

Abstract
The present study aimed to evaluate different dosage forms, emulsions, emulgels, lipogels, and thickened microemulsion-based hydrogel, as fluconazole topical delivery systems with the purpose of determining a formulation with the capacity to deliver the whole active compound and maintain it within the skin so as to be considered a useful formulation either for topical mycosis treatment or as adjuvant in a combined therapy for Cutaneous Leishmaniasis. Propylene glycol and diethyleneglycol monoethyl ether were used for each dosage form as solvent for the drug and also as penetration enhancers. In vitro drug release after application of a clinically relevant dose of each formulation was evaluated and then microemulsions and lipogels were selected for the in vitro penetration and permeation study. Membranes of mixed cellulose esters and full-thickness pig ear skin were used for the in vitro studies. Candida albicans was used to test antifungal activity. A microemulsion containing diethyleneglycol monoethyl ether was found to be the optimum formulation as it was able to deliver the whole contained dose and enhance its skin penetration. Also this microemulsion showed the best performance in the antifungal activity test compared with the one containing propylene glycol. These results are according to previous reports of the advantages of microemulsions for topical administration and they are very promising for further clinical evaluation.
KEY WORDS: fluconazole, in vitro drug permeation, lipogel, percutaneous absorption, topical microemulsion
INTRODUCTION
Fluconazole, an antimycotic agent, is a hydrophilic bis-triazole with a broad spectrum, approved by the Food and Drug Administration (US FDA) in 1990. The pharmacokinetic properties of fluconazol differ from the other azol derivatives due to the presence of two triazol rings that make it less lipophilic and with less affinity to proteins (1). It is readily absorbed after oral administration with systemic bioavailability over 90% (1,2). The distribution profile of fluconazole can be explained by its moderate lipophilicity (log P = 0.5), its low protein binding (±12%) and its neutral charge at plasma pH (pKa = 2.03) (3). After oral dosing, fluconazole is delivered to the skin where it diffuses and accumulates rapidly and extensively in the stratum corneum (SC). Fluconazole concentration in the skin is higher than in the serum and its elimination from the SC is considerably slower than from the serum or plasma. The concentration within the skin is much higher than the minimum inhibition concentration for most dermatophytes (4,5). The prolonged skin retention of fluconazole has been attributed to its high affinity to the SC due to an interaction between fluconazole and keratin (6). However, fluconazole skin distribution after topical administration has not been investigated except with the use of limited models to simulate human skin (3).
Fluconazole is able to produce a high selective inhibition of the fungal cytochrome P450 system and also an inhibition of the C-14 α esterol demetilation process, avoiding in this way membrane ergosterol synthesis (1,7). It has shown activity against Candida spp., Blastomyces dermatitidis, Cryptococcus neoformans, Epidermophytom spp., Histoplasma, Microsporum spp., and Trichophyton spp., and it has been extensively used in the treatment of dermatophytoses by oral administration (8).
Recently, fluconazol has been used for the treatment of some Leishmania specimens, because azole antifungals can inhibit a key enzyme for the production of ergosterol, which is the sterol of the leishmanial membrane; this is a flagellated protozoan parasite belonging to the order Kinetoplastida, family Trypanosomatidae that causes different clinical diseases: cutaneous leishmaniasis (CL), diffuse cutaneous leishmaniasis, mucosal leishmaniasis, and visceral leishmaniasis (9). Oral administration of fluconazole has also been tested as a possible treatment for CL and the drug has shown activity against Leishmania spp.; Alrajhi et al. presented a trial in which oral fluconazole was compared to the use of placebo, and it was shown that a 6-week course of oral fluconazol can shorten the healing time of localized Leishmania major cutaneous lesions (median 8.5 weeks for fluconazol group, as compared with 11.2 weeks in the placebo group), concluding that it is a safe and useful treatment for CL caused by L. major (10–13). However, high doses (200 mg/day for 6 weeks) were used in these studies, and concerns about potential side effects are relevant.
Some topical dosage forms studied for CL topical treatment that should be mentioned are a paromomycin (PA) ointment (14), and a PA/urea ointment (15), but no significant cure of the lesions was observed with these treatments. In animals experimentally infected by Leishmania amazonensis or L.major, the activity of formulations containing only PA is usually low (16,17). Another ointment containing PA and methylbenzethonium chloride has proven efficacy, but it was found to be toxic and irritating (18,19). Also it is important to mention that Leishmania brazilensis and Leishmania panamensis have been demonstrated to be less sensitive to PA (13).
Considering that the in vitro skin permeation of fluconazole from emulsions was high (20), topical fluconazole as compared to oral administration would be an interesting alternative for the treatment of CL with lower toxicity. However, literature has demonstrated that the in vitro anti-leishmanial activity of fluconazole is poor (21) and studies performed regarding experimentally infected animals are lacking.
Recently, lipogels—semisolid ointment-like preparations—have been investigated as vehicles for topical drug delivery. Lipogels are based on fatty components and are obtained by gelling an oily phase with a lipophilic structure, using non-ionic surfactants (22). Generally, they are opaque thermoreversible semisolids that are stable at room temperature for weeks (23).
A microemulsion is defined as a system of water, oil, and surfactants that is a transparent, single optically isotropic and thermodynamically stable liquid solution. In a recent publication, Kreilgaad presented an increasing interest in microemulsion as a potential type of formulation that can increase cutaneous drug delivery of both hydrophilic and lipophilic drugs compared to conventional vehicles (24). Talegaonkar indicated the potential comparative advantages of this type of formulations for topical administration (25); Rozman et al. have confirmed this hypothesis recently (26).
The present study aimed to evaluate the topical delivery of fluconazole after application of a clinically relevant dose of traditional topical dosage forms such as emulsion, lipogels, and emulgel and an innovative topical form that is a thickened microemulsion-based hydrogel. The influences of two skin penetration enhancers, propyleneglycol (PG), and diethyleneglycol monoethyl ether (TCL, Transcutol P®) were also investigated in each of the selected dosage forms. In vitro liberation and skin percutaneous absorption studies were carried out in Franz diffusion cells using both synthetic membranes and hairless pig ear skin, respectively. The main objective of the present work was to determine a formulation with the capacity to deliver the whole active compound and maintain it within the skin so as to be considered a real benefit either for topical mycosis treatment or as adjuvant in a combined therapy for CL.
MATERIALS AND METHODS
Materials
Fluconazole was a gift of Unifarma, Argentina; diethyleneglycol monoethyl ether (Transcutol P®, TCL) Gattefossé, France, was gift of Ferromet S.A.; glyceryl monostearate (Cutina MD®) and Polyoxyl (40) hydrogenated castor oil was kindly supplied by Cognis, Argentina; polyoxyethylene (10) oleyl ether (BRIJ 97®, ICI Américas); castor oil, carboxymethyl cellulose (CMC), and PG were purchased from Fabriquimica S.R.L., Argentina.; non-ionic self-emulsifying wax, cetostearyl alcohol, liquid paraffin, isopropyl miristate, jojoba oil, and distilled water were all of pharmaceutical grade. All reagents used for preparation of receptor medium were of analytical grade. Membranes of mixed cellulose esters type Millipore HA 0.45 µm were used for in vitro drug permeation; full-thickness skin (thickness of 0.5-1.0 mm) excised from 4-month-old domestic pig ears obtained from a local commercial supplier (Nueva Granja Burzaco; Rauch, Buenos Aires, Argentina) was used for percutaneous absorption.
Methods
Fluconazole solubility in water is low (5.5 mg/mL) but PG dissolves 147 ± 1.0 mg/mL and TCL 146 ± 5.9 mg/mL (20); therefore, solubility in PG and TCL is similar and these two enhancers were selected for dissolution and incorporation of fluconazole into the four different topical dosage forms assayed. For all compositions, the active compound concentration was 1% w/w.
Preparation of Lipogels
The compositions of lipogels are shown in Table I. The appropriate amount of Cutina MD and jojoba oil were weighed, mixed, and heated to 70°C in a water bath. After the mixture melted, it was stirred and cooled to a temperature of 40°C; then fluconazole which was previously dissolved in PG or TCL, was added. Agitation continued for another 30 min until cooling and total homogenization was obtained.
Table I.
Composition of Lipogels (% w/w)
| Component | Lipogel 1 (L1) | Lipogel 2 (L2) |
|---|---|---|
| Cutina MD® | 20 | 20 |
| PG | 10 | – |
| TCL® | – | 10 |
| Jojoba oil | 69 | 69 |
| Fluconazole | 1 | 1 |
PG: propyleneglycol; TCL®: diethyleneglycol monoethyl ether, Transcutol P®
Preparation of Microemulsions
The microemulsions were prepared according to formula shown in Table II. The appropriate amount of Polyoxyl (40) hydrogenated castor oil (CRH 40) was heated to a temperature of 40°C; at that temperature castor oil was added, and stirring was continued for 10 min. Then, fluconazole, which was previously dissolved in TCL or PG, was added to the surfactant-oil mixture; agitation continued until homogenization and the mixture was taken out of the bath. Finally, a CMC-based hydrogel was weighed and added to the preparation; stirring continued for another 30 min, until total homogenization was observed.
Table II.
Composition of Microemulsions (% w/w)
| Component | Microemulsion 1(M1) | Microemulsion 2 (M2) |
|---|---|---|
| CRH 40 | 15 | 15 |
| Castor oil | 5 | 5 |
| PG | 10 | – |
| TCL® | – | 10 |
| CMC | 3 | 3 |
| Fluconazole | 1 | 1 |
| Distilled water qs | 100 | 100 |
CRH40: Polyoxyl (40) hydrogenated castor oil; PG: propyleneglycol; TCL®: diethyleneglycol monoethyl ether, Transcutol P®; CMC: Carboxymethyl cellulose
Preparation of Emulgels
Compositions are shown in Table III. The required amount of CMC was weighed and dispersed in water; it was left for hydratation under continuous stirring, for 60 min. Separately, oily components were melted at 65°C and then added to the CMC gel, which had been previously heated at the same temperature. The preparation was cooled to a temperature of 40°C and fluconazole, which was previously dissolved in PG or TCL was added. Magnetic stirring was continued until homogenization for 30 min.
Table III.
Composition of Emulgels (% w/w)
| Component | Emulgel 1 (Eg1) | Emulgel 2 (Eg2) |
|---|---|---|
| Cetostearyl alcohol | 10 | 10 |
| IPM | 10 | 10 |
| PG | 10 | – |
| TCL® | – | 10 |
| Brij 97® | 5 | 5 |
| Liquid paraffin | 10 | 10 |
| CMC | 1 | 1 |
| Fluconazole | 1 | 1 |
| Distilled Water qs | 100 | 100 |
IPM: isopropyl miristate; PG: propyleneglycol; TCL®: diethyleneglycol monoethyl ether, Transcutol P®; Brij 97®: Polyoxyethylene (10) oleyl ether; CMC: Carboxymethyl cellulose
Preparation of Emulsions
According to the compositions shown in Table IV, aqueous and oily phases were prepared separately and heated to 70°C. Then phases were mixed using a paddle agitator at 900 rpm for 15 min. Then, the emulsion was cooled to a temperature of 40°C. Subsequently, fluconazole, which was previously dissolved in PG or TCL, was added, and stirring was applied for 30 min.
Table IV.
Composition of Emulsions (% w/w)
| Component | Emulsion 1 (E1) | Emulsion 2 (E2) |
|---|---|---|
| Cetostearyl alcohol | 5 | 5 |
| IPM | 10 | 10 |
| PG | 10 | – |
| TCL® | – | 10 |
| Liquid paraffin | 10 | 10 |
| Non ionic self emulsifying wax | 10 | 10 |
| Fluconazole | 1 | 1 |
| Distilled Water qs | 100 | 100 |
IPM: isopropyl miristate; PG: propyleneglycol; TCL®: diethyleneglycol monoethyl ether, Transcutol P®
Evaluation of Stability of Dosage Forms
All the prepared compositions were centrifuged at 3,000 rpm during 30 min 1 week after preparation and after 6 months of storage at room temperature; they were also evaluated by a cycling test at 4°C and 40°C (24 h at each temperature) for a week. Visual observation and microscopical examination after the mentioned procedures were done to detect physical changes, such as phase instabilization, phase separation, syneresis, change in color, drug crystallization, etc.
Determination of Fluconazole Concentration in the Formulations
The concentration of fluconazole in the prepared dosage forms was evaluated as follows: 0.5 g of sample was put into a glass vial under magnetic stirring for 3 h in 10 mL of ethanol, solubility 120 ± 3.9 mg/ml (20). The resulting dispersions were filtered and an aliquot of 1 mL was transferred into in a 10 ml volumetric flask and volume was adjusted with solvent for measuring concentration at 260 nm (UV-VIS Spectrophotometer Shimadzu UV-260) (27).The final concentration was calculated based on a calibration curve and the analyses were done by triplicate.
Viscosity Measurement
The viscosity of the different formulations was determined one week after preparation. For the viscosity test, a rotational viscosimeter was employed (Brookfield model PVT synchro-Lectric viscometer). Viscosity was measured through the rotation speed of the spindle immersed in the sample; spindle number 6 and 7 were used for microemulsions and for the rest of formulations, respectively. The test was carried out at 25 ± 1°C and the spindle was rotated at 10 rpm. The readings for each sample were made after 10 min.
In vitro Drug Release
The evaluation of in vitro drug release was performed on Franz diffusion cells using membranes of mixed cellulose esters type Millipore HA 0.45 µm (membrane surface area of 3.14 cm2).
Synthetic membranes were put in previous contact with phosphate-buffered saline (PBS; pH 7.4) 30 min before placing the samples. Sink conditions were obtained in the receptor compartment with PBS, with the volume of the receptor fluid being of 15 ml. During the experiment, the receptor compartment was continuously homogenized using a stirring magnetic bar. The temperature was kept at 32°C using a water circulation system (28–30).
To simulate usage conditions, experiments were performed by applying 500 ± 10 mg (“infinite dose”; n = 3) as the dose. The preparations were evenly distributed on the membranes. Serial sampling was performed after 0.5, 1, 2, 4, and 6 h and fresh receptor liquid was added to receptor compartment to replace the buffer; 5 mL of receptor fluid (PBS) was taken for UV determination of fluconazole concentration at 260 nm.
In vitro Skin Penetration
In vitro skin penetration and permeation experiments were performed on Franz diffusion cells using pig ear skin. Skin was excised from 4-month-old domestic pig ears, obtained from a local commercial supplier. Full thickness skin was used with a surface area of 3.14 cm2.
The pig ears were cleaned under running water immediately after excision. The hair from the outer region of the ears was removed and then the skin was carefully separated from cartilage using a scalpel. Subsequently, adipose subcutaneous tissue was removed; a thickness of 1 mm for all the samples was controlled with a vernier. After being dried with a tissue, the skin was immediately mounted on the diffusion cells or frozen at −20°C for a maximum period of 4 weeks (4,31).
The skin was placed horizontally on Franz diffusion cells, between the donor and receptor compartments. Sink conditions were obtained in the receptor compartment with PBS (pH 7.4), with a volume of receptor fluid of 15 ml. The receptor compartment was continuously homogenized using a stirring magnetic bar and the temperature was kept at 32°C using a water circulation system.
To simulate usage conditions, experiments were performed applying 500 ± 10 mg ("infinite dose"; n = 3) as the dose. The skin mounted in the cell was allowed to rest for an hour in contact with PBS before the application of the compositions. Serial sampling was performed after 0.5, 1, 2, 4, and 6 h, and fresh receptor liquid was added to receptor compartment in order to replace the buffer; fluconazole UV determination was carried out at 260 nm.
It is necessary to remark that pig ears were obtained from a local pork slaughterhouse for human feeding. No animal was killed for the purpose of these experiments. However, approval has been obtained from the Ethical Committe of the Faculty of Pharmacy and Biochemistry, University of Buenos Aires.
Evaluation of Skin Penetration
After the experiment (6 h), the remnant of the dosage form on the skin (dislodgeable dose) was put in a glass vial, skin was washed twice with ethanol; the used spatula was also washed so as to remove the dosage form completely. The final volume was adjusted to 5 mL with ethanol and the mixture was stirred (2,500 rpm) for 3 h. An aliquot of 1 mL was transferred into in a 10 ml volumetric flask and volume was adjusted with ethanol. The resulting solution was filtered and the remaining amount of fluconazole was quantified at 260 nm.
Terms recommended by the European Commission were considered for denomination of the drug location in the in vitro evaluations (32). Skin drug penetration, considered as the amount retained within the skin, was calculated by difference between the total dose applied and the amount that permeated plus the amount present in the remnant sample over the skin.
Droplet Size Determination
The particle size distribution and average droplet size of microemulsions were measured using a NanoZetasizer-zs (Malvern Instrument, Malvern, UK). No dilution of the samples was made for the test.
In vitro Antifungal Activity
Appropriate dilutions of the formulations in PBS (1/10; 1/100; 1/1,000; and 1/100,000) were inoculated with a suspension of Candida albicans (ATCC 10231) in Sabouraud Dextrose Agar, so that the final concentration of the test preparation after inoculation was 1 × 106 colony forming unit (cfu) per mililiter of sample. The samples were incubated at 22.5 ± 2.5 ° C and each container was observed for any change in appearance and sampled after 2, 24, and 48 h. The plate count procedure was used to determine the number of colony forming unit present in the test preparations at each time. The average cfu/ml was calculated taking into account dilution factors; a standard solution of fluconazole in PBS and both microemulsions without fluconazole were assayed simultaneously as standard and blank sample, respectively. The assay was performed in triplicate.
Statistical Analysis
Data shown of skin penetration and permeation represent mean ± standard deviation. Comparison among mean values was carried out using ANOVA. The differences were considered as statistically significant at p < 0.05.
RESULTS
Evaluation of Fluconazole Concentration and Stability of Formulations
All the prepared dosage forms had a concentration of FLZ 1 ± 0.15%. No significant changes that can be related with physical instability were observed for any of the different prepared dosage forms. Microscopic examination showed neither change in internal structure nor fluconazole crystals, suggesting that it was completely dissolved in the selected compositions.
Viscosity Measurement
Viscosity of formulations was as follows: emulsion > lipogel > emulgel > microemulsion; results of viscosity measurement are given in Table V. Each reading is an average of three determinations.
Table V.
Viscosity of Formulations at 25 ± 1°C
| Formulation | Viscosity (mPa.s) |
|---|---|
| L1 | 38,000 |
| L2 | 34,000 |
| E1 | 40,000 |
| E2 | 41,000 |
| Eg1 | 22,000 |
| Eg2 | 20,000 |
| M1 | 17,500 |
| M2 | 18,500 |
In vitro Drug Release
Data of the drug release evaluation are shown Fig. 1. The relationship of Q (cumulative amount released per surface area of membrane; µg/cm²) versus square root of time, shown in Fig. 2, is derived from the Higuchi model with the assumption that there is a reservoir of the drug always available to diffuse through (28,29), as follows:
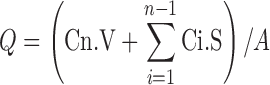 |
- Cn
concentration of FLZ determined at nth sampling interval
- V
volume of individual Franz cell
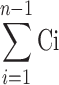
sum of concentration of FLZ determined at sampling intervals 1 through n − 1
- S
volume of sampling aliquot
- A
surface of sample cell
The amount of fluconazole released from the semisolds studied showed a linear relationship with the square root of time, accordingly to this model (correlation coefficients for Q vs. square root of time, r², ranged from 0.94 to 0.99).
Fig. 1.

Total amount of drug (%) delivered from each one of the prepared formulations evaluated after 6 h. L lipogel, M microemulsion, Eg emulgel, E emulsion
Fig. 2.

Q (cumulative amount released per surface area of membrane) profile of fluconazole from different dosage forms containing 1% w/w vs. square root of time. L lipogel, M microemulsion, Eg emulgel, E emulsion. Each point represents mean (n = 3) ± SD
Statistical analysis, ANOVA, was applied to each one of the dosage forms to evaluate if fluconazole release was significantly different for compositions containing PG and TCL. No significant difference after six hours of drug delivery was observed in any type of topical dosage forms. The only difference observed was that the total drug release from microemulsions was higher than that observed from lipogels; it was also significantly higher than that exhibited by emulsions and emulgels.
After these in vitro liberation results, microemulsions and lipogels were considered for further evaluation of percutaneous absorption through pig skin.
Evaluation of Skin Penetration
Fluconazole permeation across pig ear skin, the remaining drug of the applied dose on the skin after 6 h and the estimated penetration are shown in Table VI. All the measurements were made in triplicate.
Table VI.
Fluconazole Permeation to Receptor Fluid, Remaining Drug and Skin Penetration after 6 h of Skin Assay
| Sample | Permeation (%) | Remaining drug (%) | Penetration (%) |
|---|---|---|---|
| M1 | 13.91 ± 1.79 | 0.01 ± 0.01 | 86.09 ± 1.78 |
| M2 | 2.9 ± 4.10 | 0.01 ± 0.01 | 97.09 ± 4.09 |
| L1 | 42.65 ± 2.84 | 49.50 ± 2.12 | 7.85 ± 4.96 |
| L2 | 44.49 ± 5.78 | 31.90 ± 9.76 | 23.61 ± 15.54 |
Fluconazole permeation was low for microemulsions with significant difference between them; in the case of lipogels the amount permeated was less than half the applied dose.
When the total amount of delivered drug was evaluated, it was observed that lipogels were able to deliver only half of the applied dose, while both microemulsions completely delivered the whole applied dose (delivery enhancement ratio M1/L1 1.99 and M2/L2 1.47). Both microemulsions and lipogels showed more release of fluconazole in the pig skin assay than that observed in the synthetic membrane assay.
Comparing drug penetration from lipogels, the one containing transcutol was able to keep a higher amount of drug in skin with a statistically significant difference.
Microemulsion resulted to be the dosage form with highest ability to penetrate pig skin (penetration enhancement ratio M1/L1 2.20 and M2/L2 4.11). This dosage form has also shown an important capacity to keep the drug within the skin layers. Transcutol® showed a statistically significant difference in pig skin penetration of fluconazole from microemulsions. The total amounts of drug that penetrates the skin from microemulsions were ten and four times greater than those found with lipogels, comparing compositions containing propilenglycol and transcutol, respectively, being TCL enhancement ratio compared to PG 1.13 for the microemulsions (M2/M1) and 3.01 for the lipogels (L2/L1).
Droplet Size Determination
Size distribution by intensity showed three particle populations for microemulsion M1, whereas the analysis of distribution by volume and number showed one population of particles. Microemulsion M2 size distribution by intensity, volume, and number showed two population of particles. Mean droplet size was 131.0 nm (polidispersity index 0.564) and 237.6 nm (polidispersity index 0.558) for M1 and M2, respectively.
In vitro Antifungal Activity
The average colony forming unit per mililiter (cfu/ml) in the samples at the different times of the test are shown in Table VII. Microemulsion containing TCL resulted to be the most effective one, being as effective as the standard solution. On the other hand, microemulsion containg PG was only able to reduce by one log10 the number of cfu/mL at the end of the assay. Antifungal activity of fluconazole in a PBS solution with 10% of PG (the same concentration as in the microemulsions) was also tested and it was shown that under the conditions of our trial in vitro antifungal activity of the drug in the solution containing PG is reduced in comparison with the activity of the drug in PBS solution (Table VIII), but it showed more activity than in M1.
Table VII.
Antifungal Activity of Fluconazole Microemulsions; cfu/ml after Incubation of Samples at 22.5 ± 2.5°C
| Initial inoculum | 2 h | 24 h | 48 h | |
|---|---|---|---|---|
| Standard | 1,100,000 | <10 | <10 | <10 |
| Blank M1 | 1,099,000 | 1,120,000 | 1,280,000 | |
| M1 | 300,000 | 315,000 | 318,000 | |
| Blank M2 | 1,100,000 | 1,100,000 | 1,090,000 | |
| M2 | <10 | <10 | <10 |
Table VIII.
Antifungal Activity of Fluconazole Solution with Propyleneglycol; cfu/ml after Incubation of Samples at 22.5 ± 2.5°C
| Initial inoculum | 2 h | 24 h | 48 h | |
|---|---|---|---|---|
| Standard | 1,030,000 | <10 | <10 | <10 |
| Blank solution | 1,050,000 | 1,130,000 | 1,210,000 | |
| Fluconazole in PBS/PG | 893,000 | 500,000 | 49,800 |
DISCUSSION
Topical formulations for the treatment of skin infections must provide proper concentrations of the drug in the target site for therapeutic activity. In the case of superficial fungal skin infections, in which the main location of the pathogen is the epidermis, the drug must penetrate into the SC in proper concentrations to inhibit the fungus growth (33). Yang has recently established the need of drug delivery strategies for improving FLZ chemical potential, for targeting high concentrations to the infection sites (34).
In the present work, different dosage forms, emulsions, emulgels, lipogels, and thickened microemulsion-based hydrogel were assayed as fluconazole topical delivery systems. The main objective was to find a formulation with the capacity to deliver the whole active compound and maintain it within the skin so as to be considered a real benefit either for topical mycosis treatment or as adjuvant in a combined therapy for CL. For the purpose of this work, all the systems remained stable; no significant changes were observed for any of the different prepared dosage forms.
The amount of fluconazole released from the semisolds studied showed a linear relationship with the square root of time, accordingly to Higuchi model. Drug release was not significantly different for compositions containing PG and TCL in any type of the dosage forms. Emulsions and lipogels are the most viscous systems and that could have an influence on the release capacity, nevertheless in the case of emulgels, which have a viscosity similar to the one of microemulsions, drug release was much lower than for microemulsions. Viscosity showed not to be the main parameter that governs fluconazol release in these systems.
The total drug release from microemulsions and from lipogels was higher than from other types of formulations so they were considered for evaluation of percutaneous absorption through pig skin. In vitro release data as well as the percutaneous absorption studies showed an important enhancement effect in fluconazole skin absorption when it is carried in microemulsions. Tenfold improvement in drug absorbed was achieved in comparison to cutina lipogels when PG was the observed enhancer, and for compositions containing transcutol, the shown improvement was around four times higher. In addition, the enhancer effect of Transcutol® contained in a microemulsion is more relevant than the one promoted by PG in the same kind of dosage forms.
For skin penetration, the analysis of the remnant amount of drug in microemulsions indicate an extremely proven ability to deliver the whole dose for both compositions, while lipogels resulted in a less appropriate dosage form for fluconazole delivery. This can be explained by the affinity of microemulsion components to skin structure that enhances drug absorption (22); the pharmacokinetic characteristics of the drug might have also contributed to the effect (6).
According to literature, a dermally applied microemulsion is expected to penetrate the stratum corneum and to exist intact in the whole horny layer. The drug dissolved in the lipid domain of the microemulsion can directly partition into the lipids of SC, or the lipids contained in the microemulsion can intercalate between the lipid chains of SC (35). On the other hand, the hydrophilic domain of a microemulsion can hydrate the stratum corneum to a greater extent; consequently, the carried active compound can permeate more easily through the pathways of the SC (22). The small size of microemulsion particles makes this type of formulation an excellent carrier for promoting drugs percutaneous uptake of drugs. It is also known that Transcutol® shows enhancement of absorption by increasing the solubility in the SC barrier (36). The obtained results show a remarkable influence of the kind of dosage form used when a considerable absorption of drug is desirable. These results are in agreement with other authors that have also indicated the proven enhancement effect observed for microemulsions (24–26). It is also promising that the inclusion of Transcutol® enhanced topical administration, and it was observed, as other literature also reflects, that the effect of skin penetration enhancers depends on the vehicle (37).
If this work is compared with the few previous reports about topical delivery and percutaneous absorption of fluconazole, some issues have to be considered. First, it is necessary to point out that these experiments were performed with a dose of 500 mg of dosage forms containing 1% of fluconazole. It has to be considered as “infinite dose”, but it is near the real dose used in clinical practice. The broad differences in the rheological properties shown by the selected compositions obligated us to use this amount so as to allow for an easy application in all cases; in vivo and in vitro experiments have demonstrated there is an inverse relation between concentration (area dose) and percentage of absorption. At low concentrations, the absorbed test substance expressed as percent of applied dose per time interval is in general higher than the percentage absorption at high concentrations (32). Additionally, it is necessary to remark that mice skin is a less representative membrane; therefore, it is important to point out that experiments carried out in pig skin are a relevant model for human skin, as regulatory authorities have established (31,32).
Microemulsions have shown to be a proper dosage form for fluconazol penetration into the skin, but we have found that antifungal activity of the drug may be influenced by system components. In vitro antifungal activity was reduced for the presence of propylene glycol in the formulation. The lower activity observed for M1 was not due to an interation with the other components of the microemulsion as this solvent also affects the drug antifungal activity in a PBS solution. More investigation is needed to understand this possible interaction, as propylene glycol is a good solvent for the drug and is commonly used as enhancer for topical dosage forms.
The parasite Leishmania resides in macrophages of viable epidermis and dermis, so the results are promising for this pathology, which requires the drug to reach the intracellular environment. So far, good results have not been found when creams containing increasing concentrations of fluconazole (1%, 2%, or 10% w/w) were evaluated in mice infected by Leishmania (L) major, but it is to remark that these investigators used propylene glycol in their formulations (38).
CONCLUSION
In this work, in vitro fluconazol release was assayed for different topical dosage forms and microemulsion was the form that exhibited the largest amount of released drug. Using pig ear skin, a model that can be related to human skin, our data suggest that high skin concentrations could be obtained after topical administration of fluconazole from microemulsions applied at a clinically relevant dose. There are few antecedents of formulations which could provide proper concentrations of the drug in the skin.
Finally, microemulsion with propylene glycol showed to have less antifungal activity in vitro than the one with Transcutol®, so in order to improve fungal topical treatment and complementary topical therapy for cutaneous leishmaniasis, further research should be developed on the composition of microemulsions and their performance in animal models of these pathologies.
Acknowledgment
This work was financially supported with funds from Project UBACyT B003(2008-2010) from University of Buenos Aires.
The authors would like to thank Mrs. Daniela Sanchez (pharmacist and biochemist) from Edyafe Laboratories for her support in the in vitro microbiological tests.
References
- 1.Dash AK, Elmquist WF. Analytical profiles of drug substances and excipients. Harry G Britain, Founding Florey Academic Press 2001; Vol 27 pp 70-113.
- 2.Martindale The Extra Pharmacopea. 31st Ed p. 404.
- 3.Mathy FX, Ntivunwa D, Verbeeck RK, Pre’at V. Fluconazole distribution in rat dermis following intravenous and topical application: a microdialysis study. J Pharm Sci. 2005;94(4):770–780. doi: 10.1002/jps.20290. [DOI] [PubMed] [Google Scholar]
- 4.Wildfeuer A, Faergemann J, Laufen H, Pfaff G, Zimmermann T, Seidl HP, Lach P. Bioavailability of fluconazole in the skin after oral medication. Mycoses. 1994;37(3–4):127–130. doi: 10.1111/j.1439-0507.1994.tb00788.x. [DOI] [PubMed] [Google Scholar]
- 5.Faergemann J. Pharmacokinetics of fluconazole in skin and nails. J Am Acad Dermatol. 1999;40(6 Pt 2):S14–S20. doi: 10.1016/S0190-9622(99)70393-2. [DOI] [PubMed] [Google Scholar]
- 6.Klimke K, Schäfer-Korting M. Effect of keratin on the efficacy of fluconazole. Mycoses. 1997;40(Suppl 1):43–46. doi: 10.1111/j.1439-0507.1997.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 7.Remington. The science and practice of pharmacy. A R Gennaro Editor. 2000. 20th Ed, p. 1552.
- 8.Lesher JL. Oral therapy of common superficial fungal infections of the skin. J Am Acad Dermatol. 1999;40:S31–S34. doi: 10.1016/S0190-9622(99)70395-6. [DOI] [PubMed] [Google Scholar]
- 9.Reithinger R, Dujardin JC, Louzir H, Pirmez C, Alexander B, Brooker S. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7(9):581–596. doi: 10.1016/S1473-3099(07)70209-8. [DOI] [PubMed] [Google Scholar]
- 10.Laffitte E, Genton B, Panizzon RG. Cutaneous leishmaniasis caused by Leishmania tropica: treatment with oral fluconazole. Dermatology. 2005;210:249–251. doi: 10.1159/000083797. [DOI] [PubMed] [Google Scholar]
- 11.Baron S, Laube S, Raafat F, Moss C. Cutaneous leishmaniasis in a Kosovan child treated with oral fluconazole. Clin Exp Dermatol. 2004;29:546–547. doi: 10.1111/j.1365-2230.2004.01561.x. [DOI] [PubMed] [Google Scholar]
- 12.Alrajhi AA, Ibrahim EA, De Vol EB, Khairat M, Faris RM, Maguire JH. Fluconazole for the treatment of cutaneous leishmaniasis caused by Leishmania major. N Engl J Med. 2002;346:891–895. doi: 10.1056/NEJMoa011882. [DOI] [PubMed] [Google Scholar]
- 13.Croft SL, Seifert K, Yardley V. Current scenario of drug development for leishmaniasis. Indian J Med Res. 2006;123:399–410. [PubMed] [Google Scholar]
- 14.Soto J, Toledo JT, Gutierrez P, Arboleda M, Nicholls RS, Padilla JR, et al. Treatment of cutaneous leishmaniasis with a topical antileishmanial drug (WR279396): phase 2 pilot study. Am J Trop Med Hyg. 2002;66:147–151. doi: 10.4269/ajtmh.2002.66.147. [DOI] [PubMed] [Google Scholar]
- 15.Armijos RX, Weigel MM, Calvopina M, Mancheno M, Rodriguez R. Comparison of the effectiveness of two topical paromomycin treatments versus meglumine antimoniate for New World cutaneous leishmaniasis. Acta Trop. 2004;91:153–160. doi: 10.1016/j.actatropica.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 16.Neal RA, Allen S, McCoy N, Olliaro P, Croft SL. The sensitivity of Leishmania species to aminosidine. J Antimicrob Chemother. 1995;35:577–584. doi: 10.1093/jac/35.5.577. [DOI] [PubMed] [Google Scholar]
- 17.Grogl M, Schuster BG, Ellis WY, Berman JD. Successful topical treatment of murine cutaneous leishmaniasis with a combination of paromomycin and gentamicin. J Parasitol. 1999;85:354–359. doi: 10.2307/3285646. [DOI] [PubMed] [Google Scholar]
- 18.Soto J, Fuya P, Herrera R, Berman J. Topical Paromomycin/methylbenzethonium chloride plus parenteral meglumine antimonate as treatment for American cutaneous leishmaniasis: controlled study. Clin Infect Dis. 1998;26:56–58. doi: 10.1086/516267. [DOI] [PubMed] [Google Scholar]
- 19.Arana BA, Mendoza CE, Rizzo NR, Kroeger A. Randomized, controlled, double-blind trial of topical treatment of cutaneous leishmaniasis with paromomycin plus methylbenzethonium chloride ointment in Guatemala. Am J Trop Med Hyg. 2001;65(5):466–470. doi: 10.4269/ajtmh.2001.65.466. [DOI] [PubMed] [Google Scholar]
- 20.Ayub AC, Gomes ADM, Lima MVC, Vianna-Soares CD, Ferreira LAM. Topical Delivery of fluconazole: in vitro skin penetretion and permeation using emulsions as dosage forms. Drug Dev Ind Pharm. 2007;33:273–280. doi: 10.1080/03639040600829989. [DOI] [PubMed] [Google Scholar]
- 21.Al-Abdely HM, Graybill JR, Loebenberg D, Melby PC. Efficacy of the triazole SCH 56592 against Leishmania amazonensis and Leishmania donovani in experimental murine cutaneous and visceral leishmaniases. Antimicrob Agents Chemother. 1999;43:2910–2914. doi: 10.1128/aac.43.12.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Laithy HM, El-Shaboury KM F. The development of cutina lipogels and gel microemulsion for topical administration of fluconazole. AAPS PharmSciTech. 2002;3(4)35:3-9. [DOI] [PMC free article] [PubMed]
- 23.Murdan S, Gregoriadis G, Florence AT. Novel sorbitan monostearate organogels. J Pharm Sci. 1999;88(6):608–614. doi: 10.1021/js980342r. [DOI] [PubMed] [Google Scholar]
- 24.Kreilgaard M. Influence of microemulsions on cutaneous drug delivery. Adv Drug Deliv Rev. 2002;54(Suppl 1):77–98. doi: 10.1016/S0169-409X(02)00116-3. [DOI] [PubMed] [Google Scholar]
- 25.Talegaonkar S, Azeem A, Ahmad FJ, Khar RK, Pathan SA, Khan ZI. Microemulsions: a novel approach to enhanced drug delivery. Recent patents on drug delivery and formulation. 2008; 2(3):238-57. [DOI] [PubMed]
- 26.Rozman B, Gasperlin M, Tinois-Tessoneaud E, Pirot F, Falson F. Simultaneous absorption of vitamins C and E from topical microemulsions using reconstructed human epidermis as a skin model. Eur J Pharm Biopharm. 2009;72(1):69–75. doi: 10.1016/j.ejpb.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Göger NG, Aboul-Enein HY. Quantitative determination of fluconazole in capsules and IV solutions by UV spectrophotometric methods. Anal lett. 2001;34(12):2089–2098. doi: 10.1081/AL-100106841. [DOI] [Google Scholar]
- 28.Thakker KD, Chwern WH. Development and validation of in vitro release test for semisolid dosage forms- Case study. Dissolution Technol. May 2003;10-15.
- 29.Zatz JL, Segers JD. Techniques for measuring in vitro release from semisolids. Dissolution Technol. February 1998;3-17.
- 30.FDA Guidance for Industry: SUPAC-SS Nonsterile Semisolid Dosage Forms. scale-up and postappraoval changes: chemistry,manufacturing, and controls; in vitro release testing and in vivo bioequivalence documentation. http://www.fda.gov/cder/guidance.htm. May 1997.
- 31.European Comission Scientific Committee On Consumer Products (Sccp). Opinion on basic criteria for the in vitro assessment of dermal absorption of cosmetic ingredients. http://ec.europa.eu/health/ph_risk/committees/04_sccp/docs/sccp_s_03.pdf. Updated March 2006.
- 32.European Commission Health & Consumer Protection Directorate General Sanco/222/2000 rev. 7 19 March 2004 Guidance Document on Dermal Absorption. http://www.efsa.europa.eu/cs/BlobServer/DocumentSet/SANCO_222_2000_rev7.pdf?ssbinary = true
- 33.Alberti I, Kalia YN, Naik A, Bonny J, Guy RH. Effect of ethanol and isopropyl myristate on the availability of topical terbinafine in human stratum corneum, in vivo. Int J Pharm. 2001;219:11–19. doi: 10.1016/S0378-5173(01)00616-0. [DOI] [PubMed] [Google Scholar]
- 34.Yang W, Wiederhold NP, Williams RO. Drug delivery strategies for improved azole antifungal action. Expert Opin Drug Deliv. 2008;5(11):1199–1216. doi: 10.1517/17425240802457188. [DOI] [PubMed] [Google Scholar]
- 35.Thachrodi D, Panduranga Roo K. Transdermal absorption of nifedipine from microemulsiions of lipophilic skin penetration enhancers. Int J Pharm. 1994;111:235–240. doi: 10.1016/0378-5173(94)90346-8. [DOI] [Google Scholar]
- 36.Harrison JE, Watkinson AC, Green DM, Hadgraft J, Brain K. The relative effect of Azone and Transcutol on permeant diffusivity and solubility in human stratum corneum. Pharm Res. 1996;13(4):542–546. doi: 10.1023/A:1016037803128. [DOI] [PubMed] [Google Scholar]
- 37.Trottet L, Merly C, Mirza M, Hadgraft J, Davis AF. Effect of finite doses of propylene glycol on enhancement of in vitro percutaneous permeation of loperamide hydrochloride. Int J Pharm. 2004;274:213–219. doi: 10.1016/j.ijpharm.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 38.Vidal Mussi S, Fernandes AP, Miranda Ferreira LA. Comparative study of the efficacy of formulations containing fluconazole or paromomycin for topical treatment of infections by Leishmania (Leishmania) major and Leishmania (Leishmania) amazonensis. Parasitology Res. 2007. doi:10.1007/s00436-006-0394-6http://www.springerlink.com/content/b7002028121u8273/fulltext.html. [DOI] [PubMed]