

Abstract
Ritonavir is an antiretroviral drug characterized by low solubility and high permeability which corresponds to BCS class II drug. The purpose of the study was to develop solid dispersion by different methods and investigate them for in vitro and in vivo performance for enhancing dissolution and bioavailability, respectively. Since the drug possesses food-related absorption, the effect of biorelevant media (FaSSIF and FeSSIF state) on dissolution behavior was also studied. The solid dispersion was prepared using Gelucire as carrier in 1:4 ratio by different methods and were characterized for differential scanning calorimetry (DSC), X-ray diffractometry, scanning electron microscopy, and FT-IR. Oral bioavailability of 10 mg of ritonavir in solid dispersion prepared by solvent evaporation (SE1) and melt method (MM1) was compared with pure drug after oral administration of solid dispersion and pure drug to Albino Wistar rats of either sex. The results suggested formation of eutectic solid dispersion. In vitro dissolution studies was performed in 0.1 N HCl and biorelevant media showed enhanced dissolution rate as compared to pure drug in both FeSSIF media and 0.1 N HCl. The apparent rate of absorption of ritonavir from SE1 (Cmax 20221.37 ng/ml, tmax 0.5 h) was higher than that of MM1 (Cmax 2,462.2, tmax 1 h) and pure drug (Cmax 1,354.8 ng/ml, tmax 0.5 h). On the basis of the result obtained, it was concluded that solid dispersion is a good approach to enhance solubility and bioavailability of poorly water-soluble ritonavir.
Key words: bioavailability, gelucire, poorly soluble drug, ritonavir, solid dispersion
INTRODUCTION
Most of the newly discovered chemical entities, in spite of high therapeutic activity, have low aqueous solubility and poor bioavailability, leading to poor absorption in the gastrointestinal tracts (1). For absorption of the drug from the gastrointestinal tracts (GIT), the drug should be present in solution state in GI fluid, which forms a critical requirement for absorption of poorly water-soluble drugs. Therefore, there is a great interest to develop efficient, reliable, economical, and scalable method to increase the oral bioavailability of poorly water-soluble drugs. Common approaches used to tackle this challenge are particle-size reduction which includes microsizing and nanosizing, salt formation, solubilization, and complexation with β cyclodextrins. All these methods suffer from drawbacks. The formulation of drugs having low aqueous solubility using solid dispersion method has been an active area of research since 1960 (2). The term solid dispersion refers to the dispersion of one or more active ingredients in a hydrophilic inert carrier matrix at molecular level. It is prepared by the melt (fusion) method and solvent evaporation technique. However, the process is individualized depending on the interaction between drug and carrier. One of the underlying principles of formulation of solid dispersion is achievement of the amorphous state which is considered to be more soluble than the crystalline state. Amorphous state in the solid dispersion system has its own advantages as well as limitations. This can be explained by the fact that in the amorphous state, no energy is required to break the crystal lattice found in the crystalline phase (3–5). Generally, polymers with high glass transition temperature are used for solid dispersion formulation. This is because the glassy state is the desired state for solid dispersion. Glass transition temperature measurement has been developed as an important tool to distinguish the characteristic amorphous regions in the solid dispersion. If the achieved glass transition temperature in the solid dispersion is higher than that of the drug, it will help to retard the process of recrystallization. At the glass transition temperature, the glassy material have higher enthalpy or internal energy as well as increased specific volume relative to crystalline state, responsible for its increased solubility and enhanced dissolution rate. The particle size is reduced to its absolute minimum during entrapment of drug which causes increased surface area and effective dissolution rate (6–8). The amorphous state is considered to be kinetically frozen, it is thermodynamically more active and possess greater chemical reactivity; because of which, they have a tendency to revert back spontaneously to the crystalline phase (8,9). Scale-up of solid dispersion technique has been the greatest limitation in its development as a formulation tool. One of the reasons for this is maintenance of physicochemical properties of the drug during processing and manufacturing. Dosage form development is also a limitation in large scale since the polymers that are used for the preparation, e.g., polyethylene glycol (PEG), of Gelucires are waxy in nature and can soften and melt during tablet compression. Various shear forces, heating, and cooling cycles at large scale tends to incorporate reversion of properties and phase transition in the dispersion (10). Some of the problems of solid dispersion may be overcome by using surface-active agent and self-emulsifying carriers. Most commonly used carriers for the preparation of solid dispersion are different grade of polyethylene glycols and polyvinylpyrrolidone (PVPs), Gelucire 44/14, Labrasol, sugars, and urea. (11–14). Recently, the advent and popularity of various surface-active carriers has increased. Complete dissolution of the drug has been achieved from solid dispersion by using a surface-active carrier. These vehicles act by dispersing or self-emulsifying the drug particles. Therefore, they prevent the formation of any water insoluble surface layers. When concentration of liberated drug exceeds its saturation solubility, the drug remains undissolved in the medium; however, with the use of surface-active agent, the drug can be emulsified into a finely divided state. The high surface area of the drug produced facilitates its dissolution (10). Law et al., in 2000 (5), prepared the solid dispersion of ritonavir by the solvent evaporation method, vacuum drying using PEG 8000 as a carrier. The main lacuna observed in this study was that 100% release of drug was obtained when the carrier was in the ratio 1:9. As the minimum titrable dose is 100 mg (total dose being 1,200 mg), at the ratio of 1:9 (drug : polymer) the total mass will amount to 1,000 mg with carrier alone and other excipients for tablet and capsule will amount more.
The main objective of the study was to develop solid dispersion of ritonavir by different methods in order to enhance bioavailability and at the same time not to have bulky formulation so that patient compliance is achieved. Apart from this, the developed formulation was investigated for its performance by in vitro dissolution studies in 0.1 N HCl and biorelevant media. The enhancement of bioavailability was established by in vivo studies in either sex of albino rats.
MATERIAL AND METHOD
Material
Ritonavir was received as a gift sample from Hetero drug limited (Hyderabad, India). Poloxamer 188 (Lutrol F 68) and PEG-PVA Co-Carrier were kind gifts from BASF (Aktiengesallscheft ludwigshafen, Germany) and Gelucire 44/14 from Colorcon Asia Pvt. Ltd. (Goa, India). Polyethylene glycol 4000 and 6000, PVP K-30, and HPMC (15 cps) were procured from S.D. Fine Chemicals Ltd. (Mumbai, India). Sorbitol was purchased from Thomas Baker (Mumbai, India). Other chemicals and reagents used were purchased from Merck Limited (Mumbai, India) and were of analytical grade.
Phase Solubility Study
In order to determine the most suitable carrier for ritonavir, phase solubility studies with different carrier like Poloxamer, PVP K30, PEG 4000 and 6000, PEG-PVA copolymer, sorbitol, HPMC and Gelucire 44/14 were carried out. Carrier solutions of different concentrations were prepared with distilled water as shown in Table I. Excess of drug was added to each carrier solution in glass vials and kept in biological shaker (Nirmal International, Delhi, India) for 72 h at room temperature. The contents were centrifuged at 4,000 rpm for 15 min and filtered with a 0.45-µm membrane filter; absorbance was taken with UV spectrophotometer at λmax of 238 nm after dilution. Since Gelucire was insoluble at higher concentrations in water, solubility was determined only for 0.5% concentration.
Table I.
Solubility Data of Ritonavir with Various Carriers at Different Concentration
| (%) Carrier concentration | 0.5a | 2a | 5a | 10a | 15a |
|---|---|---|---|---|---|
| PVP K-30 | 70 | 70 | 120 | 180 | 231 |
| PEG 4000 | 60 | 120 | 210 | – | – |
| PEG 6000 | – | – | 12 | 30 | 60 |
| PEG-PVA | – | – | – | 0 | 0 |
| HPMC (15 cps) | 180 | 258 | –b | –b | –b |
| Sorbitol | 360 | 480 | 720 | 1140 | 1980 |
| Gelucire | 220 | – | – | – | – |
aSolubilty (μg/ml) in respective carrier concentration
bToo viscous
Preparation of Physical and Kneaded Mixture
In order to optimize drug to carrier ratio, physical mixture and kneaded mixture of drug with sorbitol and Gelucire in different ratios were prepared.
Physical mixture
Physical mixtures of drug with sorbitol and Gelucire were prepared separately at four different ratios 1:1, 1:2, 1:3, and 1:4. Accurately weighed 50 mg drug was taken and mixed with 50, 100, 150, and 200 mg of sorbitol and Gelucire, respectively. Sorbitol physical mixtures were prepared by triturating the powder mix in pestle mortar for 3 to 5 minutes. Subsequent to this, the mixture was passed through sieve number 60 having mesh size of 250 μm. Gelucire physical mixture could not be passed through sieve, since its physical mixture was hard wax, so it was used as it is. All the physical mixtures were filled into hard gelatin capsule (Size 2) and dissolution studies were carried out with USP apparatus 2 (Paddle type), 0.1 N HCl, 900 ml, 50 rpm, for 45 min at 37 ± 0.5°C.
Kneaded mixture
Kneaded mixture with two carriers was prepared in the same ratios as in the physical mixture. First, the drug and carrier were mixed together by trituration in pestle–mortar (glass). To this mixture, ethanol (99.9% pure) was added in a small quantity to make a coherent mass. Now, the coherent mass was passed through sieve number 22 having mesh size of 841 μm to obtain granules. These granules were dried in an oven for 20 min at 60°C. The granules for each ratio were filled into hard gelatin capsules (size 2) for dissolution studies at a similar condition to the physical mixture.
Preparation of Solid Dispersion
Ritonavir–Gelucire
Solvent Method (SE1)
The drug was weighed accurately (50 mg) and dissolved in 2 ml acetone (class 3 solvent) to obtain a clear transparent solution. Similarly, Gelucire was dissolved in 2 ml acetone separately. Two solutions were mixed and vortexed for 30 min. Subsequently, the solution was transferred to rotary evaporator (Kyoungki, South Korea) and vacuum dried at 45–50°C, 22 rpm. The dried mass was scrapped and the obtained semisolid waxy solid dispersion was stored in dessicator.
Melt Method (MM1)
Gelucire (200 mg) was melted in a china dish at 50°C. To the molten Gelucire, 50 mg of ritonavir was added and mixed thoroughly at same temperature. Cooling was done at two rates; firstly at room temperature and secondly by rapid cooling in an ice bath containing sodium chloride. However, the sample was allowed to cool rapidly which resulted in phase separation. The sample was then scrapped and was obtained as semisolid waxy substance. It was stored in desiccators.
Ritonavir–Sorbitol
Solvent Method (SE2)
Sorbitol (200 mg) was dissolved in 10% ethanol (class 3 solvent). Fifty milligrams ritonavir was dissolved in water:ethanol mixture (2:3). Sorbitol solution was added to the drug solution. Drug precipitated due to water, so more ethanol (0.5 ml) was added to it to make the solution clear. This clear solution was then vortexed for 30 min. Subsequently, it was vacuum dried in a rotary evaporator (Kyoungki, South Korea). The dried sample was scrapped, pulverized sieved with sieve no. 22 and kept in a dessicator.
Melt Method (MM2)
Sorbitol (200 mg) was melted at 120°C in a china dish and then 50 mg of the drug was added to molten mass with mixing for 20 min at 120°C. It was then allowed to cool at room temperature. When the mass solidified, it was scrapped, pulverized, sieved, and stored in a dessicator.
In Vitro Release Profile of Solid Dispersion
In vitro release profile of solid dispersion prepared by a different method was performed in dissolution apparatus (Hanson Research, CA, USA). Solid dispersion equivalent to 10 mg of ritonavir was filled into hard gelatin capsule. The dissolution test of capsules was performed at 37 ± 0.5°C by using 900 ml of 0.1 N HCl for 45 min in USP apparatus 2 (Paddle type) at 50 rpm and sinkers were used. At predetermined time intervals of 10, 20, 30, and 45 min, 5 ml of sample was withdrawn from vessels with the help of calibrated pipette and refreshed with the same amount of media. The sample withdrawn was filtered through the 0.45 µm filter and assayed spectrophotometrically (UV1601 Shimadzu, Kyoto, Japan) at 238 nm.
Characterization of Solid Dispersions
Differential Scanning Calorimetry
Thermal investigations of solid dispersion were performed by using Perkin Elmer pyres 6 DSC (MA, USA). In an aluminum pan, 7–10 mg of sample was placed and crimped with a lid containing a pin hole and kept in the differential scanning calorimetry (DSC) unit along with a similar pan as a reference. The sample was heated at the rate of 10°C/min from the temperature range of 30–200°C. Nitrogen was used as a purge gas and flow was adjusted to 50 ml/min.
FT-IR Spectroscopy
FT-IR spectra of solid dispersion were obtained with Shimadzu Biorad FT-IR system (Kyoto, Japan). The sample was dispersed in dry potassium bromide (5 wt.% of sample), ground well in mortar and pestle and potassium bromide (KBr) disk was prepared at a pressure of 1,000 psig. The disk was placed in the FT-IR sample holder and IR spectra, in absorbance mode, were obtained in the spectral region 4,000 to 500 cm−1 using the resolution 1 cm−1. The FT-IR spectra were also performed for the drug, carrier and physical mixture by same method.
Rapid Heat-Cooled DSC
Thermal transitions were also measured with rapid heat-cooled DSC (RHC-DSC; TA Instruments, New Castle, USA). Pure nitrogen gas, i.e., without water vapor, was purged through the sample cell continuously. The samples, weighing 5–7 mg, were analyzed in pierced-lid aluminum pans (Mettler-Toledo) which allows for removal of water from sample. Residual moisture was removed from the samples by pre-heating them to a temperature of about 10–20°C below the first glass transition for 30 min. Control experiments revealed that this procedure resulted in complete evaporation of all moisture and completely dried samples. The dried samples were scanned from 40 to 180°C with a heating rate of 10°C min−1 under nitrogen atmosphere. The inflection point in the step change visible in the reversing heat flow was taken as the Tg
Transmission Electron Microscopy
For the morphology and particle-size analysis of the solid dispersions, transmission electron microscopy (TEM; CM-10, Philips, Netherlands) was conducted, operating at 60–80 kV and capable of point-to-point resolution. Combination of bright field imaging at increasing magnification and diffraction modes were used to reveal the form and size of solid dispersion. In order to perform the TEM observations, solid dispersion was applied on carbon-coated grid with 2% phospho-tungestic acid (PTA) and was left for 30 s. The dried coated grid was taken on a slide and covered with a cover slip. The slide was observed under the electron microscope at different magnifications.
X-ray Diffraction
Lattice spacings were measured by X-ray diffraction using a Guinier camera XDC-700 (Stockholm, Sweden). A small sample in a rotating holder was exposed to CuKa (151.540598 Å) radiation (40 kV, 320 mA) from a PW solution 1720 X-ray generator (Philips-Eindhoven, The Netherlands) with a crystal monochromator. The experiment was carried out at room temperature. The direct beam was held for 2 s at 10 kV and 5 mA.
HPLC Analysis
HPLC analysis was performed using a reverse phase column (Supelco, 150×2.5 mm, C-18). The mobile phase consisted of acetonitrile (25 mM) and sodium acetate (25 mM) in the ratio of 70:30 v/v. Hexane sulfonic acid (25 mM) was added in the mobile phase to suppress the ionization of drug ritonavir. The pH of the mobile phase was adjusted to 4.0 with 37% HCl. Flow rate was 1 ml/min and retention time was 4.8 min. Analysis of ritonavir was performed at 239 nm with UV detector.
Dissolution Profile in FaSSIF and FeSSIF Media
In order to determine the effect of food on the absorption of ritonavir in the intestinal tract, the dissolution profile of solid dispersion was performed in FaSSIF and FeSSIF media.
FaSSIF Media
Blank fasted-state simulated intestinal fluid (FaSSIF) was prepared by dissolving 1.74 g of NaOH (pellets), 19.77 g of NaH2PO4.H2O, 30.93 g of NaCl in 5 L of purified water and pH was adjusted to 6.5 using 1 N NaOH. Sodium taurocholate (3.3 g) was dissolved in 500 mL blank FaSSIF. A volume of 11.8 mL of a solution containing 100 mg/mL lecithin in methylene chloride was added to this solution forming an emulsion. The methylene chloride was removed under vacuum at about 40°C. This resulted in a clear, micellar solution, having no perceptible odor of methylene chloride. After cooling to room temperature, the volume was adjusted to 2 L with blank FaSSIF.
FeSSIF Media
Blank fed-state simulated intestinal fluid (FeSSIF) media was prepared by dissolving 20.2 g of NaOH (pellets), 43.25 g of glacial acetic acid, and 59.37 g of NaCl in 5 L of purified water. The pH was adjusted to 5.0 with 1 N HCl. Sodium taurocholate (16.5 g) was dissolved in 500 mL of blank FeSSIF. A volume of 59.08 mL of a solution containing 100 mg/mL lecithin in methylene chloride was added to above solution, forming an emulsion. The methylene chloride was eliminated under vacuum at about 40°C. This resulted in a clear, micellar solution having no perceptible odor of methylene chloride. After cooling to room temperature, the volume was adjusted to 2 L with blank FeSSIF.
The dissolution was performed using a USP paddle apparatus, 500 ml FaSSIF and 1,000 ml of FeSSIF media at 50 ± 5 rpm, 37 ± 0.5°C (Hanson, CA, USA). A 5-ml sample was withdrawn at different time intervals of 10, 20, 30, 45, 60 min and refreshed with the same amount of media, filtered through 0.45 µm filter and analyzed spectrophotometrically at 252.5 nm for FaSSIF and 307.5 for FeSSIF condition.
In Vivo Study (Pharmacokinetics Parameter)
The in vivo studies were performed in order to compare the plasma profile of solid dispersion and pure drug in order to establish that the enhanced bioavailability was obtained with preparation of solid dispersion when compared with the drug. The study was conducted in Albino Wistar rats of either sex which weighed 150–200 g following oral administration. The research protocol of the animal experimentation was approved by the Institutional Animal Ethics Committee, Hamdard University, New Delhi, India. The animals were obtained from Central Animal House, Hamdard University and housed in animal house, Faculty of Pharmacy, Hamdard University, New Delhi, India. The animals were housed in polypropylene cages with free access to standard laboratory diet and water. The animals were divided into three groups, each having six Albino Wistar rats. The routine animal handling was performed according to Good Laboratory Practices.
The animal dose was calculated as 9.99 ≃ 10 mg/kg for rat for current study. The solid dispersions and drug were dissolved in 1 ml of gum acacia solution (2% w/v) and given orally using oral feeding sonde. The rats were anesthetized using ether, and blood samples (1–1.5 ml) were withdrawn from the tail vein at time intervals of 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h. The samples were collected in microcentrifuge tubes which were first rinsed with EDTA followed by addition of small quantity (4–8 mg) of powdered EDTA to the tube. Blood collected was mixed with anticoagulant properly by shaking and then centrifuged at 4,000 rpm for 20 min. The plasma was separated and stored at −20°C until drug analysis by HPLC.
Pharmacokinetic parameters such as AUC, Cmax, tmax, Kel, and t1/2, were calculated from plasma profile curve. All pharmacokinetic parameters were calculated individually for each subject in the group and the values were expressed as mean ± SD. The area under concentration time curve was calculated according to log trapezoidal method. The half life associated with elimination rate constant (Kel) was computed by the formula
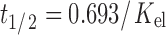 |
The pharmacokinetic data among different formulations were compared for statistical significance by the one-way ANOVA followed by Dunnett's multiple comparison tests where formulations were compared with pure drug using GraphPad Instat software (La Jolla, USA).
RESULT AND DISCUSSION
Phase Solubility Study
The solubility of ritonavir was determined in different carriers as shown in Table I. On the basis of solubility studies, two carriers ‘Gelucire and sorbitol’ were selected for formulation of solid dispersion because they showed highest increase in the solubility of the drug in aqueous solution. Sorbitol increased the solubility of ritonavir from 1 μg/ml in distilled water to 1980 μg/ml or 1.98 mg/ml in 15% solution, this indicated an increase by approximately 2,000 times. Similarly, Gelucire 44/14 increased the solubility by 220 times at only 0.5% carrier concentration which indicated that at higher concentration, more enhanced results could also be obtained.
Physical Mixture and Kneaded Mixture
For selection of drug and carrier ratio, in which the drug exhibits maximum solubility, physical mixture and kneaded mixture was prepared. Suitable ratio was optimized on the basis of dissolution studies (Figs. 1 and 2). The best release was shown by ritonavir–Gelucire and ritonavir–sorbitol in the ratio of 1:4. Thus, for the formulation of solid dispersion, 1:4 ratio was chosen. However, the physical mixture of ritonavir–sorbitol of 1:4 showed 92% release as compared to 68.2% in kneaded mixture. This might be attributed to the fact that sorbitol is insoluble in ethanol, which was used as solvent to make coherent mass. The physical mixture with sorbitol showed very high dissolution rate as compared to Gelucire. This can be because of alcoholic groups of sugar molecule, which might have given rise to strong interaction between drug and polymer and must have imparted hydrophilicity.
Fig. 1.

Dissolution profile of ritonavir:gelucire physical mixture RG1 (1:1), RG2 (1:2), RG3 (1:3), RG4 (1:4) and kneaded mixture KG1 (1:1), KG2 (1:2), KG3 (1:3), KG4 (1:4)
Fig. 2.

Dissolution profile of ritonavir:sorbitol physical mixture PS1 (1:1), PS2 (1:2), PS3 (1:3), PS4 (1:4) and kneaded mixture KS1 (1:1), KS2 (1:2), KS3 (1:3), KS4 (1:4)
In Vitro Release Profile of Solid Dispersion
The solid dispersions prepared by different methods equivalent to 10 mg ritonavir was filled into hard gelatin capsule and in vitro release profile (Fig. 3) was observed in dissolution apparatus. The result was separated into two clusters, one with solid dispersion prepared by using Gelucire as a carrier and another with sorbitol. Drug and carrier ratio were the same in both clusters (1:4). The maximum drug release was obtained 85.43% with SE1, using Gelucire as a carrier. The solid dispersion prepared with sorbitol (SE2) showed minimum release of 45.5%. It was also observed that the release profile of solid dispersion was dependent on the method of preparation. The SD prepared with the melt method for Gelucire showed less release (80.9%) while in case of sorbitol it shows maximum release (60.9%) as compared to solvent evaporation method. Now, for remaining studies, ritonavir–Gelucire preparations were considered because they showed better release profile as compared to ritonavir–sorbitol.
Fig. 3.

Dissolution profile of ritonavir and different solid dispersion prepared by solvent method containing gelucire ritonavir (SE1), sorbitol ritonavir (SE2) and melt method containing gelucire ritonavir (MM1), sorbitol ritonavir (MM2)
Characterization of Solid Dispersions
Differential Scanning Calorimetry
DSC scans of various solid dispersion of ritonavir were taken (Fig. 4). It was concluded on the basis of scans that DSC of the sample prepared by the solvent method and melt method showed absence of any drug peak at 120°C but presence of prominent peak of carrier with no change in melting point. This suggested the formation of a monotectic system where the melting point of the carrier is unchanged in the presence of drug. Significant change in delta H as compared to pure drug (96 J/g to 127 J/g for solvent evaporation and 96 J/g to 54 J/g for melt method) indicated interaction between carrier and drug, which also indicated formation of solid dispersion. The results suggested formation of the eutectic solid dispersion where the drug is present as ultrafine crystals in polymer matrix.
Fig. 4.

DSC of ritonavir (A), ritonavir–Gelucire solid dispersion by solvent method (B) and melt method (C)
FT-IR Spectroscopy
When FT-IR was performed, the drug spectrum indicated characteristic peaks at 3,480 cm−1 (N–H stretching amide group), 2,964 cm−1 (hydrogen-bonded acid within the molecule), 1,716 cm−1 (ester linkage), 1,645, 1,622, and 1,522 cm−1 (–C═C– stretching aromatic carbons). The FT-IR spectra (Fig. 5) of solid dispersion of ritonavir prepared by solvent evaporation and melt method was taken and analyzed. Two main band formations were observed between drug and carrier on the basis of FT-IR spectra. Intermolecular hydrogen bonding was observed due to the peak at 3,380 cm−1. A number of indistinguishable peaks appeared in the region of 3,400–4,000 cm−1 in the solid dispersion which was absent in the crystalline drug. A broad and strong absorption peak at 1,108 cm−1 indicated formation of secondary hydrogen bond which was absent in pure drug. The presence of strong absorption peak at 1,735 cm−1 and 2,720 cm−1 were indicative of characteristic C–H stretching band and C═O stretching band of aliphatic aldehydic group. This band was absent in the spectra of the crystalline drug. The hydrogen bonding and aldehydic group formation between drug and carrier indicated interaction between the two, which could be attributed to the cause of enhanced dissolution rate of the solid dispersion as compared to pure drug. In case of melt method, both the aldehydic group and hydrogen bond peaks were present but were very broad as compared to that obtained in solvent evaporation. The interaction between drug and carrier was due to hydrogen bond and aldehydic linkages resulting in the increase solubility of drug.
Fig. 5.

FT-IR spectra of ritonavir (A) ritonavir solid dispersion prepared by solvent evaporation (B) and melt method (C)
Rapid Heat-Cooled DSC
RHC-DSC has been designed specifically for operation at high scanning rates, i.e., up to 2,000°C/min in heating with similarly high cooling rates. The 6.3% weight loss detected between room temperature and 35°C is due primarily to water evolution. A clear step transition was visible in the scan of both the dispersions. The glass transition temperature of dispersion prepared by melt method was 95.11°C (Fig. 6) and for vacuum-dried product, it was 111.3°C which was much greater than the amorphous ritonavir having transition temperature of 51.5°C. This indicates that Gelucire was able to increase the transition temperature of ritonavir thereby increasing its stability.
Fig. 6.

Rapid heat-cooled DSC of ritonavir solid dispersion prepared by solvent method (A) and melt method (B)
Transmission Electron Microscopy
In order to determine the crystal shape and crystal size of eutectic solid dispersions, TEM analysis of the samples were performed. The drug in solid dispersion may be present in two forms: molecular dispersed, a more desirable state that yields high dissolution rate due to enhance solubility, or nanodispersion having particle size preferably lower than 500 nm (15). The particle size of solid dispersion prepared by melt method was in the range of 600–800 nm (Fig. 7) and in case of formulation prepared by solvent evaporation it was up to 60 nm (Fig. 8). The particle size of SD obtained with solvent method was lesser as compared to melt method, which was responsible for the enhanced dissolution of solid dispersion.
Fig. 7.

TEM scan of solid dispersion prepared by melt method (MM1)
Fig. 8.

TEM scan of solid dispersion prepared by solvent evaporation (SE1)
X-ray Diffraction
In order to investigate both the polymorphic transformation, which occurs during preparation of the solid dispersion, and the influence of carrier in the possible phase transformation, X-ray diffraction (Fig. 9) was performed for the physical mixture (a), solid dispersion prepared by solvent evaporation (b), melt method (c), and for ritonavir (d). Sharp and intense crystalline peaks at 2θ of 21.51° and 18.13° were observed in the diffraction spectrum of drug substance (ritonavir) and less intense and broadened peaks were observed in physical mixture as compared to the drug indicate transformation into an amorphous form. On the other hand, solid dispersion of solvent evaporation method and melt method showed the distinct broad peak that is observed in amorphous and highly disordered materials which correspond to diffraction pattern of Gelucire. The peaks of ritonavir were less intense, showing that some crystalline part of the drug was present in solid dispersion. This indicated that the solid dispersion were eutectic in nature where drug was present as ultrafine crystals in the carrier matrix.
Fig. 9.

X-ray diffraction patterns of different solid dispersion formulations (a) physical mixture (1:4), (b) ritonavir solid dispersion prepared by solvent evaporation, (c) ritonavir solid dispersion prepared by melt method (d) Pure drug ritonavir
Dissolution in FaSSIF and FeSSIF Media
In vitro dissolution in biorelevant media was performed in order to observe the effect of food on the absorption of ritonavir. These two biorelevant media, FaSSIF and FeSSIF (Fig. 10) were developed to simulate the condition in the intestine in the fasted and fed states. Dissolution results were expressed in terms of percent drug release to the time. The release of drug from solid dispersion of ritonavir and Gelucire prepared by solvent evaporation method showed maximum dissolution rate. The FeSSIF media indicated better percent drug release as compared to FaSSIF media, indicating the effect of food on the absorption of drug. So it was concluded that the formulation will be better absorbed when given with food. This study is concordant with the early study of Sekar et al., that the drug has food-related absorption. (16)
Fig. 10.

Percent release obtained from formulations MM1 and SE1 and ritonavir in FeSSIF* media and FaSSIF media
In Vivo Study
The pharmacokinetic parameters (Table II) and profile (Fig. 11) of ritonavir were determined after oral administration of pure ritonavir and solid dispersion prepared by solvent evaporation and melt method to Wistar rats. In the process of absorption of a drug dissolution is the rate-limiting step. Once the drug is available in the intestinal fluid as a dissolved form, it follows the transport across the gastrointestinal epithelium membrane. The peak plasma concentration (Cmax) of ritonavir in MM1, SE1, and pure drug were 2,462.2 + 36.51; 20,221.37 + 35.72; 1,354.8 + 39.47 ng/ml, respectively; while AUC0–t and AUC0–∞ were found to be 5,379.286, 12,649.9, 2,064.44 ngh/ml and 5,403.56, 12,688.12, 2,071.96 ngh/ml, respectively. These values indicated maximum plasma concentration and area under the curve was achieved by solid dispersion formulation prepared by solvent evaporation. The differences in bioavailability between solid dispersion prepared by solvent evaporation and melt method may be attributed to the differences in surface area of dispersed drug particles. The mean particle size of MM1 was 15–25-fold larger in comparison to SE1. The mean plasma concentration values obtained from different formulations were analyzed using one-way ANOVA and the values were found to be insignificant (p > 0.05). Thus, it is suggested that solid dispersion could exhibit enhanced bioavailability compared with pure ritonavir.
Table II.
Pharmacokinetic Parameters of Different Formulations and Pure Drug
| Pharmacokinetic Parameters | MM1 | SE1 | Pure drug |
|---|---|---|---|
| t max (hr) | 1 | 0.5 | 0.5 |
| C max (ng/ml) | 2,462.2 | 20,221.37 | 1,354.8 |
| AUC0-t (ng/ml h) | 5,379.286 | 12,649.9 | 2,064.45 |
| AUC0-∞ (ng/ml h) | 5,403.56 | 12,688.12 | 2,071.96 |
| K el (h−1) | 0.495 | 0.407 | 0.301 |
| t 1/2 (h) | 1.4 | 1.7 | 2.3 |
Fig. 11.

Plasma concentration profile of ritonavir after oral administration of different formulations and pure drug
The pharmacokinetic profile and parameters clearly show enhanced bioavailability of solid dispersion of ritonavir as compared to pure drug. The time taken to reach the Cmax was same for both formulation SE1 and pure drug, i.e., 0.5 h while it was more for MM1 i.e., 1.0 h. Maximum concentration in plasma was achieved by formulation SE1 which was approximately eight times more than the MM1 formulation and fifteen times more than pure drug indicating better release from formulation. Maximum area under the curve value for time 0 to t was found for SE1, i.e., 12,649.9 ngh/ml as compared to 5,403.56 ngh/ml from MM1 formulation indicating maximum bioavailability from SE1. The time taken for concentration of drug in plasma to decrease to 50% was 1.4 h for MM1, 1.7 h for SE1, and 2.3 h for pure drug. The elimination rate constant was found to be maximum for MM1 which was 0.495 h–1, and minimum for pure drug. The presence of surface-active carrier Gelucire which has bonded with the drug through hydrogen bonding and aldehydic linkages as well as the reduction in the drug particle size to nanometer range can be the reasons attributed to enhanced bioavailability in vivo.
CONCLUSION
Ritonavir and Gelucire form a eutectic mixture in the ratio of 1:4. In vitro dissolution studies show that release of ritonavir from solid dispersion is more than that of pure drug, which might be due to the nano size of dispersion. When the dissolution was performed in FaSSIF and FeSSIF media, maximum dissolution were obtained with FeSSIF media, which confirmed food-related absorption of drugs. The result indicated that the solid dispersion prepared by solvent evaporation method is a promising approach for the bioavailability enhancement of ritonavir and can be used for the solid dosage form development for oral use in order to commercialize.
Acknowledgments
The authors are thankful to Department of Science and Technology, Government of India for financial assistance to this project (SR/FT/L-28/2006)
References
- 1.Lipinski CA, Lombardo F, Dominyl BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 2.Dannenfelser R, He H, Joshi Y, Bateman S, Serajuddin ATM. Development of clinical dosage forms for a poorly water soluble drug I: Application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93:1165–75. doi: 10.1002/jps.20044. [DOI] [PubMed] [Google Scholar]
- 3.Mooter GVD, Augustijns P, Blaton N, Kinget R. Physico-chemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm. 1998;164:67–80. doi: 10.1016/S0378-5173(97)00401-8. [DOI] [Google Scholar]
- 4.Habib MJ. Pharmaceutical solid dispersion technology. Washington: CRC; 2000. pp. 35–78. [Google Scholar]
- 5.Law D, Krill SL, Schmitt EA, Fort JJ, Qiu Y, Wang W, Porter WR. Physicochemical considerations in the preparation of amorphous ritonavir-poly (ethylene glycol) 8000 solid dispersions. J Pharm Sci. 2000;90:1015–25. doi: 10.1002/jps.1054. [DOI] [PubMed] [Google Scholar]
- 6.Pignatello R, Ferro M, Guidi GD, Salemi G, Vandelli MA, Guccione S, Geppi M, Forte C, Puglisi G. Preparation, characterisation and photosensitivity studies of solid dispersions of diflunisal and Eudragit RS100 and RL100. Int J Pharm. 2001;218:27–42. doi: 10.1016/S0378-5173(01)00597-X. [DOI] [PubMed] [Google Scholar]
- 7.Babu GVM, Prasad DS, Murthy KVR. Evaluation of modified gum karaya as carrier for the dissolution enhancement of poorly water-soluble drug Nimodipine. Int J Pharm. 2002;234:1–17. doi: 10.1016/S0378-5173(01)00925-5. [DOI] [PubMed] [Google Scholar]
- 8.Hancock BC, Zografi G. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci. 1997;86:1–12. doi: 10.1021/js9601896. [DOI] [PubMed] [Google Scholar]
- 9.Gowariker VR, Viswanathan NV, Sreedhar J. Polymer science. New Delhi: New Age; 1986. pp. 150–79. [Google Scholar]
- 10.Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–66. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 11.Barker SA, Yap SP, Yuen KH, McCoy CP, Murphy JR, Craig DQM. An investigation in to structure and bioavailability of α-tocopherol in Gelucire 44/14. J Control Release. 2003;91:477–88. doi: 10.1016/S0168-3659(03)00261-X. [DOI] [PubMed] [Google Scholar]
- 12.Nilüfer Y, Aysegül K, Yalçın Ö, Ayhan S, Sibel AÖ, Tamer B. Enhanced bioavailability of piroxicam using Gelucire 44/14 and Labrasol: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2003;56:453–9. doi: 10.1016/S0939-6411(03)00142-5. [DOI] [PubMed] [Google Scholar]
- 13.Ozkan Y, Doganay N, Dikmen N, Isimer A. Enhanced release of solid dispersions of etodolac in polyethylene glycol. Farmaco. 2000;55:433–8. doi: 10.1016/S0014-827X(00)00062-8. [DOI] [PubMed] [Google Scholar]
- 14.Damian F, Blaton N, Naesens L, Balzarini J, Kinget R, Augustijns P, Van den Mooter G. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci. 2000;10:311–22. doi: 10.1016/S0928-0987(00)00084-1. [DOI] [PubMed] [Google Scholar]
- 15.Kanaze FI, Kokkalou E, Niopas I, Georgarakis M, Stergiou A, Bikiaris D. Novel drug delivery systems for flavonoid compounds with enhanced solubility based on solid dispersions with polyvinylpyrolidone and polyethyleneglycol. J Appl Polym Sci. 2006;102:460–71. doi: 10.1002/app.24200. [DOI] [Google Scholar]
- 16.Sekar V, Kestens D, Spinosa-Guzman S. The effect of different meal types on the pharmacokinetics of Darunavir (TMC114)/Ritonavir in HIV-Negative Healthy Volunteers. J Clin Pharmacol. 2007;47:479–84. doi: 10.1177/0091270006298603. [DOI] [PubMed] [Google Scholar]