

Abstract
Constitutive activation of signal transducer and activator of transcription 3 (Stat3) has been found in a variety of human malignancies and has been suggested to play an important role in carcinogenesis. Recently, our laboratory demonstrated that Stat3 is required for the development of skin tumors via two-stage carcinogenesis using skin-specific loss-of-function transgenic mice. To investigate further the role of Stat3 in each stage of chemical carcinogenesis in mouse skin, i.e. initiation and promotion stages, we generated inducible Stat3-deficient mice (K5.Cre-ERT2 × Stat3fl/fl) that show epidermal-specific disruption of Stat3 following topical treatment with 4-hydroxytamoxifen (TM). The epidermis of inducible Stat3-deficient mice treated with TM showed a significant increase in apoptosis induced by 7,12-dimethylbenz[a]anthracene (DMBA) and reduced proliferation following exposure to 12-O-tetradecanoylphorbol-13-acetate. In two-stage skin carcinogenesis assays, inducible Stat3-deficient mice treated with TM during the promotion stage showed a significant delay of tumor development and a significantly reduced number of tumors compared with control groups. Inducible Stat3-deficient mice treated with TM before initiation with DMBA also showed a significant delay in tumor development and a significantly reduced number of tumors compared with control groups. Finally, treatment of inducible Stat3-deficient mice that had existing skin tumors generated by the two-stage carcinogenesis protocol with TM (by intraperitoneal injection) led to inhibition of tumor growth compared with tumors formed in control groups. Collectively, these results directly demonstrate that Stat3 is required for skin tumor development during both the initiation and promotion stages of skin carcinogenesis in vivo.
Introduction
Signal transducer and activator of transcription 3 (Stat3) is one of a family of cytoplasmic proteins that participate in normal cellular responses to cytokines and growth factors as a transcription factor (1–4). Upon activation by a wide variety of cell-surface receptors via tyrosine phosphorylation, Stat3 dimerizes and translocates to the nucleus and modulates the expression of target genes that are involved in various physiological functions including apoptosis (e.g. survivin and Bcl-xL), cell-cycle regulation (e.g. Cyclin D1 and c-Myc) and tumor angiogenesis (e.g. vascular endothelial growth factor) (3,5). Studies indicate that constitutive activation of Stat3 is associated with a number of human tumors and cancer cell lines, including prostate, breast, lung, head and neck, brain and pancreas, and its inhibition can suppress growth of cancer cells by promoting apoptosis and inhibiting cell proliferation (1,2,6,7). These observations suggest that Stat3 may play a critical role in cancer cell proliferation and survival. The fact that naturally occurring mutations of Stat3 leading to its constitutive activation have not been identified indicates that aberrant growth factor signaling is the most important mechanism leading to constitutive activation of Stat3 seen in tumors (2).
Recent studies of Stat3 have suggested that it has critical roles in multistage epithelial carcinogenesis (reviewed in refs 8,9). In this regard, studies from our laboratory have shown that epidermal growth factor receptor-mediated activation of Stat3 occurred in mouse epidermis following topical treatment with diverse classes of tumor promoters, including 12-O-tetradecanoylphorbol-13-acetate (TPA), okadaic acid and chrysarobin (10). Furthermore, constitutive activation of Stat3 found in both papillomas and squamous cell carcinomas induced by two-stage carcinogenesis also occurred, at least in part, by a similar mechanism (10). Consistent with these observations, Stat3-deficient mice were completely resistant to development of skin tumors induced by the two-stage carcinogenesis regimen, and abrogation of Stat3 function by using a Stat3-specific decoy oligonucleotide inhibited the growth of skin tumors (11). These studies provided the first evidence that Stat3 is required for both the initiation and promotion stages of carcinogenesis by maintaining survival of DNA-damaged stem cells and by mediating cell proliferation necessary for the clonal expansion of initiated cells (11). More recent studies using mice in which the expression of a constitutively active/dimerized form of Stat3 (Stat3C) is targeted to the proliferative compartment of epidermis via the bovine keratin 5 promoter (referred to as K5.Stat3C transgenic mice) demonstrated heightened sensitivity of these mice to two-stage skin carcinogenesis compared with non-transgenic littermates (12). In addition, the skin tumors that developed in the K5.Stat3C mice bypassed the premalignant stage and rapidly progressed to squamous cell carcinomas that were highly vascularized, poorly differentiated and more invasive (12). These results confirmed a role for Stat3 in the early stages of epithelial carcinogenesis and revealed a novel role of Stat3 in driving malignant progression of skin tumors in vivo (12). Collectively, these studies using both skin-specific gain and loss-of-function transgenic mice have provided evidence that Stat3 plays a critical role throughout the process of epithelial carcinogenesis in mouse skin.
To more directly assess the role of Stat3 in the initiation and promotion stages of multistage carcinogenesis, we have utilized a genetic system to inducibly delete Stat3 during either the initiation or the promotion stages of two-stage skin carcinogenesis. Using this system, we provide direct evidence that Stat3 is a critical transcription factor during both initiation and promotion of skin tumor development in vivo. Finally, the results indicate that inducible abrogation of Stat3 function in predeveloped papillomas inhibited their further growth in vivo.
Materials and methods
Chemicals
7,12-Dimethylbenz[a]anthracene (DMBA) and 4-hydroxytamoxifen (TM) were purchased from Sigma–Aldrich (Milwaukee, WI) and TPA was purchased from LC Laboratories (Woburn, MA). Acrylamide:bis solution (29:1) and polyvinylidene difluoride membranes were purchased from Bio-Rad Laboratories (Hercules, CA).
Preparation of DNA construct and generation of K5.Cre-ERT2 transgenic mice
The K5.Cre-ERT2 plasmid was constructed by cloning a 2 kb EcoRI fragment isolated from pCre-ERT2 (13) into the EcoRI site of the expression vector containing the K5 promoter (14). The resulting construct was digested with KpnI, purified and injected into the pronuclei of donor embryos to generate transgenic founders on an FVB/N background. Transgenic founder mice were identified by polymerase chain reaction of genomic DNA using primers specific for the gene encoding rabbit β-globin: 5′-GTGTTGTTTAGAATGGAAGATGT-3′ and 5′-TAAAGAGAAAGGCAGGATGATGA-3′. Three founders were obtained and lines were established on an FVB/N background. Transgene copy number was checked by semiquantitative polymerase chain reaction and Cre expression was confirmed by immunostaining of skin sections taken from K5.Cre-ERT2 transgenic mice after induction of expression by treatment with TM. Of the established lines, the line (line A) that had the highest inducible Cre expression was used to generate inducible Stat3-deficient mice for experiments described herein.
Generation of K5.Cre-ERT2 × Stat3fl/fl mice
Generation of Stat3fl/fl mice (15) has been described previously. K5.Cre-ERT2 line A transgenic mice were bred with Stat3fl/fl mice to ultimately generate mice hemizygous for the K5.Cre-ERT2 transgene and homozygous for the Stat3 floxed allele (K5.Cre-ERT2 × Stat3fl/fl).
Preparation of protein lysates and western blot analysis
Dorsal back skin of each mouse was treated with a depilatory agent, excised and the epidermis removed with a razor blade and placed into RIPA lysis buffer containing 50 mM Tris–HCl (pH 8.6), 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml apoptinin, 1 mM Na3VO4, 1 mM NaF and 10 μl/ml protease inhibitor cocktail (Sigma–Aldrich). The lysates were incubated on ice for 10 min, snap frozen in liquid nitrogen, rethawed and then centrifuged at 14, 000g for 15 min at 4°C. The supernatant was separated by electrophoresis on 8–12% sodium dodecyl sulfate–polyacrylamide gels. Separated proteins were electrophoretically transferred onto polyvinylidene difluoride membranes and blocked with 5% non-fat dry milk in phosphate-buffered saline (PBS) with 0.1% Tween 20 for 1 h at room temperature. Blots were then incubated for 2 h at room temperature with specific primary antibodies for Stat3, phospho-Stat3, Bcl-xL (Cell Signaling Technology, Beverly, MA), Cyclin D1, Cyclin E, c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (Sigma–Aldrich). Blots were washed with PBS with 0.1% Tween 20 and subjected to corresponding horseradish peroxidase-conjugated secondary antibodies against rabbit or mouse (Amersham Biosciences, Arlington Heights, IL). Blots were washed with PBS with 0.1% Tween 20 and detected with ECL Western Blotting Substrate (Pierce Biotechnology, Rockford, IL). Where indicated, relative changes in protein levels were determined by densitometry and normalized to β-actin.
Immunohistochemical analysis
Formalin-fixed, paraffin-embedded tissues were deparaffinized and hydrated using standard procedures. Endogenous peroxidase activity was blocked with 0.03% hydrogen peroxide for 10 min. Sections were microwaved (10 min) in the presence of 10 mM citrate buffer (pH 6.0) containing 0.01% Tween 20 and allowed to cool for 20 min. Sections were then stained with an anti-Stat3 antibody (Cell Signaling Technology) following suggested procedures by the manufacturer.
Analysis of epidermal apoptosis following treatment with DMBA
Groups of mice (n = 3) were treated topically with 1 mg of TM or ethanol for five consecutive days. Twenty-four hours after TM or ethanol treatment, mice were treated with a single topical application of DMBA (25 nmol) or acetone (0.2 ml) on the dorsal skin and were killed 24 h later. Skin sections were stained using an In Situ Cell Death Detection Kit (Roche Diagnostics Co., Indianapolis, IN). Apoptotic keratinocytes were counted microscopically in at least three non-overlapping fields in sections from each mouse. The apoptotic index is represented as terminal deoxynucleotidyl transferase (tdt)-mediated dUTP nick end-labeling-positive cells per centimeter.
Analysis of cell proliferation following treatment with TPA
For analysis of epidermal proliferation, groups of mice (n = 3) were treated topically with 1 mg of TM or ethanol for five consecutive days. Twenty-four hours after TM or ethanol treatment, mice were treated with a single topical application of TPA (6.8 nmol) or acetone on the dorsal skin and killed at 24 h. Mice received intraperitoneal (i.p.) injections of 5-bromo-2-deoxyuridine (BrdU; Sigma–Aldrich) at 100 μg/g body wt in PBS 30 min prior to killing. Dorsal skin was then fixed in formalin and embedded in paraffin prior to sectioning of 4 μm and stained with hematoxylin and eosin. To determine epidermal cell proliferation, sections were stained with an anti-BrdU antibody (BD Pharmingen, Los Angeles, CA), followed by treatment with biotinylated anti-mouse IgG and horseradish peroxidase-conjugated ABC reagent (Vector Laboratories, Burlingame, CA). Epidermal cell proliferation, presented as the labeling index, was determined by calculating the percentage of basal cells positive for BrdU. A minimum of 500 basal cells was counted.
Disruption of Stat3 signaling at initiation during two-stage carcinogenesis
Five groups of mice (Group 1, n = 8; Group 2, n = 8; Group 3, n = 7; Group 4, n = 8 and Group 5, n = 10) were subjected to two-stage skin carcinogenesis at 8 weeks of age. The dorsal skin of each mouse was shaved 48 h prior to treatment with TM or ethanol (vehicle) alone. All solutions of DMBA and TPA were prepared in reagent grade acetone and were applied topically in a total volume of 0.2 ml. TM was prepared in reagent grade ethanol and applied topically in a total volume of 0.2 ml. Groups 1–4 were treated with either 1 mg of TM or vehicle topically for five consecutive days before initiation (see Figures 1A and 3A). Then, mice were initiated with a single topical application of 25 nmol DMBA. Mice in Group 5 (see Figure 3A) were treated with TM 1 week after initiation. Four weeks after initiation, all groups received topical applications of TPA at 6.8 nmol given twice weekly. Note that only in Groups 1 and 5 was Stat3 inducibly deleted.
Fig. 1.
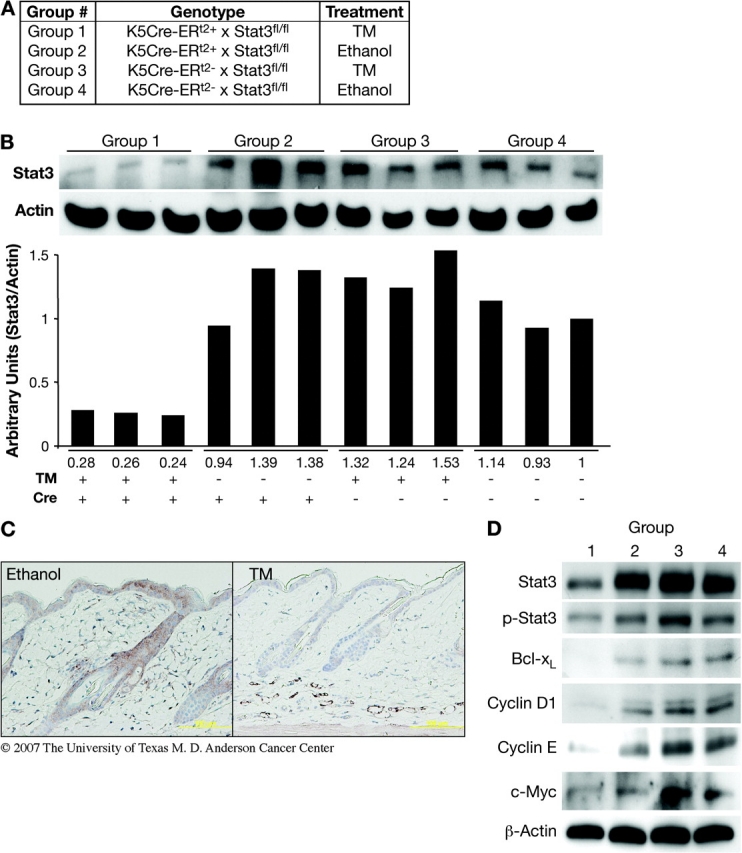
Induction of Stat3 deficiency in epidermis of K5.Cre-ERT2 × Stat3fl/fl mice by application of TM. (A) Mice were segregated by genotype into four groups and treated as shown. (B) Western blot analysis and quantification of Stat3 level in inducible Stat3-deficient mice treated with TM (Group 1) and control (Groups 2–4) mice. (C) Immunohistochemical staining for Stat3 in skin sections from inducible Stat3-deficient mice treated with ethanol or TM. (D) Western blot analysis of Stat3 and phosphorylated Stat3 levels in relation to the levels of Bcl-xL, Cyclin D1, Cyclin E and c-Myc in inducible Stat3-deficient mice treated with TM (Group 1) and the control groups (Groups 2–4).
Fig. 3.
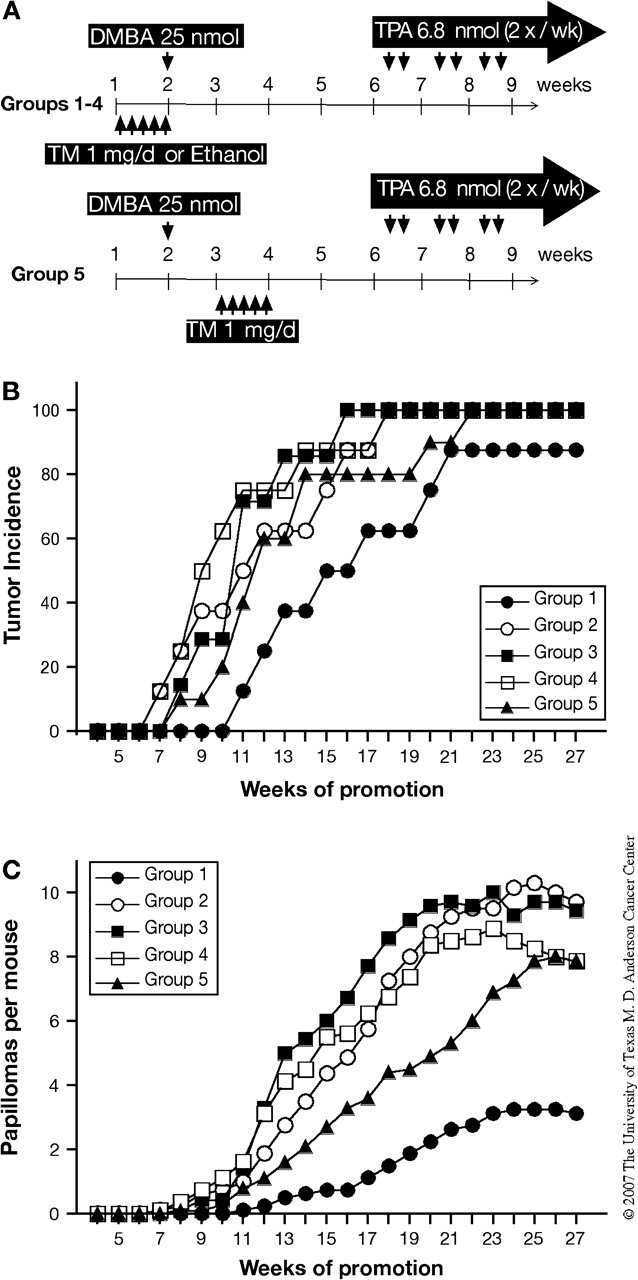
Effect of Stat3 disruption on the initiation stage of two-stage skin carcinogenesis. (A–C) Groups of mice were treated with 25 nmol of DMBA and after 4 weeks received twice-weekly applications of 6.8 nmol of TPA for the duration of the experiment. TM or ethanol was applied topically for five consecutive days either before initiation with DMBA (Groups 1–4) or after initiation with DMBA (Group 5) as shown in (A). (B) Percentage of mice with papillomas and (C) average number of papillomas per mouse [Group 1, inducible Stat3-deficient mice treated with TM (filled circles); Group 2, inducible Stat3-deficient mice treated with ethanol (open circles); Group 3, Cre− mice treated with TM (filled squares); Group 4, Cre− mice treated with ethanol (open squares) and Group 5, inducible Stat3-deficient mice treated with TM after DMBA initiation (filled triangles)].
Disruption of Stat3 signaling during the promotion stage of two-stage carcinogenesis
Four groups of mice (Group 1, n = 6; Group 2, n = 6; Group 3, n = 7 and Group 4, n = 6) were subjected to two-stage skin carcinogenesis at 8 weeks of age. Again see Figure 1A and Figure 4A for the groups and treatments. The dorsal skin of each mouse was shaved 48 h prior to treatment; only those mice in the resting phase of the hair cycle were utilized. Mice were initiated with a single topical application of 100 nmol DMBA. One week after initiation, mice were treated with either 1 mg of TM or ethanol topically for five consecutive days and this regimen was repeated every other week. Two weeks after initiation, TPA at 6.8 nmol was applied topically twice weekly.
Fig. 4.
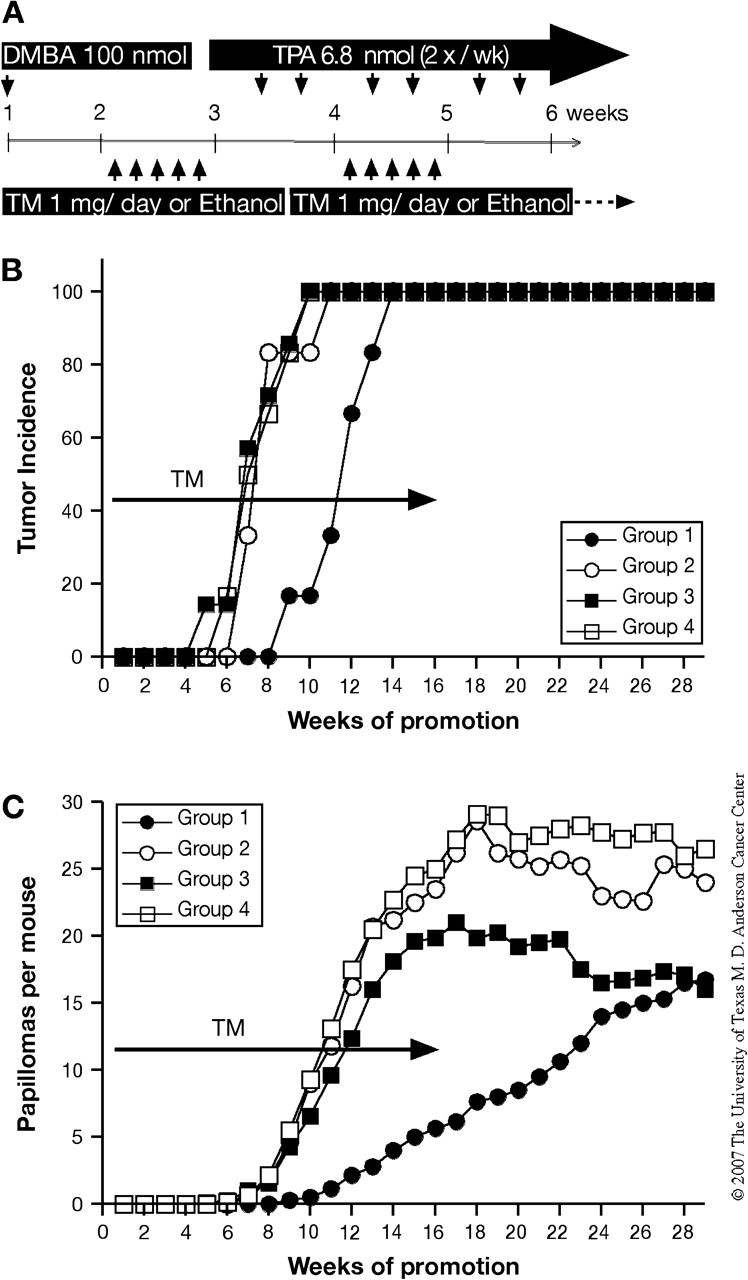
Effect of Stat3 disruption on the tumor promotion stage of two-stage skin carcinogenesis. (A–C) Groups of mice were treated with 100 nmol of DMBA and after 2 weeks received twice-weekly applications of 6.8 nmol of TPA for the duration of the experiment. TM or ethanol was applied topically for five consecutive days and repeated every 2 weeks and then stopped after 16 weeks. (A) Experimental protocol for disruption of Stat3 during the promotion stage. (B) Percentage of mice with papillomas and (C) average number of papillomas per mouse [Group 1, inducible Stat3-deficient mice treated with TM (filled circles); Group 2, inducible Stat3-deficient mice treated with ethanol (open circles); Group 3, Cre− mice treated with TM (filled squares) and Group 4, Cre− mice treated with ethanol (open squares)].
Effect of Stat3 deletion on growth of existing papillomas
Groups of mice (similar to Groups 1–4 as shown in Figure 1A; Group 1, n = 3; Group 2, n = 4; Group 3, n = 3 and Group 4, n = 3) were subjected to two-stage carcinogenesis as described above using DMBA and TPA to generate primary tumors (i.e. papillomas). Groups of mice received TPA (6.8 nmol) treatment for 26 weeks. After 2 weeks without TPA treatment, mice were treated with 1 mg of TM or ethanol i.p. for five consecutive days and this regimen was repeated every 2 weeks. To evaluate the effect of Stat3 deletion (Group 1) on papilloma growth, tumor dimensions were measured with calipers after the first week of TM treatments by i.p. and each week for the next 4 weeks. Tumor volume was calculated as [(x + y)/2]3, where x and y represent the width and the length of the tumor, respectively.
Results
Analysis of Stat3 deficiency in keratinocytes of K5.Cre-ERT2 × Stat3fl/fl mice after TM treatment
Initial experiments were conducted to validate the efficiency of Stat3 deletion in epidermis of K5.Cre-ERT2 × Stat3fl/fl mice. For these experiments, groups of 10 mice each were divided into four groups based on Cre recombinase gene expression and TM treatment (Figure 1A). Following topical application with 1 mg of TM on the dorsal skin of inducible Stat3-deficient mice for five consecutive days, the level of Stat3 expression was significantly decreased in skin epidermis 24 h after the last treatment (Figure 1B). The Stat3 protein level in epidermal lysates from the inducible Stat3-deficient mice (Group 1) was ∼25% of that in epidermis of mice from the control groups (Groups 2–4). Similar to western blot analysis, immunohistochemical staining of Stat3 revealed that its expression in the epidermis was significantly reduced following TM treatment of Group 1 mice (Figure 1C). Cre expression (assessed by immunohistochemistry) was observed only in epidermal keratinocytes of mice in Group 1 as expected (data not shown). Consistent with reduced levels of Stat3, we analyzed the level of tyrosine-phosphorylated Stat3 as well as several Stat3-regulated genes. As shown in Figure 1D, Bcl-xL, Cyclin D1, Cyclin E and c-Myc protein levels were all significantly reduced in epidermal lysates from Group 1 mice 24 h after the last treatment of TM. Note that deletion of Stat3 under the conditions employed did not alter the levels of Stat1 or Stat5 in epidermis (data not shown).
Inducible Stat3 deficiency sensitizes keratinocytes to DMBA-induced apoptosis and reduces TPA-mediated epidermal hyperproliferation
Our previous work showed that constitutive Stat3 deficiency in mouse epidermis leads to increased sensitivity to DMBA-induced apoptosis and reduced sensitivity to TPA-mediated epidermal hyperproliferation (11). To examine the effect of inducible disruption of Stat3 using the TM system on DMBA-induced epidermal apoptosis in vivo, TM-induced Stat3-deficient mice (Group 1, Figure 1A) were treated topically with DMBA (25 nmol) 24 h after the last TM treatment and the number of apoptotic keratinocytes was determined 24 h after DMBA treatment. Topical application of DMBA to TM-induced, Stat3-deficient mice resulted in a significant increase in the number of epidermal cells undergoing apoptosis compared with mice in the other control groups (Groups 2–4) as analyzed by terminal deoxynucleotidyl transferase (tdt)-mediated dUTP nick end-labeling staining (Figure 2A). These results demonstrate that temporal disruption of Stat3 was sufficient to sensitize keratinocytes to DMBA-induced apoptosis.
Fig. 2.
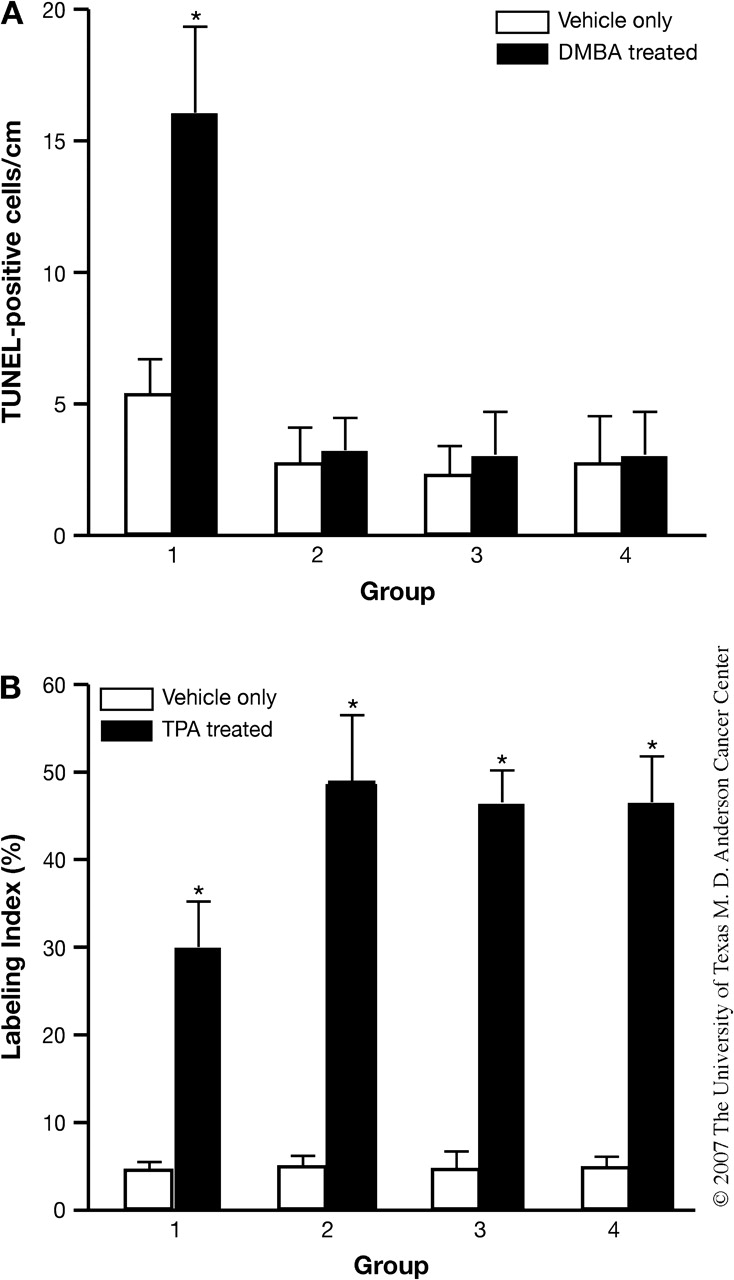
Temporal disruption of Stat3 results in increased apoptosis and decreased cell proliferation in the epidermis of inducible Stat3-deficient mice. (A) DMBA-induced apoptosis in the epidermis of inducible Stat3-deficient mice. Groups of mice (n = 3) were treated with TM or ethanol as indicated (Figure 1A) for five consecutive days, then 24 h later they received a single topical treatment of 25 nmol DMBA (black bars) or acetone (white bars). Mice were then killed 24 h later and skin sections were prepared for analysis. (B) TPA-induced cell proliferation in the epidermis of inducible Stat3-deficient mice. Groups of mice (n = 3) were treated with TM or ethanol as indicated (Figure 1A) for five consecutive days. Twenty-four hours after the last treatment of TM, they were treated with 6.8 nmol TPA (black bars) or acetone (white bars) once and were killed 24 h later. BrdU (100 μg/g body wt) was injected 30 min prior to killing. (Group 1, inducible Stat3-deficient mice treated with TM; Group 2, inducible Stat3-deficient mice treated with ethanol; Group 3, Cre− mice treated with TM and Group 4, Cre− treated with ethanol). *P < 0.01 by Mann–Whitney U-test.
To examine the effect of inducible disruption of Stat3 on tumor promoter-induced epidermal hyperproliferation in vivo, TM-induced, Stat3-deficient mice (Group 1, Figure 1A) were treated topically with a single treatment of TPA and the epidermal labeling index was determined 24 h after treatment. TM-induced, Stat3-deficient mice showed a statistically significant reduction in epidermal labeling index as revealed by a reduction (∼40%) in the number of BrdU-positive cells following treatment with TPA at a dose of 6.8 nmol (Figure 2B), compared with mice in the control groups (Groups 2–4). These results confirm previous work using Stat3-deficient mice that functional Stat3 protein is necessary for maximal TPA-induced epidermal proliferation and further demonstrate that temporal disruption of Stat3 using the TM system effectively reduces Stat3 to levels that are sufficient to reduce the proliferative response to TPA.
Stat3 disruption during initiation significantly reduces skin tumor development
To further determine the role of Stat3 during the initiation stage of chemically induced skin carcinogenesis in vivo, four groups of mice (Groups 1–4 as shown in Figure 1A) were treated with 1 mg of TM or ethanol for five consecutive days prior to initiation with DMBA. Four weeks later, mice were treated with 6.8 nmol of TPA twice weekly. In this experiment, another group of mice was included (Group 5, K5.Cre-ERT2+ × Stat3fl/fl) and was treated with 1 mg of TM for 5 days 1 week after DMBA initiation (see Figure 3A). Inducible Stat3-deficient mice treated with TM before initiation showed a delay of tumor development compared with the other control groups (Group 1, Figure 3B), and the average number of papillomas per mouse at the end of the experiment also was significantly reduced compared with mice in the control groups (P < 0.05, Figure 3C). The average number of papillomas per mouse was also slightly reduced in Stat3-deficient mice treated with TM 1 week after initiation (Group 5) compared with other control groups (Groups 2–4) from week 12 until week 24 (P ≤ 0.05) (see again Figure 3C). However, by the end of the experiment, the average number of papillomas per mouse in this group was not significantly different from Groups 2–4. These data show directly that Stat3 is required for the initiation stage of two-stage skin carcinogenesis, probably through its role in regulating keratinocyte survival in response to DMBA-induced DNA damage.
Stat3 disruption during promotion significantly inhibits skin tumor development
To further determine the role of Stat3 during skin tumor promotion, mice corresponding to Groups 1–4 (Figure 1A) were treated with 1 mg of TM or the ethanol vehicle for five consecutive days starting 1 week after initiation with 100 nmol DMBA. Mice were treated with 6.8 nmol of TPA twice a week starting the following week (Figure 4A). TM treatment for five consecutive days was repeated every 2 weeks and then stopped after 16 weeks of promotion. Inducible deletion of Stat3 during tumor promotion led to a significant delay and a significant reduction in the average number of papillomas per mouse at 16 weeks of promotion (P < 0.05, Group 1 in Figure 4B and C). TM treatment was stopped at week 16 and TPA treatment was continued for an additional 12 weeks. As shown in Figure 4C, papillomas continued to appear in Group 1 mice (TM-induced Stat3 deletion group) until the average number per mouse had reached the level seen in control Group 3 (TM treatment but no Cre expression). Thus, although deletion of Stat3 during promotion inhibited tumor development, initiated cells were still present and subject to clonal expansion via promotion by TPA once Stat3 levels recovered. These data are consistent with our previously proposed mechanism whereby Stat3 deficiency reduces the proliferative response following TPA treatment and directly show that Stat3 is required for the promotion stage of two-stage skin carcinogenesis.
Deletion of Stat3 in preexisting papillomas inhibits subsequent tumor growth
Previously, we showed that skin tumor growth could be inhibited by abrogation of Stat3 function through use of a Stat3-specific decoy oligonucleotide (11). To evaluate whether temporal Stat3 disruption effectively inhibited growth of preexisting papillomas, inducible Stat3-deficient mice (i.e. K5.Cre-ERT2+ × Stat3fl/fl mice) that had developed primary skin tumors were treated with TM by i.p. injection. TM-induced Stat3 disruption inhibited further growth of papillomas as shown in Figure 5. In this regard, the average tumor size in the three control groups increased ∼2-fold over the 4 week observation period, whereas the average tumor size in the inducible Stat3-deficient mice treated with TM was not significantly changed (P ≤ 0.05) (see again Figure 5). Further observation revealed that temporal disruption of Stat3 in primary skin tumors led to a significant reduction in the tumor volume compared with skin tumors treated with ethanol or skin tumors of Cre-negative mice treated with TM or ethanol, as shown in Table I. Among 20 papillomas from the inducible Stat3-deficient mice treated with TM, 60% underwent a reduction in tumor volume. In contrast, ∼50–80% of papillomas in the other groups of mice underwent a significant increase in tumor volume (>60% increase) (Table I). Collectively, these results indicate that inhibition of Stat3 expression by inducible disruption efficiently prevented further growth of skin papillomas.
Fig. 5.
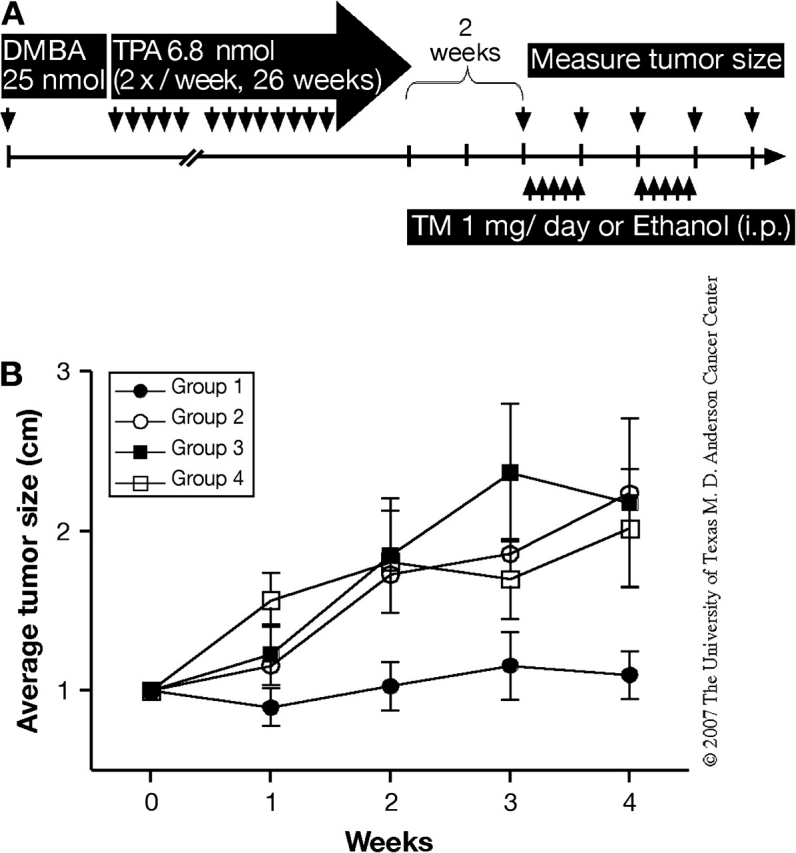
Effects of TM-induced Stat3 deficiency on growth of skin tumors. (A–B) Groups of mice were treated with 25 nmol of DMBA and 2 weeks later received twice-weekly applications of 6.8 nmol of TPA for 26 weeks. After 2 weeks without TPA treatment, the mice were then treated with TM by i.p. injection for five consecutive days on alternate weeks over the 4 week period. (A) Experimental protocol for Stat3 disruption in skin tumors. (B) Average size of skin tumors per group [Group 1, inducible Stat3-deficient mice treated with TM (filled circles); Group 2, inducible Stat3-deficient mice treated with ethanol (open circles); Group 3, Cre− mice treated with TM (filled squares) and Group 4, Cre− mice treated with ethanol (open squares)].
Table I.
Effects of TM-induced Stat3 deficiency on skin tumor volume
| Percent increase of tumor volume | Percent decrease of tumor volume | |||||
| >60 | 30–60 | 0–30 | 0–30 | 30–60 | >60 | |
| Group 1 | 4/20 | 4/20 | 7/20 | 3/20 | 2/20 | |
| Group 2 | 11/14 | 1/14 | 2/14 | |||
| Group 3 | 5/10 | 1/10 | 1/10 | 2/10 | 1/10 | |
| Group 4 | 7/15 | 3/15 | 2/15 | 3/15 | ||
Discussion
In the present study, we demonstrate critical roles for Stat3 in both the initiation and promotion stages of two-stage chemical carcinogenesis in mouse skin through use of a site- and time-specific gene targeting approach. In addition to tissue-specific gene targeting via the Cre/lox system, gene expression can be regulated at a given time during the carcinogenesis process using ligand-inducible Cre recombinase. This conditional site-specific recombination system facilitates further detailed analysis of gene function that cannot be ascertained by conventional gene targeting (16–18). For example, the peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated transcription factor that is involved in nuclear hormone receptor family and is known to play an important role in adipocyte differentiation and fat metabolism (19). However, functional analysis of PPARγ is precluded due to lethality of PPARγ knockout fetuses and tetraploid-rescued pups (20). Temporal and spatial ablation of PPARγ in adipocytes of adult mice by the TM-dependent Cre-ERT2 recombination system revealed that it is essential for the in vivo survival of mature adipocytes (21). Similarly, temporal and spatial ablation of focal adhesion kinase in skin demonstrated that it plays a critical role in malignant progression (22). This inducible recombination system can also be used to selectively activate genes (23–25).
Previous studies from our laboratory have suggested that Stat3 plays important roles in all three stages of skin carcinogenesis by using both skin-specific gain and loss-of-function transgenic mice (reviewed in refs 8,9). Stat3-deficient mice showed an increased response to initiator-induced apoptosis and a reduced response to promoter-induced epidermal hyperproliferation (11). In contrast, keratinocytes from K5.Stat3C transgenic mice showed increased survival following exposure to initiator and enhanced cell proliferation following exposure to promoter (12). Consistent with these observations, Stat3-deficient mice were resistant to skin tumor development and Stat3C mice were hypersensitive to skin tumor development compared with non-transgenic littermates (11,12). These earlier studies suggested critical roles of Stat3 in both the initiation and promotion stages of skin carcinogenesis. However, the complete lack of a tumor response in K5.Cre × Stat3fl/fl mice precluded a more detailed analysis of the specific role of Stat3 in the initiation or promotion stages of skin tumor development.
To evaluate the functional roles of Stat3 independently during the initiation and promotion stages of carcinogenesis, Stat3 was temporally disrupted using the TM-dependent inducible Cre-ERT2 system (20–26). Inducible Stat3-deficient mice (K5.Cre-ERT2+ × Stat3fl/fl mice) showed Cre recombinase expression in the basal cell layer of skin epidermis after TM treatment (data not shown), as observed previously (26). Following Cre activation, the levels of Stat3 and its downstream target genes as assessed by western blot analysis of proteins were significantly reduced in the epidermis (Figure 1B–D). The expression level of Stat3 protein in the inducible Stat3-deficient mice after 5 days of TM treatment was approximately one-fourth the level observed in the control groups based on densitometry (Figure 1B). Thus, although Stat3 protein levels were not totally depleted in the epidermal cells, this reduction was sufficient to cause significant reductions in Bcl-xL, Cyclin D1, Cyclin E and c-Myc (Figure 1D).
In previous studies, Stat3-deficient mice showed a reduced proliferative response following topical treatment of TPA and these mice were highly resistant to two-stage skin carcinogenesis (11). Consistent with these results, the epidermis of inducible Stat3-deficient mice following TM treatment showed a reduced responsiveness to TPA-mediated epidermal hyperproliferation (Figure 2B). In addition, tumor development was significantly delayed and the number of papillomas was also significantly reduced (Figure 4). Notably, the design of this experiment allowed us to show that the reduction in papillomas as a result of reduced Stat3 levels during tumor promotion was reversible once TM treatment was stopped and Stat3 levels recovered. These results support the hypothesis that Stat3 deficiency during tumor promotion primarily affected the clonal expansion of initiated cells and not the survival of already initiated cells. Thus, these results directly confirm a significant role of Stat3 during the tumor promotion stage.
As shown in Figure 3B and C, deletion of Stat3 at the time of initiation with DMBA also significantly reduced the tumor response following tumor promotion with TPA. In our previous work, constitutive deletion of Stat3 sensitized keratinocytes to DNA damage induced apoptosis by both DMBA as well as ultraviolet light (11,27). Furthermore, a significant increase in the number of apoptotic cells induced in epidermis by DMBA was seen in Stat3-deficient mice especially in the bulge region of hair follicles. Consistent with these previous results obtained using constitutive Stat3-deficient mice, TUNEL-positive cells were significantly increased in inducible Stat3-deficient mice 24 h after DMBA treatment (Figure 2A). In a two-stage skin carcinogenesis experiment, TM treatment before DMBA treatment in inducible Stat3-deficient mice led to delayed tumor development and a significantly reduced number of papillomas per mouse compared with corresponding control groups. In this experiment, one group of mice (Group 5) had Stat3 temporally disrupted after initiation with DMBA but before TPA treatment. Tumor development in this group was only slightly reduced compared with the other control groups. This group was included to determine if Stat3 deletion after initiation but before the start of TPA treatment would impact the tumor response. The slightly reduced tumor response in this group relative to the other control groups suggests one of several possibilities as follows: (i) there are still some target cells with DNA damage that may undergo apoptosis following Stat3 deletion; (ii) initiated cells (i.e. cells with Ha-ras mutations) at this early time point may be at some risk of undergoing apoptosis after Stat3 deletion or (iii) Stat3 deletion 1 week after initiation with DMBA may have affected the early stages of tumor promotion. Nevertheless, disruption of Stat3 at the time of initiation significantly reduced the tumor response, which correlated with increased DMBA-induced keratinocyte apoptosis. Stat3 is known to regulate a number of antiapoptotic genes, including Bcl-xL, survivin, Mcl-1 and Bcl-2 (28–32). One or more of these genes may play a significant role in protecting DNA-damaged keratinocytes, including bulge region keratinocytes, from apoptosis. Preliminary experiments using skin-specific Bcl-xL-deficient mice have indicated a partial role for this Stat3-regulated gene in protection of keratinocytes from DMBA-induced apoptosis in vivo (D.J.Kim, K.Kataoka, S.Sano and J.DiGiovanni, unpublished data).
Finally, our previous work showed that direct injection of a Stat3 decoy oligonucleotide into growing papillomas induced regression in some but not all tumors (11). In the current study, inducible deletion of Stat3 significantly prevented further growth of existing papillomas. A number of studies have shown that Stat3 is required for proliferation and survival of cancer cells in culture (33–36). The current results directly demonstrate a requirement for Stat3 in the continued growth of papillomas in this model of epithelial multistage carcinogenesis.
In conclusion, using a system where Stat3 was temporally disrupted in skin keratinocytes, we provide direct evidence for its role in both the initiation and promotion stages of epithelial multistage carcinogenesis. Stat3 is a unique regulator of many key proteins that play an important role throughout the carcinogenesis process (reviewed in refs 8,9). The inducible system used in the current study will allow further dissection of specific Stat3-regulated genes in the various stages of carcinogenesis. Overall, the current data provide additional support for Stat3 as an important target for cancer prevention including during the earliest stages of cancer development.
Funding
National Cancer Institute (CA76520, U01 ES11047 and U01 CA05345); The University of Texas M.D. Anderson Cancer Center (CA16672); National Institute of Environmental Health Sciences Center (ES07784); Odyssey Program and The H-E-B Award for Scientific Achievement at The University of Texas MD Anderson Cancer Center to D.J.K.
Acknowledgments
The authors would like to thank Daniel Metzger and Pierre Chambon (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch Cedex, France) for providing the Cre-ERT2 cDNA used for generating the K5.Cre-ERT2 mice.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- BrdU
5-bromo-2-deoxyuridine
- DMBA
7,12-dimethylbenz[a]anthracene
- i.p.
intraperitoneal
- PBS
phosphate-buffered saline
- PPARγ
peroxisome proliferator-activated receptor γ
- Stat3
signal transducer and activator of transcription 3
- TM
4-hydroxytamoxifen
- TPA
12-O-tetradecanoylphorbol-13-acetate
References
- 1.Bowman T, et al. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 2.Bromberg J. Stat proteins and oncogenesis. J. Clin. Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy DE, et al. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 4.Levy DE, et al. What does Stat3 do? J. Clin. Invest. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 6.Bromberg JF, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 7.Turkson J, et al. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–6626. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- 8.Kim DJ, et al. Signal transducer and activator of transcription 3 (Stat3) in epithelial carcinogenesis. Mol. Carcinog. 2007;46:725–731. doi: 10.1002/mc.20342. [DOI] [PubMed] [Google Scholar]
- 9.Sano S, et al. Impact of Stat3 activation upon skin biology: a dichotomy of its role between homeostasis and diseases. J. Dermatol. Sci. 2008;50:1–14. doi: 10.1016/j.jdermsci.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Chan KS, et al. Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004;64:2382–2389. doi: 10.1158/0008-5472.can-03-3197. [DOI] [PubMed] [Google Scholar]
- 11.Chan KS, et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 2004;114:720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan KS, et al. Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene. 2008;27:1087–94. doi: 10.1038/sj.onc.1210726. [DOI] [PubMed] [Google Scholar]
- 13.Feil R, et al. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 14.DiGiovanni J, et al. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000;60:1561–1570. [PubMed] [Google Scholar]
- 15.Sano S, et al. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999;18:4657–4668. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metzger D, et al. Site- and time-specific gene targeting in the mouse. Methods. 2001;24:71–80. doi: 10.1006/meth.2001.1159. [DOI] [PubMed] [Google Scholar]
- 17.Utomo AR, et al. Temporal, spatial, and cell type-specific control of Cre-mediated DNA recombination in transgenic mice. Nat. Biotechnol. 1999;17:1091–1096. doi: 10.1038/15073. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Otin AL, et al. Mammalian genome targeting using site-specific recombinases. Front. Biosci. 2006;11:1108–1136. doi: 10.2741/1867. [DOI] [PubMed] [Google Scholar]
- 19.Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 20.He W, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl Acad. Sci. USA. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imai T, et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl Acad. Sci. USA. 2004;101:4543–4547. doi: 10.1073/pnas.0400356101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean GW, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mallo M. Controlled gene activation and inactivation in the mouse. Front. Biosci. 2006;11:313–327. doi: 10.2741/1799. [DOI] [PubMed] [Google Scholar]
- 24.Kroll J, et al. Versatile inducible activation system of Akt/PKB signaling pathway in mice. Genesis. 2003;35:160–163. doi: 10.1002/gene.10180. [DOI] [PubMed] [Google Scholar]
- 25.Petrich BG, et al. Temporal activation of c-Jun N-terminal kinase in adult transgenic heart via cre-loxP-mediated DNA recombination. FASEB J. 2003;17:749–751. doi: 10.1096/fj.02-0438fje. [DOI] [PubMed] [Google Scholar]
- 26.Indra AK, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sano S, et al. Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 2005;65:5720–5729. doi: 10.1158/0008-5472.CAN-04-4359. [DOI] [PubMed] [Google Scholar]
- 28.Aoki Y, et al. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003;101:1535–1542. doi: 10.1182/blood-2002-07-2130. [DOI] [PubMed] [Google Scholar]
- 29.Kanda N, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23:4921–4929. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 30.Catlett-Falcone R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 31.Epling-Burnette PK, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Invest. 2001;107:351–362. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gritsko T, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006;12:11–19. doi: 10.1158/1078-0432.CCR-04-1752. [DOI] [PubMed] [Google Scholar]
- 33.Li L, et al. Autocrine-mediated activation of STAT3 correlates with cell proliferation in breast carcinoma lines. J. Biol. Chem. 2002;277:17397–17405. doi: 10.1074/jbc.M109962200. [DOI] [PubMed] [Google Scholar]
- 34.DeArmond D, et al. Autocrine-mediated ErbB-2 kinase activation of STAT3 is required for growth factor independence of pancreatic cancer cell lines. Oncogene. 2003;22:7781–7795. doi: 10.1038/sj.onc.1206966. [DOI] [PubMed] [Google Scholar]
- 35.Mora LB, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62:6659–6666. [PubMed] [Google Scholar]
- 36.Alvarez JV, et al. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]