

Abstract
BACE1 is required for the release of β-amyloid (Aβ) in vivo, and inhibition of BACE1 activity is targeted for reducing Aβ generation in Alzheimer's patients. To further our understanding of the safe use of BACE1 inhibitors in human patients, we aimed to study the physiological functions of BACE1 by characterizing BACE1-null mice. Here, we report the finding of spontaneous behavioral seizures in BACE1-null mice. Electroencephalographic recordings revealed abnormal spike-wave discharges in BACE1-null mice, and kainic acid-induced seizures also occurred more frequently in BACE1-null mice compared with their wild-type littermates. Biochemical and morphological studies showed that axonal and surface levels of Nav1.2 were significantly elevated in BACE1-null mice, consistent with the increased fast sodium channel current recorded from BACE1-null hippocampal neurons. Patch-clamp recording also showed altered intrinsic firing properties of isolated BACE1-null hippocampal neurons. Furthermore, population spikes were significantly increased in BACE1-null brain slices, indicating hyperexcitability of BACE1-null neurons. Together, our results suggest that increased sodium channel activity contributes to the epileptic behaviors observed in BACE1-null mice. The knowledge from this study is crucial for the development of BACE1 inhibitors for Alzheimer's therapy and to the applicative study of epilepsy.
Introduction
BACE1 was identified as the Alzheimer's β-secretase, which cleaves amyloid precursor protein (APP) at the N-terminal end of the β-amyloid peptide (Aβ) (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000). Aβ is the major proteinaceous component of amyloid plaques, and its excessive accumulation leads to amyloid deposition and the onset of Alzheimer's disease (AD) (Tanzi and Bertram, 2005). When AD animal models such as transgenic mice expressing familial mutant APP were bred with BACE1-null mice to produce APP transgenic mice with complete deficiency of BACE1, the production of Aβ was abolished and amyloid deposition was no longer detectable (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001). Hence, inhibition of the enzymatic activity of BACE1 is regarded as one of the most promising targets for treating AD patients.
As a type I transmembrane aspartyl protease, BACE1 has been long predicted to cleave many membrane-bound cellular substrates that may share similar membrane topology with APP (Yan et al., 2001). Indeed, it has been shown that BACE1 can process type I transmembrane α2,6-sialyltransferase (Kitazume et al., 2001), P-selectin glycoprotein ligand-1 (Lichtenthaler et al., 2003), the APP homolog proteins APLP1 and APLP2 (Li and Südhof, 2004; Pastorino et al., 2004), LRP (low-density lipoprotein receptor-related protein) (von Arnim et al., 2005), the voltage-gated sodium channel β-subunits (Kim et al., 2005; Wong et al., 2005), neuregulin-1 (Hu et al., 2006; Willem et al., 2006), and neuregulin-3 (Hu et al., 2008). Abolished cleavage of type III neuregulin-1 (and perhaps neuregulin-3) is suggested to cause hypomyelination of nerves in the developmental central (Hu et al., 2006) and peripheral (Hu et al., 2006; Willem et al., 2006) nervous systems as well as delayed remyelination of adult sciatic nerves (Hu et al., 2008). Schizophrenia-like behaviors seen in BACE1-null mice may also be related to the abolished cleavage of neuregulin-1 (Savonenko et al., 2008) because genetic mutation of neuregulin-1 is linked to the pathogenesis of schizophrenia (Buonanno et al., 2008; Mei and Xiong, 2008). Moreover, BACE1-null mice exhibit altered hippocampal synaptic plasticity as well as decreased cognitive performance (Laird et al., 2005). All of these observations imply that BACE1 plays multiple roles in physiological processes.
While exploring the biological functions of BACE1, we have found that variable but significant numbers of BACE1-null mice spontaneously develop epileptic seizures. These behavioral seizures are found in both young and old BACE1-null mice, and the duration of the observed tonic-clonic seizures in these mice lasts no more than 25 s. The finding of this phenotype prompted us to compare neuronal activities of BACE1-null mice to those in their wild-type littermates by using electrophysiological, biochemical, and immunohistochemical approaches. Here, we demonstrate that BACE1-null mice express higher surface levels of voltage-gated sodium channel Nav1.2 subunits in their hippocampal areas than their wild-type littermates. Consistently, neuronal activity and excitability were also elevated in BACE-null hippocampal neurons. More importantly, we present the evidence that hippocampal neuronal loss appears in aged BACE1-null mice. Because of these more severe phenotypes observed in BACE1-null mice, we suggest that caution is required during the development of therapeutic strategies designed to inhibit BACE1 in Alzheimer's patients, who appear to be more vulnerable to epileptic seizures.
Materials and Methods
BACE1-null mice.
Generation of BACE1-null mice was described by Cai et al. (2001). The primers (HC69, 5′-AGGCAGCTTTGTGGAGATGGTG; HC70, 5′-CGGAAATCGGAAAGGCTACTCC; and HC77, 5′-TGGATGTGGAATGTGTGCGAG) were used to genotype BACE1-null mice (Cai et al., 2001). The primer pair (5′-TGAAAAGCTGCACCTCATTG and 5′-CTGGTTGACCTGAGCTGTGA) was used to amplify a digoxigenin-labeled 544 bp DNA fragment for Southern blotting (Roche Applied Science), which was used to ensure complete deletion of BACE1 in BACE1-null mice. Animals that exhibited behavioral seizures were genotyped again by Southern blotting. All experimental protocols were approved by the Animal Care and Use Committee at the Lerner Research Institute in compliance with the guidelines established by the Public Health Service Guide for the Care and Use of Laboratory Animals.
Video-electroencephalogram/electromyogram monitoring.
Wild-type (n = 8) and BACE1-null mice (n = 8) at 6 months of age were surgically implanted for electroencephalogram (EEG)/electromyogram (EMG) monitoring according to the procedures described in the operation manual from Pinnacle Technology. Briefly, mice were anesthetized with 40 mg/kg sodium pentobarbital and held in a stereotaxic frame fitted with a mouse adaptor (David Kopf Instruments). The skull was exposed, cleaned of all connective tissue, and dried. The head mount was fixed to the dry skull with a small amount of cyanoacrylate, and four holes were drilled anterior and posterior to bregma bilaterally [anterior-posterior (AP), 2.0 mm, medial-lateral (ML), ±1.3 mm; and AP, −3.5 mm, ML, ±1.3 mm]. A pair of anterior-hole screws (0.1”) and a pair of posterior-hole screws (0.12”) were tightened down to the mouse skull. Then, two EMG wire electrodes were inserted contralaterally into the nuchal musculature using blunt dissection techniques. Then, dental ceramic compound was applied over the screws, wires, the entire base of the head mount, and the exposed skullcap. After surgery, the mouse was removed from the stereotaxic frame and allowed to recover from anesthesia. After 2 weeks of recovery, a preamplifier was connected with the mouse's head mount and a commutator, which was attached to the Data Acquisition and Control System (Pinnacle Technology) to allow video-EEG/EMG recording for 24 or 72 h while the mouse was freely moving in a plastic cage. Amplified EEG and EMG signals were digitally collected, processed, and viewed in a real-time manner with the Sirenia software package (Pinnacle Technology), which permitted the analysis of the behavioral aspects of all the seizures during each experiment. Epileptiform activity was manually scored. To test chemically induced seizures, the animals were first recorded for 10 min before the administration of kainic acid (KA) via an intraperitoneal injection. The animals receiving kainic acid (10 mg/kg, i.p. injection) were continuously recorded for 4 h immediately after the injection.
Kainic acid seizure induction.
For monitoring inducible behavioral seizures, wild-type (n = 11; females) and BACE1-null mice (n = 11; females) at 4 months of age were injected intraperitoneally with kainic acid according to previously described procedures with a slight modification to a dose of 15 mg/kg (Racine, 1972). All mice were injected between 1:00 P.M. and 5:00 P.M. to minimize behavioral variation attributable to the circadian rhythm. After injection of kainic acid, mice were observed for 2 h and scored according to the following criteria: stage 0, no response; stage 1, freezing, staring, mouth or facial movements; stage 2, rigid posture, head nodding, or isolated twitches; stage 3, tail extension, unilateral-bilateral forelimb clonus, or repetitive scratching; stage 4, rearing with one or both forepaws extended; stage 5, clonic seizures with loss of posture, jumping, and falling; stage 6, severe tonic-clonic seizures.
Western blotting and antibodies.
Protein extraction was performed according to previously described procedures (Kim et al., 2007). The hippocampal samples were homogenized in 0.5 ml of disrupting buffer [10 mm Tris-HCl at pH 6.8, 1 mm EDTA, 150 mm NaCl, and a protease inhibitor mixture (Roche)] and centrifuged at 100,000 × g for 1 h. Then, the pellets were homogenized in SDS extraction buffer (10 mm Tris-HCl at pH 6.8, 2% SDS, 1 mm EDTA, 150 mm NaCl, and a protease inhibitor mixture). Equal amounts of protein were resolved on a NuPAGE Bis-Tris Gel (Invitrogen) and transferred onto nitrocellulose membranes (Invitrogen). Subsequently, blots were incubated with primary antibodies (Nav1.1, 1:200; Nav1.2, 1:200; Nav1.6, 1:200; Millipore Bioscience Research Reagents; PS1, 1:1000; Calnexin, 1:1000; Sigma-Aldrich) overnight at 4°C. After extensive washing, the blots were reacted with HRP-conjugated secondary antibodies and visualized using enhanced chemiluminescence (Thermo Fisher Scientific).
Culture of primary neurons.
Neuronal cultures were derived from day 15 mouse embryos according to procedures previously described (Qahwash et al., 2003). Briefly, the hippocampi were dissected from embryonic brains and incubated for 15 min in trypsin/EDTA (0.05%/0.02% in PBS) at 37°C. Then, the samples were rinsed twice in PBS, centrifuged at 800 rpm for 3 min, added to the dissociation medium (DMEM with 10% FBS, 10 mm HEPES, 44 mm glucose, 100 U/ml penicillin plus streptomycin, 2 mm l-glutamine, 2 μm insulin), mechanically dissociated by repeated passage through a fire-polished Pasteur pipette followed by filtration through 40 μm nylon mesh, centrifuged (800 rpm for 3 min at 25°C), and re-dissociated in the growth medium (Neurobasal medium with 2% B27, 0.5 mm l-glutamine, 25 μm glutamate, 0.2 mm uridine, 0.1 mm deoxyuridine, 100 U/ml penicillin plus streptomycin). Dissociated neurons were plated onto poly-l-lysine-coated coverslips at a density of 2.0 × 104 cells/cm2, cultured for 10 d in growth medium at 37°C with 5% CO2, and fed three times a week by replacing one-half of the medium. After that, the cultured neurons were processed for immunofluorescence.
Immunofluorescent confocal microscopy.
Confocal experiments were performed according to standard methods as previously described (He et al., 2004). After transcardial perfusion with 4% paraformaldehyde, the mouse brain was surgically removed and immersed in 20% sucrose overnight at 4°C. Then the brain was sagittally cut into 12-μm-thick sections on a freezing microtome (Microm). Sections were permeabilized with 0.3% Triton X-100 containing 0.3% H2O2 for 30 min. After being rinsed in PBS three times to remove the detergent, the sections were treated by microwave in 0.05 m citrate-buffered saline, pH 6.0, for 5 min, blocked with 5% normal goat serum, and incubated with individual primary antibodies at the following dilutions: Nav1.1, 1:100; Nav1.2, 1:100; Nav1.6, 1:100; and NeuN, 1:1000 (Sigma-Aldrich). After washing with PBS three times, sections were incubated with secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 568 (Invitrogen).
Acute dissociation of hippocampal neurons.
Hippocampal neurons were acutely dissociated from postnatal day 21 to 30 mice by following standard procedures (Chen et al., 2002; Yu et al., 2006). Mice were decapitated under anesthesia and hippocampal slice (400 μm) was quickly cut in a low-calcium, ice-cold buffer (in mm: 140 sodium isethionate, 3 KCl, 4 MgCl2, 0.1 CaCl2, 23 glucose, 15 HEPES, pH 7.4). Slices were then incubated for 1–6 h in NaHCO3-buffered Earle's balanced salt solution (Sigma-Aldrich) oxygenated with 95% O2/5% CO2. CA1 region of hippocampus was dissected under a microscope and placed in a treatment chamber containing protease type XIV (1.5 mg/ml; Sigma-Aldrich). After 20–30 min treatment, the tissue was rinsed and triturated with a series of fire-polished glass pipettes. The cell suspension was plated on a poly-dl-lysine-precoated 12-mm-diameter glass coverslip (Thermo Fisher Scientific), and the coverslip was transferred to a recording chamber after sitting for 5 min.
Whole-cell recording.
Whole-cell recording was performed on acutely dissociated hippocampal pyramidal neurons from postnatal day 21 to day 30 mice by using EPC10 USB patch master amplifier with PatchMaster (HEKA Elektronik) and Chart5 (ADInstruments). Patch pipettes (∼3–5 MΩ) were made from borosilicate glass (G150) with a Sutter P-97 puller (Sutter Instrument). Data were collected in whole-cell voltage-clamp or current-clamp mode, sampled at 10 kHz, and filtered at 3 kHz. Access resistance was <10 MΩ before the series resistance compensation (75–80%). For voltage-clamp experiments, extracellular bath solution contained the following (in mm): 30 NaCl, 100 choline chloride, 10 tetraethylammonium chloride, 3 KCl, 1.3 MgCl2, 1.8 CaCl2, 0.1 CdCl2, 10 HEPES, 25 glucose, pH 7.4, adjusted with NaOH; patch pipette solution: 110 cesium methanesulfonate, 20 tetraethylammonium chloride, 1 NaCl, 2.5 MgCl2, 9 EGTA, 10 HEPES, 3 Na2ATP, 0.3 Na3GTP, pH 7.3, adjusted with CsOH. For current-clamp experiments, extracellular solution contained the following (in mm): 140 NaCl, 3 KCl, 1.3 MgCl2,1.8 CaCl2, 0.1 CdCl2, 10 HEPES, 25 glucose, pH 7.4, adjusted with NaOH; patch pipette solution: 130 potassium gluconate, 0.2 EGTA, 10 HEPES, 3 Na2ATP, 0.3 Na3GTP, pH 7.3, adjusted with KOH. All recordings were performed at room temperature. After establishing the whole-cell mode, pyramidal neurons (identified as cells with triangle or fusiform shape) were held at −70 mV. Peak Na+ current was recorded by the step depolarization between −60 and +20 mV in an increment of 10 mV. Peak current (in picoamperes) at each command potential was normalized to the capacitance of the cell (in picofarads) and plotted against the corresponding potential (in millivolts). The sodium conductance (G) at a given command potential (V) was calculated with the following equation: G = I/(V − Erev), and then normalized to the peak conductance (Gmax) and plotted against V. Erev, the sodium channel reverse potential, equals +33.9 mV calculated under our experimental conditions. To determine the voltage dependence of Na+ channel inactivation, neurons were held at a prepulse potential between −120 and 0 mV for 500 ms followed by a constant depolarization pulse at 0 mV for 30 ms. Data were fitted by the following Boltzmann equation: G/Gmax = 1/(1 + exp((V1/2 − V)/k)) (for activation curve); or I/Imax = 1/(1 + exp((V − V1/2)/k)) (for inactivation curve). V1/2 is the potential at which one-half of the Na+ channels are activated or inactivated, respectively, and k is the slope (in millivolts).
For current experiments, pyramidal neurons were held at −80 mV, and their intrinsic firing properties were recorded in response to 800 ms depolarization current injection with an interpulse interval of 5 s. The action potential number versus current injection, action potential threshold, half-width, peak amplitude, maximum slope, minimum voltage, and input resistance were compared. The threshold was defined as the first action potential during the depolarizing current injection as the voltage corresponding to the peak of the third differential of the action potential. Half-width was measured at half-amplitude of action potential. Input resistance was determined from the slope of I–V curve, which was generated from a series of hyperpolarizing current injection. Data analysis was performed using PatchMaster, Chart 5, and Origin 6 (OriginLab), and the results were expressed as mean ± SEM.
Extracellular field potential recording.
Brain slices were prepared following a previously described protocol (Baba et al., 2003). In brief, horizontal hippocampal slices (350 μm thickness) were prepared from the brains of 6-month-old wild-type and BACE1-null mice in ice-cold, 95% O2/5% CO2 oxygenated artificial CSF (aCSF) consisting of the following (in mm): 124 NaCl, 3 KCl, 1.24 KH2PO4, 1 MgSO4, 2.0 CaCl2, 26 NaHCO3, and 10 glucose. After a 1–2 h recovery period, the slice was placed onto the center of a MED-P515A probe (Panasonic International) with 64 embedded recording sites and perfused with aCSF. 4-Aminopyridine (4-AP)-induced extracellular field potentials were recorded using a MED64 multichannel recording system, and data were collected at a 20 kHz sampling rate. Data were collected from the dentate gyrus region and then analyzed with Mini Analysis 5.6 (Synaptosoft) to quantify burst frequency, amplitude, and the total burst area per slice by using the predefined “burst analysis” function. A burst is defined as two or more continuous positive-going discharges. Amplitude was calculated from the first positive-going discharge from the baseline within a population burst, and burst area was calculated by the predefined fitting method in Mini Analysis.
Results
Epileptic phenotypes of BACE1-null mice
To explore the physiological roles of BACE1 in vivo, we set out to examine mouse phenotypes associated with complete deficiency of BACE1. A variable but significant number of BACE1-null mice were found to have behavioral epileptic seizures. Typically, the initiation of seizures began with a tonic presentation of mice falling onto one side with stiff bodies, followed by a clonic presentation of shaking limbs. Before completely regaining control of their body movement, these mice tended to repeatedly lick their front paws for several seconds after shaking subsided, followed by eventual reorientation to a standing position (supplemental video, available at www.jneurosci.org as supplemental material). The consistent precipitating or provoking factors resulting in the generation of these seizures remain to be explored. However, sudden stress during cage changes sporadically induced seizures of BACE1-null mice. On induction under these conditions, BACE1-null mice often displayed the tonic-clonic movement pattern described above. The seizure duration typically lasted no more than 25 s. After a seizure, the animal remained responsive but relatively inactive for several minutes and then fully recovered. Similar behavioral tonic-clonic seizures were not seen in wild-type mice.
Continuous monitoring of entire colonies of BACE1-null mice for the past 4 years showed that ∼11% of BACE1-null mice developed behavioral seizures at younger than 1 month of age, 14.7% at 3–6 months of age, and 21.9% mice older than 10 months of age (Table 1). Consistent with our observation, another line of BACE1-null mice also displayed behavioral epileptic seizures during a spatial memory test (Kobayashi et al., 2008), indicating that this epileptic phenotype is unlikely an artifact. In several cases, BACE1-null mice died immediately after seizures, with these mice being younger than 3 weeks of age. We also observed that ∼30% of BACE1-null mice died shortly after birth (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). It is unclear whether the infantile seizures actually increase this premature death rate. Overall, behavioral seizures in mice older than 4 months of age were more readily captured.
Table 1.
Summary of seizure occurrence
| Age |
|||
|---|---|---|---|
| <1 month | 3–6 months | 10–24 months | |
| No. of BACE1−/− mice | 226 | 143 | 82 |
| No. of seizure mice | 25 | 21 | 18 |
| % of epileptic seizure | 11.0 | 14.7 | 21.9 |
Since behavioral epileptic seizures were only detected in a small percentage of BACE1-null mice, we performed video-EEG recording of freely moving mice during a 24 h period to monitor their electroencephalographic activities. Overall, wild-type mice exhibited a low-amplitude baseline EEG as depicted in the recording (Fig. 1). In contrast, BACE1-null mice displayed variable types of spike discharges ranging from single, double, or multiple spikes (Fig. 1A) to varying longer durations of spike-wave discharges (Fig. 1B–E). Among eight recorded BACE1-null mice, all exhibited at least single or double spikes. Six of eight BACE1-null mice exhibited spike-wave discharges lasting 22.2 ± 36.5 s, whereas none of the wild-type mice exhibited similar discharges (Fig. 1B–D). We also noticed that two BACE1-null mice exhibited exceptionally long stretches of spike-wave discharges that lasted up to 230 s with the amplitude of these long wave discharges being smaller than single or double spikes (Fig. 1E). During video recording, one BACE1-null mouse exhibited behavioral epileptic seizures that were concurrent with a long episode of spike-wave discharges (Fig. 1B). Noticeably, one BACE1-null mouse only showed single or double spikes within 72 h of video-EEG recording even though this mouse showed behavioral epileptic seizures before it was assessed with video-EEG recording.
Figure 1.

Spontaneous epileptic seizures in BACE1-null mice. Representative EEG recordings shown from both BACE1+/+ and BACE1−/− mice. Spike amplitude (in microvolts on y-axis) is plotted as a function of recorded time course (in seconds on x-axis). Variable patterns of spiking can be seen, ranging from single or multiple spikes (A) to spike-wave discharges (B–E) in BACE1-null mice. All eight of the recorded BACE1-null mice showed at least some single spikes as shown in A, whereas only two of the wild-type mice showed occasional single spikes. The pattern of discharge seen in B was recorded in one mouse with spontaneous seizures. C–E, Six of the eight BACE1-null mice showed multiple spikes (C, D) or sustained spike-wave discharge patterns (E). The scale bar is shown on the bottom right.
Quantitative analysis of EEG results revealed that single spike discharge occurred at a frequency of ∼20 times/h, whereas double or triple spikes discharges occurred at ∼2 times/h. The amplitude was usually >200 μV in BACE1-null mice (Table 2). Interestingly, a lower amplitude of spike-wave discharges (∼100 μV) occurred more frequently in BACE1-null mice (∼40 times/h), with the average duration lasting ∼22 s. In contrast, only single spikes but no spike-wave discharges occurred in wild-type littermates (4.5 times/h) (Table 2). Together, we found that BACE1-null mice mainly exhibited high-amplitude single or double spikes and low-amplitude spike-wave discharges, with only a small percentage of mice (one in eight) exhibiting high amplitude (200–400 μV) spike-wave discharges.
Table 2.
Summary of EEG results
| Single spike |
Double spikes |
Triple spikes |
Spike-wave discharges |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Amp (μV) | Fr (n/h) | Amp (μV) | Fr (n/h) | Amp (μV) | Fr (n/h) | Amp (μV) | Fr (n/h) | Duration (s) (range) | |
| BACE1−/− | 221.4 ± 61.2 | 20.0 ± 5.4 | 228.6 ± 54.3 | 2.4 ± 0.3 | 211.2 ± 54.5 | 2.0 ± 0.3 | 98.8 ± 9.2 | 40 ± 8.6 | 22.2 ± 36.5 (1–230) |
| BACE1+/+ | 178.6 ± 54.3 | 4.5 ± 1.2 | 0 | 0 | 0 | ||||
Spike-wave discharges are defined as three or more single spikes scored within the period of 1 s. Mean ± SD of amplitude (Amp), frequencies (Fr), and duration (in seconds) of the discharges are displayed. Frequencies are defined as the average occurrence of discharges per hour. The baseline of EEG in BACE1-null mice was ∼50 μV, whereas that of wild-type mice was ∼20 μV.
Lowered threshold for KA-induced epileptiform activity in BACE1-null mice
The finding of silent epileptic seizures in BACE1-null mice prompted us to examine whether BACE1-null mice were more susceptible to kainate-induced seizures. We injected 4-month-old BACE1-null mice and their age-matched wild-type littermates intraperitoneally with KA, a kainate glutamate receptor agonist, according to a previously described procedure (Wu et al., 2005). After the administration of KA, mice were observed for 2 h, and seizure severity was assessed using a modified Racine scale in which a score of “0” indicates no response and a score of “6” indicates severe tonic seizures (Racine, 1972). At the dose of 15 mg/kg KA, 10 of 11 treated BACE1-null mice progressed to stage 5 of the scale, which is generalized clonic seizures, within 20 min; 8 further progressed to stage 6, defined as severe tonic seizures, within the 2 h limit (Table 3) (n = 11). The remaining BACE1-null mouse showed only spasms in its rearing limbs. In contrast, only 4 of 11 wild-type mice reached stage 6 within 2 h of observation, and the remaining 7 mice were rated between stages 2 and 5 (Table 3). Clearly, BACE1-null mice scored significantly higher than wild-type controls (5.6 ± 0.2 vs 4.8 ± 0.3; n = 11; p < 0.001).
Table 3.
Increased susceptibility of BACE1-null mice to kainic acid-induced seizures
| Genotype | Behavior | ||||||
|---|---|---|---|---|---|---|---|
| Response stage | |||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| No. of BACE1+/+ mice | 0 | 0 | 1 | 1 | 1 | 4 | 4 |
| No. of BACE1−/− mice | 0 | 0 | 0 | 0 | 1 | 2 | 8 |
Kainic acid (15 mg/kg) was injected intraperitoneally in 4-month-old wild-type (n = 11) and BACE1-null mice (n = 11). Scoring of kainic acid-induced seizures is based on the following criteria: 0, no response; 1, freezing, staring, mouth or facial movements; 2, rigid posture, head nodding, or isolated twitches; 3, tail extension, unilateral-bilateral forelimb clonus, or repetitive scratching; 4, rearing with one or both forepaws extended; 5, clonic seizures with loss of posture, jumping, and falling; 6, severe tonic seizures.
To further evaluate the KA-induced epileptic seizures, we performed video-EEG recording of 6-month-old BACE1-null mice and their age-matched wild-type littermates after administration of a lower dose of KA (10 mg/kg). After intraperitoneal injection, animals were immediately recorded for 4 h to monitor their EEG/EMG activities. EEG recording showed that BACE1-null mice displayed a typically longer duration of discharges compared with their wild-type littermates (Fig. 2A) (n = 6). Although quantitative analysis revealed no apparent difference in the latency of first convulsive discharges and first nonconvulsive discharges among both genotypes of mice (Fig. 2B), the duration of nonconvulsive discharges was significantly longer in BACE1-null mice compared with wild-type controls (Fig. 2C) (108 ± 11 vs 60 ± 9 s; n = 6; p < 0.01). Similarly, the duration of convulsive discharges in BACE1-null mice was significantly longer than that of wild-type littermates (Fig. 2C) (236 ± 65 vs 96 ± 8 s; n = 6; p < 0.01). Moreover, the intraburst frequency was also higher in BACE1-null mice than in their littermate controls (Fig. 2D) (4.1 ± 0.2 vs 2.9 ± 0.2 Hz; p < 0.01). Together, our results demonstrate that BACE1-null mice tend to have a lowered threshold for epileptiform activity after KA administration and that these mice are more susceptible to KA-induced seizures.
Figure 2.

Analysis of the EEG results from kainic acid-induced epileptiform activity. A, Female BACE1-null mice and wild-type controls at 6 months of age were surgically implanted for long-term EEG/EMG monitoring using a Pinnacle mice EEG/EMG recording system. After a recovery period of 2 weeks, mice were injected with kainic acid (10 mg/kg) and immediately recorded to monitor video-EEG/EMG activity for 4 h. Typical examples of EEG recordings from either genotype of mice are shown. The duration of spike-wave discharges in BACE1-null mice was visibly longer than in wild-type controls (top panels). The bottom panels are an enlarged view from top panels at the marked region, lasting for 10 s. B–D, The latency of the first nonconvulsive discharge and the first convulsive discharge, the duration of nonconvulsive discharges and convulsive discharges, as well as the frequency of spike-wave discharges were quantified and compared. BACE1-null mice exhibited a significantly longer duration of both convulsive and nonconvulsive discharges (C) and a significantly higher spike wave of discharges (D). N = 6; p < 0.01, Student's t test. Error bars indicate SEM.
Increased neuronal surface expression of voltage-gated sodium channels
To explore causal factors that lead to epileptic seizures in BACE1-null mice, we asked whether genetic deletion of BACE1 alters voltage-gated sodium channel activity, as abnormal sodium channel activity is often linked to behavioral seizures (Catterall, 2002; Stafstrom, 2007). Voltage-gated sodium channels consist of a heterotrimeric complex of one 260 kDa α-subunit and one or two auxiliary β-subunits (Catterall, 2000). The type I transmembrane β-subunits are suggested to be BACE1 substrates (Kim et al., 2005; Wong et al., 2005). The β-subunits are known to modulate channel gating and surface expression of the α-subunit, which forms an ion-conducting pore and senses voltage charges to gate the channel (Isom, 2002; Yu et al., 2005). More relevantly, a recent study suggests that the BACE1-cleaved intracellular domain of the β-subunit can regulate expression of α-subunits (Kim et al., 2007).
To determine whether genetic ablation of BACE1 potentially affects the expression and/or trafficking of sodium channel α-subunits, we first examined immunoblots of hippocampal lysates from 4-month-old mice with antibodies specific to three sodium channel proteins expressed in adult hippocampus. The initial Western blots of brain lysates extracted with 2% SDS showed significantly reduced sodium channel proteins in BACE1-null brain lysates (Fig. 3A,B). This reduction was repeatedly observed and appeared specific to the sodium channel proteins, because another multispanning transmembrane protein presenilin 1 exhibited no obvious differences under the same extraction conditions. Since the expression of sodium channel proteins is elevated in BACE1-overexpressing transgenic mice (Kim et al., 2007), it appears consistent that the total levels of sodium channel proteins in BACE1-null mice were lower than wild-type controls.
Figure 3.

Altered expression of Nav1.1, Nav1.2, and Nav1.6 in BACE1-null mice. A, Membrane proteins were extracted from mouse hippocampi by 2% SDS extraction buffer and equal amounts of protein from two genotypes of samples were resolved by electrophoresis for Western blot analysis with specified antibodies. The levels of sodium channel proteins were lower in BACE1−/− samples than in BACE1+/+ samples. The levels of the multispanning transmembrane protein PS1 were similar with both treatment conditions. Calnexin was used to verify the loading of each sample. B, Bar graphs represent a relative quantification of Nav1.1, Nav1.2, and Nav1.6 protein levels based on the results in A. Error bars indicate SEM; p < 0.01.
More intriguingly, this previous study demonstrates that the surface expression of sodium channel proteins is actually reduced even though the total protein levels are increased in BACE1-overexpressing transgenic mice (Kim et al., 2007). To examine whether genetic deletion of BACE1 alters the surface expression of sodium channel proteins, we performed confocal staining of brain sections with antibodies to sodium channel proteins. We reproducibly observed a visible increase in staining of NaV1.2 in the BACE1-null hippocampal mossy fibers compared with wild-type controls (Fig. 4A) (n = 6). As demonstrated in Figure 4A, the overall intensity of NaV1.2 immunoreactivity in the cell body was comparable among two genotypes of brain samples, but the increased staining intensity was obviously manifested in the axonal area highlighted with dashed lines. Sodium channel NaV1.2 is normally localized in dendrites, unmyelinated and premyelinated axons (Westenbroek et al., 1992; Boiko et al., 2001; Kaplan et al., 2001). The detection of NaV1.2 in mossy fibers is consistent with its axonal localization. Hence, our result suggests higher expression of NaV1.2 in this region.
Figure 4.
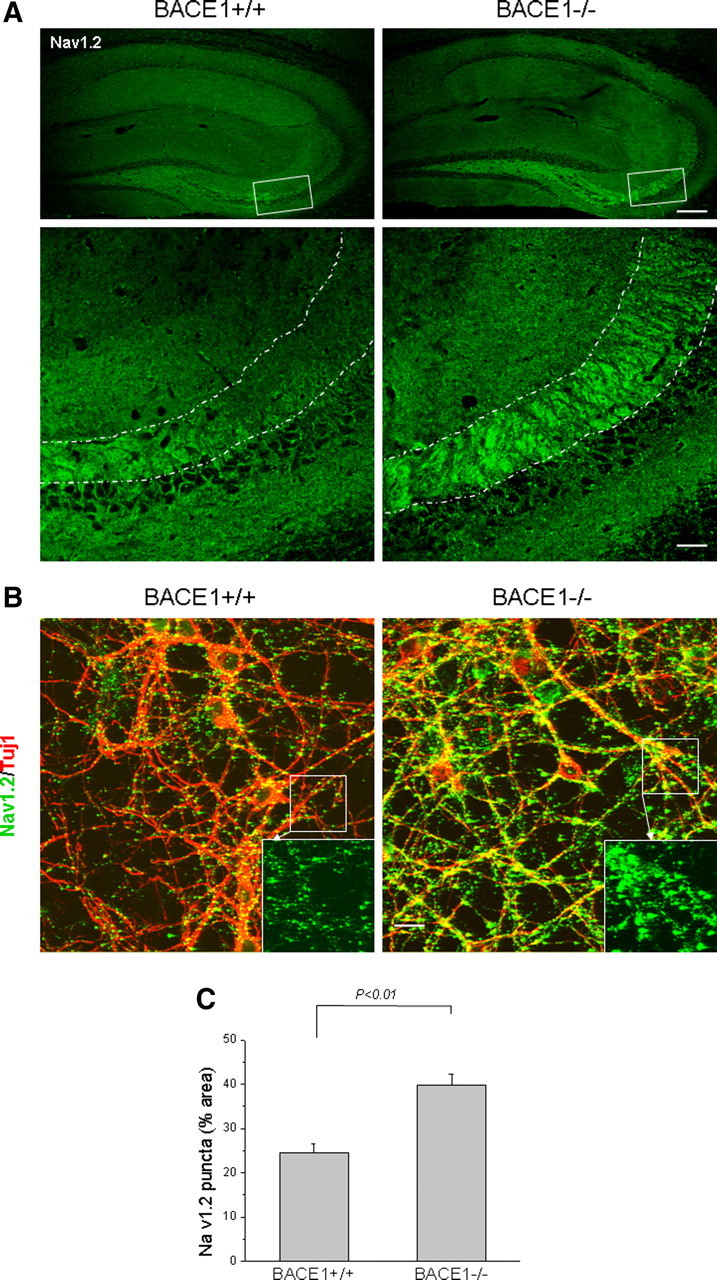
Confocal staining of Nav1.2 protein. A, Fixed brain slices were immunoreacted with Nav1.2 antibody followed by confocal examination. The boxed areas in top panels were enlarged and presented in the respective bottom panels. Nav1.2 is mostly localized in dendrites and unmyelinated axons. The dotted lines highlight areas of mossy fibers, which are clearly different between the two genotypes. Scale bars: top row, 125 μm; bottom row, 30 μm. B, Neurons were cultured from embryonic hippocampus on coverslips for confocal examination with Nav1.2 antibody. Antibody Tuj1 was used to mark axons in cultured neurons. The enlarged views of each specified inset are shown in the corresponding right bottom corner. Scale bar, 15 μm. C, ImageJ software was used to quantify the Nav1.2 immunoreactivity and percentage of the area occupied by the green puncta over the entire neuronal area that was plotted. N = 6 cultured coverslips; p < 0.01, Student's t test. Error bars indicate SEM.
To further confirm this observation, we performed detergent-absent staining of primary neurons cultured from mouse hippocampus with NaV1.2 antibody to detect surface NaV1.2. We found that the density of NaV1.2-immunoreactive puncta (shown in green on the neuronal surface), particular along the axon, were visibly increased, and these puncta were larger in size in BACE1-null mice compared with wild-type controls (Fig. 4B). When percentage of puncta area to total neuronal area was calculated using ImageJ software for quantitative comparison, it became evident that the percentage of NaV1.2 puncta area was significantly increased in BACE1-null neurons compared with wild-type controls (Fig. 4C). Hence, our results suggest that surface levels of sodium channel proteins are elevated on genetic ablation of BACE1.
Increased neuronal activity in BACE1-null mice
Altered expression of both NaV1.2 and NaV1.6 proteins in the hippocampus of BACE1-null mice is expected to alter their neuronal activity. To compare sodium channel properties between BACE1-null mice and their wild-type littermates, we recorded sodium channel activities in hippocampal pyramidal neurons acutely dissociated from mice between postnatal day 21 and day 30 using whole-cell patch-clamping method (Chen et al., 2004; Yu et al., 2006). Neurons dissociated from BACE1-null mice yielded larger normalized peak current compared with wild-type mice (Fig. 5A,B) (181 ± 13 pA/pF, n = 8; vs 136 ± 21 pA/pF, n = 10 neurons; p < 0.05). Although isolated BACE1-null and wild-type neurons showed an identical activation curve plotted against the command potential (Fig. 5C), a rightward shift of the inactivation curve toward depolarization was obviously displayed in isolated BACE1-null neurons compared with that in wild-type neurons (Fig. 5D,E) (V1/2, −60 ± 1 mV, n = 11; vs −69 ± 1 mV, n = 12 neurons; p < 0.05). This rightward shift of an inactivation curve implies more sodium channels will be available for firing at a given holding potential in mature neurons. We also recorded sodium channel-mediated fast currents using hippocampal neurons cultured from either wild-type or BACE1-null embryos. Whole-cell patch-clamp recording performed in hippocampal neuronal culture [day in vitro 11 (DIV11) to DIV15] revealed a similar rightward shift of inactivation curve and an increase in current density (data not shown). Hence, our results recorded from both cultured and isolated neurons showed altered sodium channel currents, which are consistent with our immunoreactivity quantification.
Figure 5.

Deletion of BACE1 causes modification of sodium channel function in hippocampal neurons. A, Sample sodium current from acutely dissociated hippocampal neurons from postnatal day 21–30 mice. Peak Na+ current was elicited by the step depolarization between −60 and +20 mV in an increment of 10 mV. Calibration: 5 ms, 500 pA. B, Normalized peak Na+ current density (in picoamperes/picofarad) versus voltage in acutely dissociated hippocampal neurons. BACE1−/− neurons yielded significant greater Na+ current density (181 ± 13 pA/pF; n = 8) compared with that of wild-type neurons (136 ± 21 pA/pF; n = 10). *p < 0.05. C, The activation curve of sodium channel recorded from acutely dissociated hippocampal neurons. The activation curve was fitted by the Boltzmann equation: G/Gmax = 1/(1 + exp((V1/2 − V)/k)). V1/2 is the voltage of half-maximal Na+ conductance, and k is a slope factor. D, Sample sodium current traces elicited by prepulse potential between −120 and 0 mV, followed by the depolarization command potential at 0 mV. Calibration: 5 ms, 500 pA. E, Steady-state inactivation curve of sodium channel. Peak Na+ current (I) at such prepulse potential was normalized to the maximum peak current (Imax) and fitted by the following Boltzmann equation: I/Imax = 1/(1+exp((V − V1/2)/k)). V1/2 is the voltage of half-maximal availability, and k is the slope. Note that inactivation curve obtained from BACE1-null neurons displays a shift in the depolarization direction compared with that of wild-type controls (V1/2: BACE1-null, −60 ± 1 mV, vs wild-type, −69 ± 1 mV). *p < 0.05. Error bars indicate SEM.
To compare the intrinsic neuronal firing properties, we further recorded action potentials from similarly dissociated wild-type and BACE1-null neurons under current clamp (Yu et al., 2006). It appeared that intrinsic firing properties of two genotypes of neurons were obviously different. First, to reach the maximum frequency of discharge, isolated BACE1-null neurons needed a greater total current injection when compared with their wild-type controls (Fig. 6A,B) (27 AP at 140 pA injection vs 24 AP at 100 pA injection; n = 12 and 11 neurons, respectively). Second, the action potential amplitude elicited from hippocampal pyramidal neurons was larger in isolated BACE1-null neurons than in wild-type neurons (Table 4, Fig. 6C) (101.4 ± 3.3 vs 90.3 ± 2.4 mV; p < 0.05). Third, the maximum slope during the rising phase (dV/dt) was greater in isolated BACE1-null neurons (94.7 ± 2.6 vs 86.3 ± 2.3 mV/ms in wild type; p < 0.05). This observation was, perhaps, not surprising because the conductance during the rising phase of action potentials depends on the number of open fast sodium channels, and BACE1-null neurons indeed have more sodium channels available than the wild type at same holding potential. Other than the above differences, the threshold, half-width, and action potential minimum amplitude were not obviously altered (Table 4). Together, our results suggest that neurons from BACE1-null mice can be more susceptible to the occurrence of epileptic activity because of significant changes in sodium channel properties and activity.
Figure 6.

Action potential properties of acutely dissociated hippocampal neurons. A, Action potential elicited by depolarizing current injection of 20, 100, 140, and 190 pA. Calibration: 100 ms, 50 mV. B, Action potential number in relation to the current injection. Note that action potential discharge in wild-type neurons (BACE1+/+) reached the maximum frequency at 100 pA injection and decreased afterward, whereas BACE1−/− neurons reach the maximum firing at 140 pA injection. Error bars indicate SEM. C, Single action potential recorded from wild-type (BACE1+/+) neurons (gray) compared with that of BACE1−/− neurons (black). Calibration: 2 ms, 25 mV.
Table 4.
Action potential parameters from hippocampal pyramidal neurons
| Threshold (mV) | Amplitude (mV) | Max dV/dt (mV/ms) | AP min (mV) | Half-width (ms) | Input resistance (MΩ) | |
|---|---|---|---|---|---|---|
| BACE1+/+ | −44.1 ± 1.2 | 90.3 ± 2.4 | 86.3 ± 2.3 | −54.6 ± 2.7 | 2.36 ± 0.34 | 400 ± 57 |
| BACE1−/− | −45.2 ± 1.4 | 101.4 ± 3.3* | 94.7 ± 2.6* | −56.3 ± 2.0 | 2.63 ± 0.39 | 450 ± 65 |
*p < 0.05, Student's t test, significantly different from BACE1+/+.
Elevated neuronal excitability in BACE1-null mouse brains
In addition to the patch-clamping experiments, we also compared the neuronal firing pattern of BACE1-null mice with their wild-type littermates by performing extracellular field recording of brain slices that were exposed to 100 μm 4-AP, a potassium channel blocker used for inducing intense electrical discharges in slices (Luhmann et al., 2000). We detected synchronous epileptiform-like activity within the whole hippocampal area after bath application of 4-AP (Fig. 7). Noticeably, the dentate gyrus region of slices from BACE1-null mice consistently showed higher bursting activity when compared with wild-type controls (Fig. 7B–D). Our recording of hippocampal slices from 6-month-old mice showed that average discharge amplitude (0.07 ± 0.01 mV in wild-type vs 0.11 ± 0.01 mV in BACE1-null mice; p < 0.01), average burst frequency (0.08 ± 0.01 Hz in wild type vs 0.14 ± 0.02 Hz in BACE1-null mice; p < 0.05), and the total area of burst per slice (23.74 ± 5.39 mV×ms in wild type vs 50.09 ± 5.77 mV×ms in BACE1-null mice; p < 0.05) were significantly different in the hippocampal region of BACE1-null mice compared with wild-type controls. Together, these data show that genetic deletion of BACE1 in mice leads to increased neuronal excitability and enhanced synchronous neuronal firing, consistent with our biochemical and morphological results.
Figure 7.

Increased depolarized-evoked firing in BACE1-null hippocampal slices. A, Synchronous firing in the dentate gyrus region was induced by treatment of brain slices from 6-month-old mice with 100 μm 4-AP. In a given recording period, BACE1-null mice displayed population spikes with a larger amplitude than the control mice. Calibration: 0.05 mV, 0.2 s. B, Comparison of burst frequency between wild-type and BACE1-null mice. Burst frequency was analyzed with Mini Analysis and calculated from interburst interval. Wild type, 0.08 ± 0.01 Hz; BACE1-null, 0.14 ± 0.02 Hz; *p < 0.05. C, Comparison of burst amplitude between wild-type and BACE1-null mice. Burst amplitude was analyzed from the first positive-directed discharge within a population spike. Wild type, 0.07 ± 0.01 mV; BACE1-null, 0.11 ± 0.01 mV; **p < 0.01. D, Comparison of total burst area per slice between wild-type and BACE1-null mice. Wild type, 23.74 ± 5.39 mV × ms; BACE1-null, 50.09 ± 5.77 mV × ms; *p < 0.05. n = 12 slices from 5 wild-type mice and 7 BACE1-null mice. Error bars indicate SEM.
Neurodegeneration in aged BACE1-null mice
Seizure-induced neuronal loss is one of the commonly described consequences of chronic epilepsy (Henshall and Murphy, 2008). Genetic deletion of BACE1 causes elevated sodium channel-mediated fast currents and neuronal excitability. In BACE1-null mice, loss of neurons (specified with arrowheads) was found in the dentate gyrus and CA3 regions in all three BACE1-null mice examined at 2 years of age (Fig. 8A). This neuronal loss was not obvious in BACE1-null mice younger than 8 months of age (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) or in age-matched wild-type littermates. Quantification analysis showed a reduction of total pyramidal neurons by ∼22% in aged BACE1-null mice compared with their wild-type littermates, but showed no obvious neuronal loss in young BACE1-null mice (Fig. 8B). Perhaps sustained asynchronous stimulation arising from abnormally higher neuronal activity may have triggered neurodegeneration during the aging process.
Figure 8.
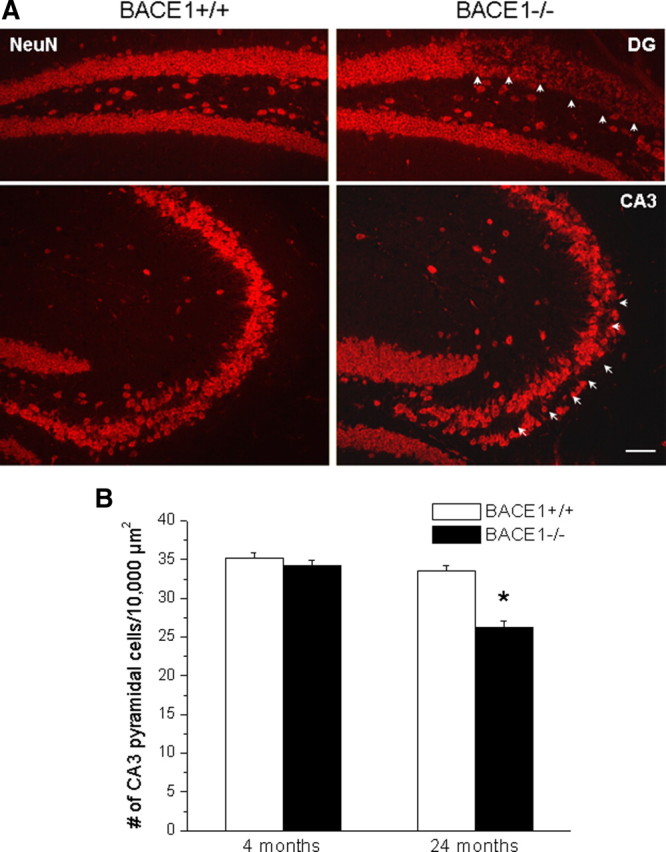
Neurodegeneration in aged BACE1-null mice. A, Fixed brain slices were immunostained with NeuN to label neurons. Neuronal loss in both hippocampal CA1 and dentate gyrus was evident and is shown by arrowheads. Scale bar, 60 μm. B, Total pyramidal neurons in CA3 were quantified based on the numbers of neurons occupied in 10,000 μm2. BACE1-null mice showed a significantly smaller cell count than wild-type mice at 24 months of age, but not at 4 months of age. N = 3 animals per group; *p < 0.05. Error bars indicate SEM.
Discussion
BACE1 is widely regarded as an important therapeutic target for AD, and it is anticipated that BACE1 inhibitors will be therapeutically used to stop the progression of AD. Because of this importance, the knowledge of phenotypes exhibited in BACE1-null mice is particularly informative for monitoring potential mechanism-based toxicity. The finding of epileptic seizures together with the previously reported hypomyelination in BACE1-null mice indicates that BACE1 plays multiple roles in cellular processes. In this study, our results suggest that the altered sodium channel activity in BACE1-null mice may contribute to the occurrence of spontaneous epileptic seizures. Hence, careful monitoring of changes in sodium channel activity on long-lasting inhibition of BACE1 activity may need to be considered during the development of BACE1 inhibitors for therapeutic applications.
It has been demonstrated that abnormal neuronal excitability associated with altered sodium channel activity can cause epileptic seizures (Catterall et al., 2008). For example, a variable percentage (but not all) of animals with deletion of a single copy of NaV1.1 (Scn1a+/− heterozygotes) develop behavioral seizures (Yu et al., 2006). A persistently elevated sodium current seen in a transgenic mouse model expressing a “gain-of function” mutation in NaV1.2 (Scn2a; Q54) produces an epilepsy phenotype (Kearney et al., 2001). In BACE1-null mice, both behavioral and silent epileptic seizures are detected; expression of sodium channel proteins and sodium currents are altered. These alterations are likely attributable to the abolished cleavage of the sodium channel β-subunit at the site juxtaposed to the transmembrane region by BACE1 because this alteration potentially leads to reduced release of an intracellular domain of the β-subunit, a fragment known to regulate gene expression of sodium channel α-subunits (Kim et al., 2007). We showed that sodium channel α-subunits in total 2% SDS-extracted protein lysates were indeed reduced in BACE1-null mice (Fig. 3).
Reduced expression of sodium channel α-subunits is conventionally expected to result in less surface expression. Intriguingly, we found that NaV1.2 expression on the neuronal surface or in axons was clearly increased in BACE1-null mice based on three pieces of evidence: (1) the levels of NaV1.2 in mossy fibers are higher (Fig. 4A); (2) the amount of NaV1.2 immunoreactivity is greater and the area is larger in cultured BACE1-null neurons than in wild-type neurons (Fig. 4B); and (3) sodium channel current density is consistently increased in BACE1-null neurons (Fig. 5). Altered surface expression of α-subunits is likely mediated by the full-length sodium channel β-subunit because it can bind to the α-subunits and mediates their cellular trafficking (Isom, 2002). In BACE1-overexpressed neurons, the reduced levels of the full-length sodium channel β-subunit correlate with the reduced expression of α-subunits on the cell surface (Kim et al., 2007). When the cleavage of sodium channel β-subunit is abolished in BACE1-null mice, the levels of the full-length sodium channel β-subunit will be increased and trafficking of α-subunits to the surface will therefore be increased. In another study, it is suggested that even noncatalytic BACE1 molecule will affect sodium channel activity (Huth et al., 2009).
However, other phenotypic changes in BACE1-null mice may additionally contribute to the increased surface expression of sodium channel NaV1.2 to maintain sustained functional excitability. For example, hypomyelination has been shown to increase surface expression of sodium channel NaV1.2 in previous studies (Noebels et al., 1991; Westenbroek et al., 1992) and BACE1-null mice exhibit hypomyelination in their central nerves (Hu et al., 2006). This contribution can be tested in a model with hypomyelination rescued. In addition, it is likely that altered synaptic plasticity in mossy fiber region (Wang et al., 2008) may also trigger changes in neuronal excitation that can contribute to the seizure phenotype.
Finding elevated sodium channel NaV1.2 on embryonic hippocampal neurons suggests that this elevation occurs as early as developmental embryonic stages. Impaired function and altered surface expression of NaV1.2 have been found in inherited epilepsies including benign familial neonatal-infantile seizures (Heron et al., 2002; Berkovic et al., 2004; Scalmani et al., 2006; Striano et al., 2006). As previously mentioned, we observed severe behavioral epileptic seizures in a number of BACE1-null mice at an age around postnatal day 21, suggesting the occurrence of neonatal-infantile-like seizures in these mice. In addition, we also observed that some BACE1-null young mice died immediately after clonic seizures. Approximately 30% of BACE1-null mice died before postnatal day 15 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). It is unclear whether the occurrence of severe epileptic seizures actually contributes to this high rate of premature death on genetic deletion of BACE1.
Altered expression of sodium channel α-subunits in BACE1-null mice can affect their normal electrophysiological properties. Normalized sodium current density in BACE1-null neurons was significantly larger than that in wild-type controls (Fig. 5), indicating that BACE1-null neurons display higher neuronal activity. This result is consistent with greater neuronal excitability as manifested by more frequent firing with larger amplitude in BACE1-null brain slices (Fig. 7). In addition, a significant shift of the inactivation curve in the direction of depolarization was recorded from BACE1-null neurons, and a significant shift in steady-state inactivation of the sodium channel toward more depolarized potentials was also seen in BACE1-null pyramidal neurons acutely isolated from slices of mouse neocortex (Dominguez et al., 2005). This shift in steady-state inactivation indicates increased availability of total sodium channels at a depolarization potential in BACE1-null neurons, which is believed to modify the generation and propagation of the action potential. Indeed, BACE1-null neurons did have an intrinsically different action potential firing pattern compared with that of wild-type neurons (Fig. 6). The small yet significant increase in both amplitude and maximum slope of action potential is primarily attributable to the opening of available sodium channels on surface. If BACE1 is overexpressed in transgenic mice or neuroblastoma cell lines, a dramatic decrease of sodium current density is correspondingly observed (Kim et al., 2007). These consistent results lead us to conclude that BACE1 regulates sodium channel function and properties in vivo.
Noticeably, epileptic seizures appear to occur more frequently in AD patients and increased Aβ production may be a culprit (Palop and Mucke, 2009). Since inhibition of BACE1 can reduce the formation of Aβ, it is expected that BACE1 inhibitory drugs will be highly beneficial in both cases. However, the finding of epileptic seizures in BACE1-null mice raises an unmet question of whether significant inhibition of BACE1 will increase the incidence of epileptic seizures. Potentially, proper dosing is crucial to avoid likely elevation of epileptic incidence during drug development of BACE1 inhibitors for AD therapy.
Footnotes
This work was partially supported by National Institutes of Health (NIH) Grant AG025493 (R.Y.); awards from Ralph Wilson Foundation, American Health Assistance Foundation, and Alzheimer's Association (R.Y.); and NIH Grant HL081622 (C.G.W.). We thank Drs. M. Prior and Q. Shi for the discussion during this study and Chris Nelson for critical reading of this manuscript; Imad Najm (Cleveland Clinic Epilepsy Center, Cleveland, OH), Lori Isom and Luis Lopez-Santiago (University of Michigan, Ann Arbor, MI), Stefan Herlitze and Takashi Maejima (Case Western Reserve University, Cleveland, OH), and Lin Mei and Yongjun Chen (Medical College of Georgia, Augusta, GA) for the helpful discussions; and C. Mayer, R. Foglyano, and S. Reed (Case Western Reserve University, Cleveland, OH) and Paromita Das and Elizabeth I. Tietz (University of Toledo, Toledo, OH) for their technical assistance in whole-cell patch-clamp and acute dissociation experiments.
The authors declare no competing financial interests.
References
- Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci. 2003;23:7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, Gambardella A, Steinlein OK, Grinton BE, Dean JT, Bordo L, Hodgson BL, Yamamoto T, Mulley JC, Zara F, Scheffer IE. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–557. doi: 10.1002/ana.20029. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Buonanno A, Kwon OB, Yan L, Gonzalez C, Longart M, Hoffman D, Vullhorst D. Neuregulins and neuronal plasticity: possible relevance in schizophrenia. Novartis Found Symp. 2008;289:165–177. doi: 10.1002/9780470751251.ch13. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Molecular mechanisms of gating and drug block of sodium channels. Novartis Found Symp. 2002;241:206–218. discussion 218–232. [PubMed] [Google Scholar]
- Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci. 2008;28:11768–11777. doi: 10.1523/JNEUROSCI.3901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Bharucha V, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ, 3rd, Kazen-Gillespie KA, Kazarinova-Noyes K, Shrager P, Saunders TL, Macdonald RL, Ransom BR, Scheuer T, Catterall WA, Isom LL. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci U S A. 2002;99:17072–17077. doi: 10.1073/pnas.212638099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL. Mice lacking sodium channel β1-subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004;24:4030–4042. doi: 10.1523/JNEUROSCI.4139-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D'Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B. Phenotypical and biochemical analysis of BACE1 and BACE2 deficient mice. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat Med. 2004;10:959–965. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Murphy BM. Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol. 2008;8:75–81. doi: 10.1016/j.coph.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, Shevell M, Mercho S, Seni MH, Guiot MC, Mulley JC, Berkovic SF, Scheffer IE. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–852. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB, Trapp BD, Yan R. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 2008;22:2970–2980. doi: 10.1096/fj.08-106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Huth T, Schmidt-Neuenfeldt K, Rittger A, Saftig P, Reiss K, Alzheimer C. Non-proteolytic effect of beta-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol Dis. 2009;33:282–289. doi: 10.1016/j.nbd.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Isom LL. Beta subunits: players in neuronal hyperexcitability? Novartis Found Symp. 2002;241:124–138. [PubMed] [Google Scholar]
- Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of Ranvier. Neuron. 2001;30:105–119. doi: 10.1016/s0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- Kim DY, Ingano LA, Carey BW, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J Biol Chem. 2005;280:23251–23261. doi: 10.1074/jbc.M412938200. [DOI] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, Kovacs DM. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9:755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer's beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A. 2001;98:13554–13559. doi: 10.1073/pnas.241509198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D, Zeller M, Cole T, Buttini M, McConlogue L, Sinha S, Freedman S, Morris RG, Chen KS. BACE1 gene deletion: impact on behavioral function in a model of Alzheimer's disease. Neurobiol Aging. 2008;29:861–873. doi: 10.1016/j.neurobiolaging.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Südhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279:10542–10550. doi: 10.1074/jbc.M310001200. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;278:48713–48719. doi: 10.1074/jbc.M303861200. [DOI] [PubMed] [Google Scholar]
- Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ, Dzhala VI, Ben-Ari Y. Generation and propagation of 4-AP-induced epileptiform activity in neonatal intact limbic structures in vitro. Eur J Neurosci. 2000;12:2757–2768. doi: 10.1046/j.1460-9568.2000.00156.x. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, Marcom PK, Jalilian-Tehrani MH. Sodium channel density in hypomyelinated brain increased by myelin basic protein gene deletion. Nature. 1991;352:431–434. doi: 10.1038/352431a0. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino L, Ikin AF, Lamprianou S, Vacaresse N, Revelli JP, Platt K, Paganetti P, Mathews PM, Harroch S, Buxbaum JD. BACE (beta-secretase) modulates the processing of APLP2 in vivo. Mol Cell Neurosci. 2004;25:642–649. doi: 10.1016/j.mcn.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Qahwash I, Weiland KL, Lu Y, Sarver RW, Kletzien RF, Yan R. Identification of a mutant amyloid peptide that predominantly forms neurotoxic protofibrillar aggregates. J Biol Chem. 2003;278:23187–23195. doi: 10.1074/jbc.M213298200. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalmani P, Rusconi R, Armatura E, Zara F, Avanzini G, Franceschetti S, Mantegazza M. Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci. 2006;26:10100–10109. doi: 10.1523/JNEUROSCI.2476-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;7:15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striano P, Bordo L, Lispi ML, Specchio N, Minetti C, Vigevano F, Zara F. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia. 2006;47:218–220. doi: 10.1111/j.1528-1167.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- von Arnim CA, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, Spoelgen R, Hshieh TT, Ranganathan S, Battey FD, Liu CX, Bacskai BJ, Sever S, Irizarry MC, Strickland DK, Hyman BT. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem. 2005;280:17777–17785. doi: 10.1074/jbc.M414248200. [DOI] [PubMed] [Google Scholar]
- Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci. 2008;28:8677–8681. doi: 10.1523/JNEUROSCI.2440-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Noebels JL, Catterall WA. Elevated expression of type II Na+ channels in hypomyelinated axons of shiverer mouse brain. J Neurosci. 1992;12:2259–2267. doi: 10.1523/JNEUROSCI.12-06-02259.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P, Nukina N. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Wang J, Wang Y, Xie X, Meyenhofer MF, Ledeen RW. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane-permeant analog of GM1. J Neurosci. 2005;25:11014–11022. doi: 10.1523/JNEUROSCI.3635-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yan R, Han P, Miao H, Greengard P, Xu H. The transmembrane domain of the Alzheimer's beta-secretase (BACE1) determines its late Golgi localization and access to beta-amyloid precursor protein (APP) substrate. J Biol Chem. 2001;276:36788–36796. doi: 10.1074/jbc.M104350200. [DOI] [PubMed] [Google Scholar]
- Yu EJ, Ko SH, Lenkowski PW, Pance A, Patel MK, Jackson AP. Distinct domains of the sodium channel beta3-subunit modulate channel-gating kinetics and subcellular location. Biochem J. 2005;392:519–526. doi: 10.1042/BJ20050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]