extrusion, kinetisol dispersing, solid dispersion, thermally labile
INTRODUCTION
Roche Research Compound A (ROA) is a developmental stage compound manufactured by Hoffman-La Roche. Preformulation studies determined that ROA is a BCS class II drug, with reported aqueous solubilities in acidic and neutral media of approximately 0.002 and 0.39 mg mL−1, respectively. Being classified as a class II drug, ROA exhibits high permeability in the gastrointestinal lumen (1). The need to formulate a compound such as ROA into a viable dissolution-enhanced oral dosage form is becoming increasingly common. It is estimated that 25% to 30% of newly developed drug substances exhibit poor solubility characteristics, resulting in poor dissolution performance and low bioavailability (2). The development of processing methods to enhance the dissolution rate and degree of supersaturation achieved with these compounds has become a major focus in the pharmaceutical industry. The formation of solid dispersions has emerged as a relatively inexpensive and effective method of improving these characteristics (3).
Solid dispersions are defined as being a dispersion of one or more active ingredients in an inert solid matrix and can be broadly classified as those containing a drug substance in the crystalline state or in the amorphous state (3). Solid dispersions containing drug substance in the crystalline state provide dissolution enhancement by simply decreasing surface tension, reducing agglomeration, and improving the wettability of the drug substance (4). While crystalline systems are more thermodynamically stable than their amorphous counterparts, the crystalline structure must be interrupted during the dissolution process, requiring energy. Solid dispersions containing a drug dissolved at the molecular level, known as amorphous solid solutions, can result in a significant increase in dissolution rate and extent of supersaturation (5–7). While these systems have several advantages, physical instability can be problematic due to molecular mobility and the tendency of the drug to recrystallize. Polymeric carriers with high glass transition temperatures such as Eudragit® L100-55 and HPMCAS are well suited to stabilize these systems by limiting molecular mobility.
The commercial preparation of these systems can be broadly classified as those utilizing solvents and those that require the melting of one or more substances. Techniques that utilize solvents to form amorphous solid solutions include solvent evaporation (8,9), co-precipitation (10,11), freeze drying (9), supercritical fluid technologies (12,13), and spray drying (2,7,14). These methods require the use of a solvent system, often organic in nature, to dissolve an inert carrier and active drug substance (15). Once a solution is formed, the solvent is subsequently removed by a mass transfer mechanism dependent on the manufacturing technique chosen. Although solvent-based techniques such as spray drying are relatively common, they suffer from several disadvantages. Selection of a solvent system that is compatible with the active substance and carrier polymer may prove to be difficult or require very large amounts of organic solvent. This presents a safety hazard at the manufacturing facility as organic solvents must be collected and disposed of properly to limit the environmental impact (16). Furthermore, organic solvents may be difficult to fully remove from processed materials. This solvent removal step may require prolonged times at elevated temperatures, presenting an additional cost to the manufacturer. For these reasons, fusion processing has gained increased acceptance over solvent-based techniques and has become the method of choice for the large-scale manufacture of amorphous solid solutions (17).
Although hot melt extrusion, a fusion processing technique, has been used in the food and plastics industry for more than a century, it has only recently gained acceptance in the pharmaceutical industry for the preparation of these systems. In this method, a thermoplastic carrier is combined with a drug substance and optional inert excipients. The mixture is introduced into rotating screws that convey the powder into a heated zone where shear forces are imparted into the mixture, compounding the materials until a molten mass is achieved. While this manufacturing method has many advantages over solvent-based methods, it does have significant limitations. During processing, drug substances are exposed to elevated temperatures for prolonged periods of time. Although a variety of factors can affect the residence time distribution of an extruded substance, these times typically fall within the 1- to 2-min range but have been reported to be as long as 10 min (18–20). This prolonged exposure to elevated temperatures can induce decomposition of thermally labile compounds or accelerate decomposition of chemically unstable compounds. When these processing issues are encountered, the addition of processing aids such as plasticizers may allow processing to be carried out at a lower temperature (21–23). However, the addition of a plasticizer can affect the solid-state physical stability of the solid dispersion once formed. That is, the increased molecular mobility may allow the drug substance to transition to the more thermodynamically stable state when the glass transition temperature of the resulting amorphous solid solution is within 50°C of the storage temperature (24).
KinetiSol® Dispersing (KSD) is a new fusion-based process that was recently developed to rapidly form solid dispersions by imparting high shear and frictional forces without external heat input. Previous studies conducted by DiNunzio et al. have illustrated clear advantages of the technology over HME, including the preparation of plasticizer free Eudragit® L100-55 compositions leading to enhanced physical stability and the successful processing of hydrocortisone, a thermally labile drug substance (25).
The present study focuses on improving the dissolution characteristics of ROA, a compound exhibiting poor aqueous solubility and instability at elevated temperatures and acidic pH environments. More specifically, it was desirable to form amorphous solid solutions of ROA in Eudragit® L100-55 and HPMCAS. These polymers have been shown to facilitate the supersaturation of other drug substances during in vitro dissolution testing (6,7). In this study, solid dispersions of ROA were prepared by HME and KSD processing techniques. It was hypothesized that the KSD process would allow for improved ROA recovery values and increased amorphous character due to the characteristics of each processing method. Preformulation studies were conducted using thermogravimetric and pH stability analyses at elevated temperatures in order to identify the route of decomposition. Once prepared, solid dispersions were evaluated for crystalline drug content by X-ray powder diffraction (XRPD) and characterized by near-infrared imaging in order to evaluate homogeneity. Finally, compositions prepared by both techniques were evaluated for ROA recovery, impurities, and non-sink dissolution performance.
MATERIALS AND METHODS
ROA was manufactured and kindly donated by Hoffman-La Roche, Inc. (Nutley, NJ, USA). Eudragit® L100-55 was donated by Evonik (Piscataway, NJ, USA), AQOAT® LF (hypromellose acetate succinate, HPMCAS) was donated by Shin-Etsu Chemical Company (Tokyo, Japan), and triethyl citrate was donated by Vertellus (Indianapolis, IN, USA). High-performance liquid chromatography grade acetonitrile was purchased from EMD Chemicals (Darmstadt, Germany). All other chemicals were of ACS grade.
Hot Melt Extrusion
HME studies were conducted on a HAAKE Minilab II Microcompounder (Thermo Electron Corporation, Newington, NH, USA) equipped with 5/14-mm conical screws. Compositions were manually fed into the feed hopper and forced into the processing area. The recirculation valve was placed in the flush position unless recirculation was utilized. Actual processing conditions are detailed in following sections. All extrudates were allowed to exit directly from the extruder without a die. A Laboratory L1A Fitzmill (Fitzpatrick Inc., Elmhurst, IN, USA), equipped with a 0.020-in. screen in a knives forward configuration, operating at 9,000 rpm, was used to mill compositions.
Kinetisol® Dispersing
A pharmaceutical grade machine designed by DisperSol Technologies, LLC (Austin, TX, USA) was utilized to compound pre-mixed compositions by KSD. This compounding unit is comprised of a circular processing chamber containing a rotating shaft with blades that protrude toward the chamber wall. The composition was loaded into the processing chamber at room temperature where a computer control module was utilized to set the desired rotational processing speed and ejection set point. As the blades rotated at high speeds, heat was generated through shear and friction within the chamber. This method of heat generation is different from that of other fusion processing methods in that no external heat was applied. Actual rotational speeds and temperatures inside the processing chamber were monitored and recorded in real time by the computer control module and are detailed in subsequent sections. After reaching the predetermined processing temperature, the compounder ejected molten material directly into liquid nitrogen to rapidly quench the material. Compounded material was placed under vacuum for 30 min to prevent moisture absorption. A Laboratory L1A Fitzmill (Fitzpatrick Inc., Elmhurst), equipped with a 0.020-in. screen in a knives forward configuration, operating at 9,000 rpm, was used to mill compositions.
Micronization
To obtain micronized drug substance, a ball mill (U.S. Stoneware, East Palestine, OH, USA) was utilized. Approximately 70 g of raw drug substance was placed in a sealed high-density polyethylene container with 0.5-in. diameter ceramic beads. The container was placed on the rotating mill, operating at 75% capacity, for 72 h.
Particle Size Analysis
To evaluate the particle size of the drug substance, ROA was dispersed in deionized water by sonication. The aqueous drug dispersions were placed in a Mastersizer X (Malvern Instruments, Malver, UK) and analyzed by laser diffraction with lenses of 300- and 1,000-mm focal length. Mastersizer Version 2.18 software was utilized to analyze data and determine particle size. It was found that 90% of the total fraction of micronized ROA, D90, was below 11.1 μm.
Thermogravimetric Analysis
To evaluate the behavior of ROA and polymeric matrices at elevated temperatures, a Perkin-Elmer Thermogravimetrical Analyzer (Norwalk, CT, USA) was utilized. Prior to analysis, bulk materials were placed under a vacuum for 48 h at room temperature. Samples were weighed to approximately 10 mg and placed in 100 µL aluminum crucibles. Under a constant flow of nitrogen at 65 mL min−1, samples were heated from 50°C to 350°C at a rate of 10°C min−1 and monitored for weight loss.
pH Stability Analysis
To evaluate the decomposition of ROA as a function of pH, a stability analysis was conducted at 60°C. Solutions of ROA with pH values of 1.0, 2.0, 3.0, 4.0, 5.0, 7.0, 9.0, and 11.0 were prepared by dissolving ROA in a 1:1 mixture of acetonitrile/pH buffer until a concentration of 400 µg mL−1 was obtained. Solutions were placed in sealed borosilicate glass containers and stored in a Blue M 0V-718 Electric Oven (Blue Island, IL, USA) at 60.0 ± 1.0°C. At selected time points, the sealed containers were removed and allowed to cool to room temperature. From each container, 4.0 mL was removed, placed in a culture tube, and diluted at a 1:1 (v/v) ratio with pH 11.0 buffer to prevent further reaction. All samples were filtered through 13 mm 0.2-µm PTFE filters (Whatman, Piscataway, NJ, USA) and transferred to 1-mL vials (VWR International, West Chester, PA, USA) for analysis. A Waters (Waters Corporation, Milford, MA, USA) high-performance liquid chromatography (HPLC) system was utilized for analysis. A Waters 717 Autosampler was used to accurately inject 10-µL samples. The system was operated under gradient flow with an aqueous mobile phase consisting of deionized water and 0.05% trifluoroacetic acid. The organic phase consisted of acetonitrile and 0.05% trifluoroacetic acid. A Phenomenex Luna 5 µm C18(2) 100 Å, 150 mm × 4.6 mm (Phenomenex, Torrance, CA, USA) HPLC column, equipped with guard column efficiently separated chemical entities. A Waters 996 photodiode array detector, extracting at 275 nm, was utilized to detect the presence of ROA and impurities. Empower Version 5.0 software processed all chromatography data.
Aqueous Polymer Suspension pH Determination
To determine the approximate micro-environment pH of Eudragit® L100-55 and HPMCAS, a pH analysis was conducted, as described by Riedel et al. (26). A 6% (w/v) suspension of each polymer was prepared in deionized water and allowed to stir for 1 h. The pH value was measured (n = 3) with a calibrated pHTestr 3+ electrode (Oakton Instruments, Vernon Hills, IL, USA).
ROA Recovery and Impurity Testing
Processed compositions were weighed such that a theoretical 20.0 mg of ROA was present. A 65:35 (v/v) mixture of acetonitrile/water was used to dissolve samples in 100-mL volumetric flasks. Samples were then filtered through 13-mm, 0.2-µm PTFE filters (Whatman) and transferred to 1-mL vials (VWR International) for analysis. High-performance liquid chromatography was utilized to measure ROA recovery and related impurities, as described in previous sections. ROA recovery values were adjusted for the recorded sample weight and compared to a known standard containing 20.0 ± 0.5 mg in 100 mL of 65:35 (v/v) acetonitrile/water. ANOVA calculations were conducted in MiniTab Release 14 (Minitab, Inc., State College, PA, USA) to determine if results were statistically significant (p < 0.05).
X-ray Powder Diffraction
ROA exhibits several strong characteristic peaks at 2θ values of 18.5, 20.5, 22.2, and 24.8, which are apparent in physical mixtures of ROA and polymeric excipients (Eudragit® L100-55 and HPMCAS). A Philips Model 1710 X-ray diffractometer (Philips Electronic Instruments Inc., Mahwah, NJ, USA) measured the degree of crystallinity in compositions by XRPD analysis. Operating voltage and amperage were set to 40 kV and 30 mA, respectively. Aluminum sample holders held samples in place while the diffractometer scanned over a 2θ range of 5° to 30° with a step size of 0.05° and a dwell time of 3 s. Microsoft Excel 2007 software processed all XRPD data
Near-Infrared Chemical Imaging
A SapphireTM Near Infrared Chemical Imaging (NIR CI) system (Malvern, Columbia, MD, USA) was utilized to determine the regions of amorphous ROA dispersed throughout compositions. Images were collected at wavelengths between 1,200 and 2,400 nm at 10-nm intervals. The objective was set to 35 µm pixel−1 with a field of view of 8.96 × 11.2 mm. Spectral units were converted from reflectance to absorbance, and Savitzky-Golay smoothing was utilized (nine-point window, second-order polynomial, zero derivative). A discriminant partial least square regression was used to differentiate ROA from polymer in each sample matrix (27).
Non-sink Dissolution Analysis
Non-sink dissolution analysis of processed compositions was performed according to the USP 29 apparatus 2 paddle method at 50 rpm with a VK 7000 dissolution apparatus (Vankel Technology Group, Cary, NC, USA) equipped with a VK 8000 autosampler. The dissolution medium (750 mL of 0.1 N HCl) was allowed to equilibrate at 37 ± 0.5°C prior to the analysis. Processed compositions were corrected for ROA recovery values and weighed such that each vessel contained 20 mg of ROA (∼10× equilibrium solubility). After 2 h has elapsed, 250 mL of 0.2 M Na3HPO4 equilibrated at 37 ± 0.5°C was added to each vessel to adjust the pH to 6.8. During analysis, 5-mL samples were removed, without medium replacement at 60, 120, 125, 130, 135, 150, 180, 240, 300, 360, and 1,440 min. After sampling, solutions were immediately filtered with 13-mm, 0.2 µm PVDF membrane filters to remove any particulate material. An aliquot of each sample was subsequently diluted at a 1:1 ratio (v/v) with HPLC mobile phase (65:35 ACN/H2O) and transferred into 1-mL HPLC vials for analysis.
RESULTS AND DISCUSSION
Preformulation Characterization
The chemical decomposition of an active drug substance during fusion processing can be due to a number of factors. The heat that is inherent to these processes can accelerate chemical decomposition. Thermogravimetric analysis (TGA) was utilized in order to better understand the chemical decomposition of ROA alone and in the presence of Eudragit® L100-55 and HPMCAS, as illustrated in Fig. 1a.
Fig. 1.

Thermogravimetric analysis of solid dispersions and physical mixtures containing a Eudragit® L100-55 and b HPMCAS
The drug substance was found to decompose rapidly after reaching its melting point at 230°C as evidenced by significant weight loss. Inspection of Fig. 1a shows that Eudragit® L100-55 began to decompose at approximately 170°C to 180°C, due to a combination of side and main polymer chain decomposition (28). Based on individual weight loss measurements, the predicted weight loss (Wp) of a 1:2 ROA/polymer physical mixture was estimated with the following equation:
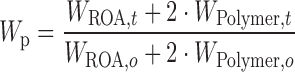 |
Where WROA,t is the weight of ROA at time t, WPolymer,t is the weight of the polymer at time t, WROA,o is the initial weight of ROA, and WPolymer,o is the initial weight of polymer. The observed weight loss of the 1:2 ROA/Eudragit® L100-55 physical mixture was significantly higher than the predicted weight loss based on the individual components, indicating an incompatibility between the drug substance and Eudragit® L100-55 at elevated temperature. The observed weight loss due to the interaction may be decomposition of the polymer, drug substance, or combination thereof. As shown in Fig. 1b, decomposition of HPMCAS began at approximately 200°C, well before the onset of ROA decomposition. It was apparent that the 1:2 ROA/HPMCAS physical mixture also began to decompose at 200°C, but at a higher rate than the predicted weight loss based on individual components, indicating a chemical interaction between ROA and HPMCAS.
In order to better understand the mechanism by which ROA decomposed, a study was conducted to evaluate the decomposition over the pH range from 1.0 to 11.0 at 60°C. The decomposition of ROA in the presence of aqueous buffers was described by pseudo-first-order kinetics. Pseudo-first-order rate constants (k′) were determined from linear first order plots at each pH based on the following equation:
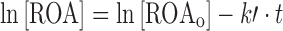 |
where the time-dependent concentration of ROA is [ROA], the initial concentration of ROA is [ROAo], the pseudo-first-order rate constant is k′, and the time is t. The resulting pH rate profile is shown in Fig. 2. Corresponding half-lives (t1/2) are outlined in Table I.
Fig. 2.

Pseudo-first-order rate profile of ROA decomposition as a function of pH at 60°C
Table I.
Half Lives of Roche Research Compound A at 60°C
| pH | t 1/2 (days) |
|---|---|
| 1.0 | 1.1 |
| 2.0 | 14.5 |
| 3.0 | 23.7 |
| 4.0 | 23.4 |
| 5.0 | 20.0 |
| 7.0 | 26.2 |
| 9.0 | 73.2 |
| 11.0 | 143.5 |
t 1/2 half life of ROA
The chemical stability of ROA was found to be strongly dependent on pH with significant decomposition occurring more readily at acidic pH values. This finding further demonstrated that ROA may be incompatible with enteric polymers, which inherently have acidic pH values due to the presence of free carboxyl groups. Eudragit® L100-55 and HPMCAS specifications and measured aqueous dispersion pH values are detailed in Table II. The high percentage of free carboxyl groups present in Eudragit® L100-55 are responsible for the low acidic pH measured. Similarly, HPMCAS has fewer free carboxyl groups and a pH value higher than that of Eudragit® L100-55. However, both substances provide an acidic pH micro-environment that may facilitate decomposition of ROA.
Table II.
Characteristics of Enteric Polymers According to Specifications and Measured Values
| Polymer | Free carboxyl groups (%) | Measured aqueous dispersion pH |
|---|---|---|
| Eudragit® L100-55 | 46–50 | 2.90 |
| HPMCAS | 14–18 | 3.23 |
Based on preformulation characterization results, it can be concluded that the decomposition of ROA is a function of temperature and pH. In order to successfully prepare compositions of ROA in Eudragit® L100-55 and HPMCAS, it is apparent that exposure to elevated temperatures during processing must be limited in order to minimize decomposition. That is, increased processing times and temperatures may increase the rate of decomposition. It was hypothesized that the KSD process would provide higher ROA recovery values due to rapid processing times and reduced processing temperatures.
Solid Dispersions Prepared by Hot Melt Extrusion
In the case of HME, it is sometimes necessary to select a processing temperature that is above the melting point of the drug substance in order to render compositions amorphous. In doing so, the drug substance readily converts to its amorphous state and thus does not rely solely on mixing effects or shear rates. However, processing a thermally labile drug substance such as ROA is problematic in that rapid decomposition is observed upon melting. In a case such as this, extending residence times at minimized extrusion temperatures may provide the time necessary for dissolution of the drug substance into the viscous polymer, rendering it amorphous. However, it is possible for decomposition to occur in this scenario as well. Furthermore, the selected processing temperature must be above the glass transition temperature of the polymeric system being extruded in order to achieve melt flow.
Eudragit® L100-55 Compositions
Eudragit® L100-55 has a glass transition temperature of approximately 120°C and is a thermally labile carrier (22). Based on the collected TGA data for Eudragit® L100-55, the onset of decomposition is between 150°C and 170°C, allowing for a small processing window below the melting point of ROA and above the glass transition temperature of the polymer. Initial attempts to extrude 1:2 ROA/Eudragit® L100-55 (w/w) compositions were unsuccessful, both with and without recirculation within the extruder. At temperatures between 150°C and 170°C, compositions exhibited a high viscosity that prevented continuous processing. When processing at temperatures above 170°C, the extrudate was visually decomposed, as evidenced by a foam-like structure indicative of mass loss due to off-gassing. This result was not predicted by the TGA data, which indicated stability below 150°C. This deviation was likely due to intimate mixing and shearing effects within the extruder barrel, resulting in increased contact between ROA and the polymer. Furthermore, micro-environmental temperatures within the extruder can exceed the temperature set point due to localized shear forces.
In order to lower melt viscosity sufficiently to allow for processing, a plasticizer was incorporated. Triethyl citrate (TEC) was chosen because of its known compatibility with acrylic polymers during extrusion processing (29,30). Blends of 1:1.6:0.4 ROA/Eudragit® L100-55/TEC (w/w/w) were processed by HME at the lowest temperature at which melt flow could be achieved. The processing conditions and resulting ROA recovery values and impurity levels are outlined in Table III.
Table III.
Hot Melt Extrusion Manufacturing Conditions and Resulting Potency Values
| Polymer | Particle size | Temperature (°C) | Screw speed (rpm) | Recirculation time (min) | Recovery (%) | Impurities (%) |
|---|---|---|---|---|---|---|
| Eudragit® L100-55 | Unmicronized | 140 | 300 | 2 | 22.7 ± 0.5 | 55.9 |
| Eudragit® L100-55 | Micronized | 140 | 300 | 0 | 69.1 ± 0.5 | 17.3 |
| HPMCAS | Unmicronized | 170 | 300 | 2 | 70.9 ± 0.3 | 10.2 |
| HPMCAS | Micronized | 170 | 300 | 0 | 78.4 ± 0.1 | 8.9 |
Recirculation was utilized in order to prolong residence time at elevated temperatures in order to render ROA amorphous. As the extrudate exited the die, a foam-like structure was observed visually. Upon analysis, it was determined that decomposition occurred as evidenced by a low ROA recovery value and high impurity values. A mass balance between measured ROA recovery and impurity values was not obtained. This was likely due to the nature of the impurities. Insoluble complexes were formed during decomposition. Furthermore, the foam-like structure of compositions indicates that there is a mass loss due to off-gassing, which may also account for the discrepancy.
When processing below the melting point of a drug substance, a solid solution is formed by dissolution of the drug substance into the polymer. It was hypothesized that increasing the surface area of ROA by micronization prior to the extrusion process would facilitate the formation of amorphous compositions by reducing the time required for solvation. However, it is important to note that while the drug substance may convert from the crystalline to amorphous state more readily, residence times typical of the hot melt extrusion process may accelerate decomposition.
Blends of 1:1.6:0.4 micronized ROA/Eudragit® L100-55/TEC (w/w/w) were prepared and processed at the conditions outlined in Table III. The composition containing micronized ROA was not subjected to recirculation in the hot melt extruder in order to minimize decomposition. The extrudate appeared to have a foam-like structure, again indicating decomposition of ROA. This was confirmed by a ROA recovery analysis, which showed high levels of impurities. However, a significant reduction in impurity level was seen in the composition containing micronized ROA, indicating that decomposition was proportional to residence time at elevated temperatures. Additionally, the level of crystallinity was reduced with the incorporation of micronized ROA, as described in subsequent sections.
HPMCAS Compositions
HPMCAS has a glass transition temperature of approximately 120°C under dry conditions (2). The TGA data collected indicated that the physical mixture of ROA/HPMCAS (w/w) began to decompose at a high rate once reaching 200°C. A processing temperature of 160°C was found to be the lowest temperature melt flow could be achieved and was utilized for extrusion studies without plasticizer. Compositions containing 1:2 ROA/HPMCAS were processed as outlined in Table III. Significant decomposition occurred during processing of compositions containing unmicronized ROA as evidenced by impurity levels, although foaming of the extrudate was not observed. The resulting recovery was improved by reducing the extruder residence time and utilizing micronized ROA, again demonstrating that decomposition of ROA was dependent on the time exposed to elevated temperature. The level of crystallinity was not reduced with the incorporation of micronized ROA, as described in the solid-state characterization section.
Solid Dispersions Prepared by KinetiSol® Dispersing
KinetiSol® Dispersing is a fusion processing technology that has recently been reported for the production of solid dispersions (25,31). Furthermore, this technology has shown to be effective in processing thermally labile compounds and plasticizer free compositions (30,32). During KSD processing, a blend is subjected to shear forces much greater than those achieved in HME processes. Temperatures of processed compositions are rapidly increased through frictional forces, without external heat input, until the desired temperature is reached. It is through both rapid heat generation and efficient particle size reduction that solid solutions are formed. This process allows not only for very short residence times when compared to HME but also very efficient mixing. Compositions containing 1:2 ROA/polymer (w/w) were processed by KSD without utilizing a plasticizer or micronized drug substance for both Eudragit® L100-55 and HPMCAS compositions. Processing conditions, associated ROA recovery values, and impurity levels are outlined in Table IV.
Table IV.
KinetiSol® Dispersing Manufacturing Conditions and Resulting Potency Values
| Polymer | Particle size | Speed (rpm) | Temp. (°C) | Recovery (%) | Impurities (%) |
|---|---|---|---|---|---|
| Eudragit® L100-55 | Unmicronized | 1,450 | 100 | 70.9 ± 0.8 | 12.9 |
| HPMCAS | Unmicronized | 2,400 | 112 | 99.4 ± 1.2 | 1.6 |
Temp. temperature
A significant improvement in ROA recovery was observed in the KSD processed Eudragit® L100-55 composition containing unmicronized ROA when compared to the HME processed composition containing unmicronized ROA. This improvement in recovery is due to the short processing times inherent to the KSD process. Furthermore, the use of micronized ROA in the KSD process was not required to obtain significantly higher recovery values, presenting a reduction in processing costs. When compared to the HME processed composition utilizing micronized ROA, the KSD processed material provided only a slight improvement in ROA recovery. This result was expected as the HME residence time was reduced during processing, effectively reducing decomposition. However, the use of micronized ROA was required to obtain similar recovery when processed by HME.
When comparing the HME and KSD processes for HPMCAS-based compositions, it was apparent that ROA recovery was significantly higher in the KSD processed composition, and very little decomposition was observed. This result confirmed the hypothesis that reduced processing time significantly improves ROA recovery. It is also important to note that KSD processing was carried out without the addition of a plasticizer in each case, which can lead to improved physical stability.
Chromatograms obtained by HPLC analysis of all compositions containing Eudragit® L100-55 and HPMCAS are illustrated in Fig. 3. It can clearly be seen that the impurity eluting at approximately 4 min is reduced significantly in KSD processed compositions. This measured impurity is related to the thermal decomposition of ROA, as determined in preformulation studies. Impurities related to the acidic decomposition of ROA were not measured. However, insoluble material was observed during recovery and impurity analysis, indicating that insoluble complexes were formed during processing. The amount of insoluble complex was proportional to the level of decomposition measured during ROA recovery testing.
Fig. 3.

HPLC chromatograms of processed compositions containing a Eudragit® L100-55 and b HPMCAS
Examination of processing profiles in Fig. 4 revealed that the composition containing Eudragit® L100-55 was processed in under 17 s and was exposed to a temperature greater than 100°C for less than 1 s. Similarly, HPMCAS compositions were processed in under 14 s and were exposed to temperatures above 100°C for less than 4 s. This relatively short processing time at elevated temperatures is directly responsible for the reduced thermal decomposition observed in Fig. 3.
Fig. 4.

KSD processing profiles demonstrating the rapid increase in temperature during the manufacture of ROA solid dispersions
KSD compositions were ejected at temperatures well below temperatures utilized for HME, indicating that the high frictional forces inherent to the KSD process are effective in allowing processing at reduced temperatures while rendering materials substantially amorphous. This capability is due to the high torque output of the KSD manufacturing equipment.
Solid-State Characterization of Solid Dispersions Prepared by HME and KSD
HME and KSD compositions were characterized by XRPD, and NIR chemical imaging to determine the amorphous character as differential scanning calorimetry was not a feasible analytical technique due to decomposition at elevated temperatures. The XRPD profiles of the compositions containing Eudragit® L100-55 are illustrated in Fig. 5.
Fig. 5.

XRPD profiles of Eudragit® L100-55 physical mixture, HME processed compositions, and KSD processed composition
The results indicated that when unmicronized ROA was incorporated into the HME composition with recirculation, a significant amount of crystallinity was detected when compared to the physical mixture. This result is particularly interesting due to the extended residence time and resulting ROA recovery value (22.7 ± 0.5%). An improvement in amorphous content was observed in the KSD processed Eudragit® L100-55 composition containing unmicronized ROA. The KSD processed composition containing unmicronized ROA resulted in a substantially amorphous solid solution, with only minor crystallinity detected. This is an important finding due to the differences in processing time and temperature between the two manufacturing methods. This is due to the ability of the KSD process to rapidly reduce the particle size of ROA in situ, rendering the material amorphous through high shear forces and efficient mixing. In order to duplicate the results by HME, a screw design utilizing a high degree of dispersive mixing would be required. However, it should be noted that increasing the dispersive mixing component in a HME process generally leads to a higher residence time distribution, which can negatively impact ROA recovery.
Similarly, the composition containing Eudragit® L100-55 prepared by HME with micronized ROA was found to be substantially amorphous, indicating that a reduction in particle size facilitated the formation of amorphous compositions. This critical finding has not been previously reported in the literature and may have applications for high melting point compounds that are difficult to render amorphous. Based on these results, it is apparent that the high shear rates inherent to the KSD process are effective in reducing the particle size of processed materials and provides an obvious advantage over HME.
The XRPD profiles of the compositions containing HPMCAS are illustrated in Fig. 6. When comparing the HME and KSD processes for HPMCAS-based compositions containing unmicronized ROA, it was apparent that the amorphous character was significantly improved in the KSD processed composition. The addition of micronized ROA to HME compositions did not reduce the crystalline content. The KSD process effectively rendered compositions amorphous while maintaining ROA recovery values. Again, this finding indicated that high shear rates inherent to the KSD process are instrumental in rendering material amorphous while maintaining high ROA recovery values.
Fig. 6.

XRPD profiles of ROA/HPMCAS physical mixture, HME processed compositions, and KSD processed composition
A partial least square discriminant analysis (PLS DA) model was developed using amorphous ROA and polymer to further characterize the solid dispersions. This model allowed homogeneity of compositions to be determined by differentiating amorphous ROA and polymer by color and intensity. The resultant images of compositions containing Eudragit® L100-55 after PLS DA analysis are demonstrated in Fig. 7. The high-intensity pixels represent localized amorphous ROA, while the low-intensity pixels represent localized Eudragit® L100-55.
Fig. 7.

PLS DA images illustrating regions of localized amorphous ROA (red) and Eudragit® L100-55 (blue) in solid dispersions prepared by a HME and b KSD
It appeared that ROA was more uniformly distributed within the solid dispersion prepared by HME than the dispersion prepared by KSD. However, a statistical analysis of the data indicated that each processing method provided similar levels of ROA homogeneity within the two dispersions, as demonstrated in Fig. 8.
Fig. 8.

Normalized standard deviation of amorphous ROA pixels in Eudragit® L100-55 solid dispersions prepared by a HME and b KSD. Size distribution of amorphous ROA domain sites in solid dispersions prepared by c HME and d KSD
The standard deviation of the normalized pixel value is a direct indication of amorphous ROA homogeneity. The standard deviation from the images of HME and KSD processed solid dispersions are comparable, as shown in Fig. 8. The slightly higher standard deviation value in the KSD processed material is most likely due to the usage of unmicronized ROA during processing which may inhibit the distribution of ROA within the polymer during processing to the same extent that would be achieved with micronized ROA. The sizes of the amorphous ROA domains were also similar between processing methods, as shown in Fig. 8. This demonstrated that the KSD process was capable of providing a similar end result without the need for micronized ROA.
Compositions containing HPMCAS were also analyzed by PLS DA, and the resultant images are shown in Fig. 9. ROA was distributed slightly better in samples prepared by HME processing than those prepared by KSD as evidenced by the localized areas of high-intensity pixels present in the KSD composition. As shown in Fig. 10, a statistical analysis confirmed that samples prepared by HME with micronized ROA exhibited a slightly more uniform distribution of amorphous ROA than KSD processed compositions with unmicronized ROA. This is likely due to the fact that unmicronized ROA was utilized by the KSD process. However, the composition processed by KSD contained a significantly higher amount of amorphous ROA, confirming the XRPD results.
Fig. 9.

PLS DA images illustrating regions of localized amorphous ROA (red) and HPMCAS (blue) in solid dispersions prepared by a HME and b KSD
Fig. 10.

Normalized standard deviation of amorphous ROA pixels in HPMCAS solid dispersions prepared by a HME and b KSD. Size distribution of amorphous ROA domain sites in solid dispersions prepared by c HME and d KSD
Further analysis of Fig. 10 revealed that the amorphous ROA domain size distribution was significantly different between the two processing methods. The HME processed material contained domain sizes much smaller than those found in the KSD processed material. This result may be due to the starting particle size utilized or the fact that there is more amorphous ROA present in the KSD processed composition.
These XRPD and PLS DA results indicated that the KSD process was capable of producing similar results as the HME process without the need for micronized drug substance. As previously described, the mechanism by which solid solutions are formed during HME and KSD processing below the melting point of a drug substance is dissolution of the drug into the polymer. While the dissolution of ROA into a polymer is accelerated at elevated temperatures during HME processing, decomposition is a major concern. Conversely, processing at reduced temperatures may not provide sufficiently amorphous compositions due to a reduced convective component. In this case, the formation of a solid solution would be reliant on dispersive mixing. The KSD process provides high rates of shear through dispersive mixing that effectively render compositions amorphous, relying less on convection to achieve a solid solution. High levels of shear in the KSD process effectively rendered ROA amorphous and provided acceptable levels of homogeneity.
Non-sink Dissolution Analysis of Solid Dispersions
High-energy amorphous drug compositions have the ability to exhibit dissolution rates significantly higher than their crystalline counterparts. Furthermore, a drug substance may stay in a supersaturated state for prolonged periods of time due to the concentration enhancing properties of some polymers. Eudragit® L100-55 and HPMCAS have both been characterized as polymers facilitating supersaturation of drug substances (2,6,7). Non-sink dissolution analysis was conducted on HME compositions utilizing micronized ROA and KSD compositions utilizing unmicronized ROA. The dissolution analysis of HME and KSD compositions containing Eudragit® L100-55 is presented in Fig. 11a. Critical dissolution metrics are presented in Table V.
Fig. 11.

Dissolution profiles of ROA solid dispersions prepared by HME and KSD containing a Eudragit® L100-55 b HPMCAS. The amount of dispersion placed into each vessel (n = 3) was adjusted, based on ROA recovery values, such that each vessel contained 20.0 mg of ROA. The dotted line indicates the equilibrium solubility of ROA in the studied pH range
Table V.
Critical In Vitro Dissolution Metrics with Reported Maximum Observed Amounts (A max), Observed Time to Achieve Maximum Amount (t max), and Area Under the Supersaturation Dissolution Curve (AUC) in Acidic and Neutral Media
| Composition | Process | A max (mg) | t max (min) | AUCdissolution (mg min) | ||
|---|---|---|---|---|---|---|
| Acid | Neutral | Total | ||||
| Crystalline ROA (unmicronized) | Unprocessed | 2.4 ± 0.3 | 1,440 ± 0 | 0.8 ± 0.0 | 2,833.1 ± 268.8 | 2,833.4 ± 268.8 |
| 1:1.6:0.4 ROA/Eudragit® L100-55:TEC | HME | 9.2 ± 0.4 | 125 ± 3 | 57.9 ± 5.7 | 11,215.3 ± 351.2 | 11,273.1 ± 356.9 |
| 1:2 ROA/Eudragit® L100-55 | KSD | 7.7 ± 1.0 | 180 ± 35 | 65.6 ± 2.9 | 7,823.4 ± 550.6 | 7,889.1 ± 553.5 |
| 1:2 ROA/HPMCAS | HME | 8.2 ± 0.4 | 1,440 ± 0 | 51.5 ± 6.3 | 10,374.1 ± 176.1 | 10,425.6 ± 182.4 |
| 1:2 ROA/HPMCAS | KSD | 10.5 ± 0.8 | 150 ± 0 | 59.3 ± 6.0 | 12,585.5 ± 1,462.7 | 12,644.8 ± 1,468.7 |
ROA Roche Research Compound A, KSD KinetiSol® Dispersing, HME hot melt extrusion
During the acidic phase of the dissolution study, both compositions containing Eudragit® L100-55 exhibited only a partial release of ROA due to the enteric nature of the polymer. After the pH was adjusted to neutral, both compositions exhibited rapid release, resulting in an approximate twofold increase in equilibrium solubility. The HME composition provided a slightly higher level of supersaturation than the KSD formulation. While both HME and KSD compositions contained similar levels of amorphous material and levels of concentration enhancing polymer, the HME formulation contained TEC. The increase in initial supersaturation is likely due to the pore forming ability of TEC, a water-soluble plasticizer, which allows a higher surface area for dissolution after the pH change. The ability of TEC to act as a pore former and accelerate the release of a drug substance from melt-extruded compositions containing Eudragit® L100-55 has been demonstrated in the literature (29,30). However, this does not explain the improved levels of ROA supersaturation after the initial time points. This phenomenon may be due to the presence of larger ROA domain sizes in the KSD composition or to TEC providing a solubility enhancing effect in the HME composition.
The dissolution analysis of HME and KSD formulations containing HPMCAS is presented in Fig. 11b. As observed in Eudragit® L100-55-based compositions, only a partial release was observed prior to the pH change to neutral media. While both HME and KSD compositions provided rapid supersaturation, the KSD composition provided the highest level with a 2.5- to threefold increase over the equilibrium solubility of ROA. The KSD processed composition likely provided superior performance when compared to the HME composition due to greater amorphous character, as evidenced by XRPD analysis. Based on this finding, it may be possible to detect residual amounts of crystallinity, not detected by XRPD or differential scanning calorimetry, by performing a non-sink dissolution analysis.
CONCLUSIONS
Dissolution-enhanced compositions of a drug substance exhibiting chemical instability at elevated temperatures and in acidic environments were prepared by fusion processing techniques. When comparing manufacturing techniques, it was found that KSD provided a threefold increase in ROA recovery over HME for Eudragit® L100-55 compositions, without the need for plasticizer or micronized ROA. Furthermore, compositions utilizing HPMCAS prepared by HME contained crystalline ROA and high levels of impurities, whereas KSD compositions were substantially amorphous and contained fewer impurities. Non-sink dissolution performance of HME and KSD processed compositions provided supersaturation levels approximately two to three times greater than equilibrium solubility. It was clearly demonstrated that KSD is an effective method of forming dissolution-enhanced amorphous solid solutions of chemically and thermally unstable compounds due to the ability to rapidly process compositions.
Acknowledgments
The authors wish to gratefully acknowledge the financial support of Hoffman-La Roche and DisperSol Technologies, L.L.C.
References
- 1.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JA. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Molecular Pharmaceutics. 2008;5(6):1003–1019. doi: 10.1021/mp8000793. [DOI] [PubMed] [Google Scholar]
- 3.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60(9):1281–1302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 4.Sinswat P, Gao X, Yacaman MJ, Williams RO, III, Johnston KP. Stabilizer choice for rapid dissolving high potency itraconazole particles formed by evaporative precipitation into aqueous solution. International Journal of Pharmaceutics. 2005;302:113–124. doi: 10.1016/j.ijpharm.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 5.DiNunzio JC, Miller DA, Yang W, McGinity JW, Williams RO., III Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Molecular Pharmaceutics. 2008;5(6):968–980. doi: 10.1021/mp800042d. [DOI] [PubMed] [Google Scholar]
- 6.Miller DA, DiNunzio JC, Yang W, McGinity JW, Williams RO., III Enhanced in vivo absorption of itraconazole via stabilization of supersaturation following acidic-to-neutral pH transition. Drug Dev Ind Pharm. 2008;34:890–902. doi: 10.1080/03639040801929273. [DOI] [PubMed] [Google Scholar]
- 7.Curatolo W, Nightingale JA, Herbig SM. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI Milieu. Pharm Res. 2009;26(6):1419–1431. doi: 10.1007/s11095-009-9852-z. [DOI] [PubMed] [Google Scholar]
- 8.Chowdary KPR, Suresh Babu KVV. Dissolution, bioavailability and ulcerogenic studies on solid dispersions of indomethacin in water-soluble cellulose polymers. Drug Dev Ind Pharm. 1994;20(5):799–813. doi: 10.3109/03639049409038332. [DOI] [Google Scholar]
- 9.Sekikawa H, Fukyda W, Takada M, Ohtani K, Arita T, Nakano M. Dissolution behavior and gastrointestinal absorption of dicumarol from solid dispersion systems of dicumarol-polyvinylpyrrolidine and dicumarol-beta-cyclodextrin. Chem Pharm Bull (Tokyo). 1983;31(4):1350–1356. doi: 10.1248/cpb.31.1350. [DOI] [PubMed] [Google Scholar]
- 10.Martínez-Ohárriz MC, Rodríguez-Espinosa C, Martín C, Goñi MM, Tros-Ilarduya MC, Sánchez M. Solid dispersions of diflunisal-PVP: polymorphic and amorphous states of the drug. Drug Dev Ind Pharm. 2002;28(6):717–725. doi: 10.1081/DDC-120003864. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Young TJ, Sarkari M, Williams RO, Johnston KP. Preparation of cyclosporine A nanoparticles by evaporative precipitation into aqueous solution. Int J Pharm. 2002;242:3–14. doi: 10.1016/S0378-5173(02)00147-3. [DOI] [PubMed] [Google Scholar]
- 12.Rogers TL, Johnston KP, Williams RO., III Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies. Drug Dev Ind Pharm. 2001;27(10):1003–1015. doi: 10.1081/DDC-100108363. [DOI] [PubMed] [Google Scholar]
- 13.Hu J, Johnston KP, Williams RO., III Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev Ind Pharm. 2004;30(3):233–245. doi: 10.1081/DDC-120030422. [DOI] [PubMed] [Google Scholar]
- 14.Jung J-Y, Yoo SD, Lee S-H, Kim K-H, Yoon D-S, Lee K-H. Enhanced solubility and dissolution rate of itraconazole by a solid dispersion technique. International Journal of Pharmaceutics. 1999;187(2):209–218. doi: 10.1016/S0378-5173(99)00191-X. [DOI] [PubMed] [Google Scholar]
- 15.Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 16.Lakshman JP, Cao Y, Kowalski J, Serajuddin ATM. Application of melt extrusion in the development of a physically and chemically stable high-energy amorphous solid dispersion of a poorly water-soluble drug. Molecular Pharmaceutics. 2008;5(6):994–1002. doi: 10.1021/mp8001073. [DOI] [PubMed] [Google Scholar]
- 17.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 18.Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54:107–117. doi: 10.1016/S0939-6411(02)00061-9. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Ganjyal GM, Jones DD, Hanna MA. Modeling residence time distribution in a twin-screw extruder as a series of ideal steady-state flow reactors. Journal of Food Engineering. 2008;84:441–448. doi: 10.1016/j.jfoodeng.2007.06.017. [DOI] [Google Scholar]
- 20.Verreck G, Decorte A, Heymansa K, Adriaensen J, Liu D, Tomasko D, et al. Hot stage extrusion of p-amino salicylic acid with EC using CO2 as a temporary plasticizer. Int J Pharm. 2006;327:45–50. doi: 10.1016/j.ijpharm.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 21.Schilling SU, Shah NH, Malick AW, Infeld MH, McGinity JW. Citric acid as a solid-state plasticizer for Eudragit RS PO. J Pharm Pharmacol. 2007;59(11):1493–1500. doi: 10.1211/jpp.59.11.0005. [DOI] [PubMed] [Google Scholar]
- 22.Andrews GP, Jones DS, Diak OA, McCoy CP, Watts AB, McGinity JW. The manufacture and characterisation of hot-melt extruded enteric tablets. Eur J Pharm Biopharm. 2008;69:264–273. doi: 10.1016/j.ejpb.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Repka MA, Gerding TG, Repka SL, McGinity JW. Influence of plasticizers and drugs on the physical-mechanical properties of hydroxypropylcellulose films prepared by hot melt extrusion. Drug Dev Ind Pharm. 1999;25(5):625–633. doi: 10.1081/DDC-100102218. [DOI] [PubMed] [Google Scholar]
- 24.Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm Res. 1995;12(6):799–806. doi: 10.1023/A:1016292416526. [DOI] [PubMed] [Google Scholar]
- 25.DiNunzio JC, Brough C, Miller DA, Williams RO, III, McGinity JW. Fusion processing of itraconazole solid dispersions by KinetiSol® dispersing: a comparative study to hot melt extrusion. J Pharm Sci. 2009;99(3):1239–1253. doi: 10.1002/jps.21893. [DOI] [PubMed] [Google Scholar]
- 26.Riedel A, Leopold CS. Degradation of omeprazole induced by enteric polymer solutions and aqueous dispersions: HPLC investigations. Drug Dev Ind Pharm. 2005;31:151–160. doi: 10.1081/DDC-200047787. [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Marín DC, Garrido-Varo A, Guerrero JE. Optimization of discriminant partial least squares regression models for the detection of animal by-product meals in compound feedingstuffs by near-infrared spectroscopy. Applied Spectroscopy. 2006;60(12):1432–1437. doi: 10.1366/000370206779321427. [DOI] [PubMed] [Google Scholar]
- 28.Petereit H-U, Weisbrod W. Formulation and process considerations affecting the stability of solid dosage forms formulated with methacrylate copolymers. Eur J Pharm Biopharm. 1999;47:15–25. doi: 10.1016/S0939-6411(98)00083-6. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Y, Shah NH, Malick AW, Infeld MH, McGinity JW. Solid-state plasticization of an acrylic polymer with chlorpheniramine maleate and triethyl citrate. Int J Pharm. 2002;241:301–310. doi: 10.1016/S0378-5173(02)00244-2. [DOI] [PubMed] [Google Scholar]
- 30.DiNunzio JC, Brough C, Miller DA, Williams III RO, McGinity JW. Applications of KinetiSol® Dispersing for the Production of Plasticizer Free Amorphous Solid Dispersions. Eur J Pharm Sci. 2009. doi:10.1016/j.ejps.2010.03.002. [DOI] [PubMed]
- 31.DiNunzio JC, Hughey JR, Brough C, Miller DA, Williams III RO, McGinity JW. Production of Advanced Solid Dispersions for Enhanced Bioavailability of Itraconazole Using KinetiSol® Dispersing. Drug Dev Ind Pharm. 2009. doi:10.3109/03639041003652973. [DOI] [PubMed]
- 32.DiNunzio JC, Brough C, Hughey JR, Miller DA, Williams III RO, McGinity JW. Fusion production of solid dispersions containing a heat sensitive active ingredient by hot melt extrusion and KinetiSol® dispersing. Eur J Pharm Biopharm. 2009. doi:10.1016/j.ejpb.2009.09.007. [DOI] [PubMed]