

Abstract
New blood vessel formation (angiogenesis) is not only essential for the growth of solid tumors but there is also emerging evidence that progression of hematological malignancies like multiple myeloma, acute leukemias, and myeloproliferative neoplasms, also depends on new blood vessel formation. Anti-angiogenic strategies have become an important therapeutic modality for solid tumors. Several anti-angiogenic agents targeting angiogenesis-related pathways like monoclonal antibodies, receptor tyrosine kinase inhibitors, immunomodulatory drugs, and proteasome inhibitors have been entered clinical trials or have been already approved for the treatment of hematological malignancies as well and in some instances these pathways have emerged as promising therapeutic targets. This review summarizes recent advances in the basic understanding of the role of angiogenesis in hematological malignancies and clinical trials with novel therapeutic approaches targeting angiogenesis.
Introduction
The hypothesis of tumor angiogenesis in malignancies was raised by Judah Folkman: To grow over a certain size of a few millimetres in diameter solid tumors need blood supply from surrounding vessel [1]. Up to 2-3 mm3 solid tumors can grow without blood vessel supply. Nutrition and oxygen is provided via diffusion from the surrounding tissue. Above this size, diffusion becomes insufficient due to the negative surface/volume ratio. Based on a balance between angiogenic and anti-angiogenic growth factors, a tumor of this size can stay dormant for a very long time period until the so-called angiogenic switch occurs [2]. Tumor blood vessels are generated by various mechanisms, such as expansion of the host vascular network by budding of endothelial sprouts (sprouting angiogenesis), cooption of the existing vascular network, remodeling and expansion of vessels by the insertion of interstitial tissue columns into the lumen of preexisting vessels (intussusceptive angiogenesis) and homing of endothelial cell precursors (EPC; CEP) from the bone marrow or peripheral blood into the endothelial lining of neovessels (vasculogenesis) [3].
Tight control of angiogenesis is maintained by a balance of endogenous anti-angiogenic and pro-angiogenic factors [4]. VEGF has a key, rate-limiting role in promoting tumor angiogenesis and exerts its effects by binding to one of three tyrosine kinase receptors: VEGF receptor-1 (VEGFR-1; fms-like tyrosine kinase-1, Flt-1), VEGFR-2 (human kinase domain region, KDR/murine fetal liver kinase-1, Flk-1) and VEGFR-3 (Flt-4). VEGFR-1 (ligands include VEGF-A, -B and placental growth factor [PIGF]) and VEGFR-2 (ligands include VEGF-A, -C and -D) are predominantly expressed on vascular endothelial cells, and activation of VEGFR-2 appears to be both, necessary and sufficient, to mediate VEGF-dependent angiogenesis and induction of vascular permeability [4,5]. Both receptor tyrosine kinases are expressed in all adult endothelial cells, except for the brain endothelial cells. VEGFR-1 is also expressed on hematopoietic stem cells, vascular smooth muscle cells, monocytes, and leukemic cells [6,7], while VEGFR-2 is expressed on endothelial progenitor cells and megakaryocytes [8,9]. VEGFR-3, largely restricted to lymphatic endothelial cells, binds the VEGF homologues VEGF-C and VEGF-D and may play an important role in the regulation of lymphangiogenesis. Thus, VEGF and VEGFR represent significant anti-cancer therapy targets, which elegantly bypass potential tumor-related treatment barriers [4].
A further important pathway in angiogenesis is the recently identified Delta-Notch pathway, and particularly the ligand Delta-like 4 (Dll4), was identified as a new target in tumor angiogenesis [10]. Dll4 is highly expressed by vascular endothelial cells and induced by VEGF [11]. It interacts with Notch cell surface receptors to act as a negative feedback inhibitor downstream of VEGF signaling to restrain the sprouting and branching of new blood vessels [10,12]. Inhibition of Dll4-Notch signaling induces an increase in vessel density but these blood vessels are abnormal and not perfused [13]. Therefore intratumour hypoxia is increased and leads to induction of transcription of proangiogenic genes regulated by Hypoxia inducible factor-1 (HIF-1) [10,14]. Disruption of Dll4 signaling by overexpression or inhibition of Dll4 may impair angiogenesis and blockade of Dll4-Notch signaling results in an increased density of nonfunctional vasculature and is associated with a reduction in the growth of human tumor xenografts [13,14]. Further, certain xenografts that are resistant to anti-VEGF therapy are reported to be sensitive to anti-Dll4 and combination treatment with anti-VEGF and anti-Dll4 has additive inhibitory effects on tumor growth [13-15].
This review summarizes the role of pathological angiogenesis in hematological malignancies focusing on multiple myelomas (MM), acute leukemias, and myeloproliferative neoplasms (MPN) and its therapeutic intervention with novel agents within clinical trials or already approved.
Pathophysiology of angiogenesis in hematological malignancies
Many studies suggest a role for angiogenesis not only in the pathogenesis of solid tumors but also in hematological malignancies like acute and chronic leukemia, lymphoma, myelodysplastic syndromes, myeloproliferative neoplasms, and multiple myeloma [16-21]. We and others reported an increased microvessel density and VEGF expression in the bone marrow of patients with myeloproliferative neoplasms and lymphoma [17,20]. Thereby, the extent of angiogenesis in the bone marrow often correlated with disease burden, progonosis, and treatment outcome [22,23]. In the neoplastic bone marrow there is an imbalance of the cells, cytokines and growth factors maintaining physiological angiogenesis in the normal bone marrow. The bone marrow tumor cells upregulates several factors, including interleukin-6, granulocyte-macrophage colony-stimulating factor and VEGF, have autocrine and paracrine effects acting on multiple cell types, thereby stimulating angiogenesis and leading to increased vascularity [7,24]. The role for VEGF in hematogical malignancies has been extensively studied since its isolation from the leukemia cell line HL- 60 in 1989 [25]. Apparently, this growth factor is expressed in many other leukemic cell lines [7,26] and a subset of leukemic cells also expresses VEGFR-2 which allows VEGF to act as autocrine growth factor in leukemia [26,27]. In addition to that, isolated blast cells from leukemia patients also produce VEGF [26] and the cellular level of VEGF in acute myeloid leukemia (AML) patients has been identified as independent prognostic risk factor [28]. VEGF from leukemic blasts contributes to disease progression, either as positive regulator for proliferation and apoptosis protection for the blast itself or by activating the surrounding stroma cells with subsequent induction of bone marrow angiogenesis.
Regarding the Notch pathway, Notch signals are oncogenic in hematogical malignancies in many cellular contexts [29]. Activating Notch-mutations have been shown to be present in at least 50% of human T-cell acute lymphoblastic leukaemia (T-ALL) cases and have been proved to play a unifying role in the pathogenesis of T-ALL [30]. An important role of Notch has been proposed in cell survival in several B-cell malignancies such as Hodgkin's disease [31,32] and in two B-cell non-Hodgkin lymphoma entities, chronic lymphocytic leukaemia (CLL) [33-35] and in MM [36,37].
Multiple myeloma
MM was the first hematological malignancy, in which increased angiogenesis rate was detected [21,38]. MM is characterized by proliferation of malignant plasma cells that accumulate in the bone marrow and often produce a monoclonal immunoglobulin. New vessel formation in the bone marrow seems to play an important role in the pathogenesis of MM [39,40]. Increased bone marrow microvessel density (MVD) in patients with MM appears to be also an important prognostic factor [41]. Malignant plasma cells can secrete various cytokines, including VEGF, basic fibroblast growth factor (bFGF), and hepatocyte growth factor (HGF), all known for their pro-angiogenic activity [42]. It has been shown that MM cells are capable of secreting VEGF in response to Interleukin-6 (IL-6) stimulation; in response to that VEGF stimulation microvascular endothelial cells and bone marrow stromal cells secrete in turn IL-6, a potent growth factor for malignant plasma cells, thus closing a paracrine loop [43]. Specifically, increased microvessel density (MVD) in the BM of MM patients has been correlated with disease progression and poor prognosis [21,23]. Moreover, VEGF also exerts direct effects on MM cell migration, proliferation, survival, and drug resistance. VEGF triggered effects in MM cells are predominantly mediated via VEGFR 1 and in endothelial cells, predominantly via VEGF R2 [44]. Rajkumar et al. showed a gradual increase of bone marrow angiogenesis along the disease spectrum from monoclonal gammopathy of undetermined significance (MGUS) to smoldering MM, newly diagnosed MM and relapsed MM [45], though the expression levels of VEGF, bFGF, and their receptors were similar among MGUS, smoldering MM, and newly diagnosed MM [46], rising the hypothesis that MVD increase in plasma cell neoplasias could be rather a function of chronology.
Acute leukemias
The first demonstration that leukemia progression might be accompanied by an increase of bone marrow vascularization was provided by Judah Folkman's group [47]. In their studies, it was demonstrated that the bone marrow of acute lymphoblastic leukemia (ALL) patients had increased blood vessel content, compared to normal counterparts. Moreover, it was also shown that urine and peripheral blood samples from ALL patients contained elevated levels of pro-angiogenic growth factors, namely bFGF and VEGF, which correlated with the increase of bone marrow angiogenesis [48]. The existence of an "angiogenesis switch", first proposed for solid tumors [49], was therefore suggested to apply to hematological malignancies as well. "Angiogenesis switch" in leukemia is documented by increased bone marrow MVD, increased expression of HIF-1, multiple pro-angiogenic factors (VEGF, bFGF, angiopoietin-2), soluble VEGFR, and decreased expression of endogenous angiogenesis inhibitors, such as thrombospondin-1 [50,51].
In a recent study by Norén-Nyström et al. [52] MVD, analyzed on 185 bone marrow biopsies, was higher in T-ALL compared to B-ALL. In the B-ALL group, cases with t(12;21) were characterized by a low MVD, while patients with hyperdiploid leukemia showed a high MVD. Similarly, in previously untreated acute myeloid leukemia (AML), increased levels of plasma VEGF correlate with reduced survival and lower remission rates [53]. In addition to that, isolated blast cells from leukemia patients also produce VEGF and the cellular level of VEGF in AML patients has been identified as independent prognostic risk factor [28]. In a reccent study [54] dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) was used as a non-invasive technique to measure bone marrow angiogenesis in AML. DCE-MRI was performed beforte treatment and on day 7 after induction chemotherapy. Thereby, bone marrow angiogenesis with remission, rate overall and disease-free survival.
Myeloproliferative neoplasms
The available data on angiogenesis and expression of VEGF and its receptors in the bone marrow of patients with BCR-ABL1-negative myeloproliferative neoplasms (MPN) suggest that MVD is increased, especially in primary myelofibrosis (PMF), and that increased angiogenesis might inversely correlate with survival [55-58]. In a recent study, we found a significantly increased MVD and VEGF expression in MPN compared to controls especially in cases with high JAK2-V617F mutant allele burdens [17]. The identification of an acquired somatic mutation in the JAK2 gene, resulting in a valine to phenylalanine substitution at position 617 (JAK2-V617F), has provided new insights into the pathogenesis of BCR-ABL1-negative MPN, being present in most patients with polycythaemia vera (PV) and in about 50% of patients with essential thrombocythemia (ET) and PMF [59,60]. In another study by Alonci et al. in patients with MPN, serum levels of VEGF and VEGFR-2 was examined. In MPN, VEGF levels were higher compared to controls, wheresas VEGFR-2 levels was reduced in ET but not in PV and PMF [61].
Anti-angiogenic therapies in hematological malignancies
Anti-angiogenic therapies are mostly based on inhibiting the binding of VEGF to VEGFR by neutralizing antibodies to the ligand or to the receptor, soluble receptors, small molecule inhibitors or are directed against the tyrosine kinase activity of the VEGF receptors (Figure 1). The first anti-angiogenic agent to be approved in solid tumors was bevacizumab (Avastin™, Genentech), a humanized anti-VEGF monoclonal antibody. Administration of bevacizumab, in combination with cytotoxic chemotherapy, conferred benefits to patients with metastatic colorectal cancer, non-squamous, non-small cell lung cancer and metastatic breast cancer [62-64]. Additionally, two small-molecule inhibitors targeting VEGFRs and other kinases, sorafenib (Nexavar™, Bayer and Onyx pharmaceuticals) and sunitinib (Sutent™, Pfizer), have been approved based on their efficacy in treating renal cell- and hepatocellular carcinoma [65,66]. A growing list of anti-angiogenics is now available, either in various stages of clinical development or as components of standard clinical regimens. The major classes of anti-angiogenic therapy include: (1) direct anti-VEGF acting molecules (anti-VEGF antibodies, VEGF-antisense nucleotides); (2) immunomodulatory drugs (IMIDs) with antiangiogenic properties; (3) receptor tyrosine kinase inhibitors, targeting VEGFR signaling as well as receptors of other (pro-angiogenic) factors; (4) anti-endothelial approach of metronomic therapy and (5) other new compounds, targeting signaling downstream to pro-angiogenic growth factors, such as mammalian target of rapamycin (mTOR) inhibitors, histone deacetylases' (HDAC) inhibitors and proteasome inhibitors.
Figure 1.

Therapeutic strategies to target the VEGF/VEGF receptor system. VEGF, vascular endothelial growth factor.
In our review, we will focus on several molecules interfering with the VEGF/VEGFR system, which already have been approved or are currently evaluated in clinical trials for treatment of hematological malignancies (Table 1).
Table 1.
Selection of clinical trials and approved anti-angiogenic therapies in hematological malignancies
| Drug | Target | Study entities | Approved for |
|---|---|---|---|
| Receptor tyrosine kinase inhibitors | |||
| PTK787/ZK 222584 (Vatalanib®) | VEGFR1-3, PDGFRβ, c-Kit | AML, PMF, MDS, CML, DLBCL, MM | |
| SU5416 (Semaxinib) |
VEGFR1-2, c-kit, Flt3 | AML, MDS, MM, MPN | |
| Sorafenib (Nexavar®) | VEGFR2-3, B-Raf, Faf-1, PDGFRβ | AML, ALL, MDS, CML, CLL, NHL, MM | Advanced renal cell carcinoma, HCC |
| Sunitinib (Sutent®) | VEGFR1-3, PDGFRα+β, c-kit, Flt3 | AML, MDS, CLL, Myeloma, NHL | Advanced renal cell carcinoma, GIST |
| PKC-412 (Midostaurin) | VEGFR2, PKC, PDGFR, Flt3, c-Kit | AML | |
| Cediranib (Recentin®) | VEGFR1-3, PDGFRβ, c-Kit | AML, MDS, CLL | |
| Proteasome inhibitors | |||
| Bortezomib (Velcade®) | 26S proteasome, NF-κB | AML, ALL, MDS, CML, NHL, MCL | MM, MCL |
| Anti-VEGF strategies | |||
| Bevacizumab (Avastin®) | VEGF-A | AML, MDS, CLL, CML, NHL, MM | Metastatic colorectal cancer, NSCLC, breast cancer |
| Immunomodulatory drugs | |||
| Thalidomide | bFGF, VEGF, IL-6 | AML, MDS, MPN, CLL, NHL, MM | MM |
| Lenalidomide (Revlimid®) | bFGF, VEGF, IL-6 | AML, MDS, CLL, NHL | MM, 5q- MDS |
AML, acute myeloid leukemia; bFGF, basic fibroblast growth factor; DLBCL, diffuse large B-cell lymphoma; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; GIST, gastrointestinal stromal tumors; HCC, hepatocellular carcinoma; IL-6, Interleukin-6; NHL, non-Hodgkin lymphoma; NSCLC, non-small cell lung cancer; MCL, mantle cell lymphoma; MDS, myelodysplastic syndrome; MM, multiple myeloma; MPN, myeloproliferative neoplasm; PMF, primary myelofibrosis; VEGF, vascular endothelial growth factor.
Anti-VEGF monoclonal antibodies
Bevacizumab
The humanized monoclonal anti-VEGF antibody bevacizumab (Avastin®) is the first drug targeting VEGF and is officially approved in combination with chemotherapy. Bevacizumab is a humanized murine anti-human VEGF monoclonal IgG1 antibody that blocks the binding of human VEGF to its receptors, thereby disrupting also autocrine and paracrine survival mechanisms mediated by VEGFR-1 and VEGFR-2 [67]. Bevacizumab was approved for advanced non-small cell lung cancer (NSCLC), breast cancer, and colorectal cancer. In patients with refractory AML (n = 9) bevacizumab resulted in reduction of VEGF expression in the bone marrow but without a clinical response [68]. Bevacizumab-associated side effects are generally mild to moderate in severity, although there are specific, uncommon events that are more severe and potentially lifethreatening. The most commonly observed adverse events are hypertension, proteinuria, bleeding and thrombosis, which are generally mild to moderate and manageable [68]. In a phase II clinical trial by Karp et al. bevacizumab was administered after chemotherapy to adults with refractory or relapsed AML [69]. Bevacizumab 10 mg/kg was administered on day 8 after cytarabine beginning day 1 and mitoxantrone beginning day 4. Forty-eight adults received induction therapy. Overall response was 23 of 48 (48%), with complete response (CR) in 16 (33%). Eighteen patients (14 CR and 4 partial responses) underwent one consolidation cycle and 5 (3 CR and 2 partial responses) underwent allogeneic transplant. Median overall and disease-free survivals for CR patients were 16.2 months (64%, 1 year) and 7 months (35%, 1 year), respectively. As biomarkers, the microvessel density in bone marrow biopsies, serum VEGF levels and the expression of the VEGFR-1 FLT1 by AML marrow blasts were determined. The expression of FLT-1 in pretreatment AML marrow cells was higher compared to normal bone marrow. Bone marrow samples demonstrated marked MVD decrease after bevacizumab. VEGF was detected in pretreatment serum in 67% of patients tested, increased by day 8 in 52%, and decreased in 93% (67% undetectable) 2 h after bevacizumab. Currently, bevacizumab is evaluated as treatment option for newly diagnosed AML in combination with cytarabine and idarubicin in a phase II study.
Anticalins©
The anticalins represent a novel class of human binding proteins. PRS-050 is an anticalin with extended serum half-life due to pegylation. This anticalin targets VEGF and exhibits favourable binding und functional in vitro activity profile in direct comparison to the currently approved VEGF antagonists. A strong enhanced vascular permeability was demonstrated. The wordwide "first trial in man" is running since May 2010 and first results will be expected in 2011. The potential use will be similar to Bevacizumab. Its usefulness in malignant hematological disorders has to be explored after the phase I study.
Receptor tyrosine kinase inhibitors
Small tyrosine kinase inhibitors that target VEGFR are a further important class of anti-angiogenic drugs. Their efficacy in hematological malignancies, especially in AML, might be attributable to inhibition of a lot of pathways, especially such related to c-kit and Flt3.
SU5416 (Semaxinib) is a small molecule inhibitor of VEGFR-1 and 2, c-kit and Flt3 [70-72]. In a phase II study, 42 patients with advanced AML were treated [70]. 7 patients achieved a partial response (reduction of blasts by at least 50%), with one complete morphological response lasting 2 months. Treatment was generally well tolerated. Most study drug-related adverse events were mild to moderate in severity and the most frequently reported adverse events were nausea, bone/muscoloskeletal pain, headache, insomnia, and vomiting. As biomarkers, VEGF RNA expression by leukemic blasts, bone marrow MVD, and the expression of the target receptors c-kit, Flt3, VEGFR-1, and VEGFR-2 prior to therapy was determined. Patients with AML blasts expressing high levels of VEGF mRNA by quantitative polymerase chain reaction (PCR) had a significantly higher response rate and reduction of bone marrow MVD than patients with low VEGF expression consistent with the anti-angiogenic effects of SU5416. Patients with a high c-kit expression had a lower response.
Vatalanib (formerly PTK787/ZK 222584) is an oral protein kinase inhibitor (PTK) acting as angiogenesis inhibitor that is active against VEGFR and PDGFR tyrosine kinases, thereby offering a novel approach to inhibiting tumor growth [73]. It interferes with the ATP binding sites of VEGFR. In a phase I study by us, vatalanib was well tolerated and showed clinical activity in a variety of solid tumors [74]. It is active in MM by primarily reducing the number of tumor microvessels, accompanied by dilation of the remaining vessels [75,76]. Ongoing studies evaluate the efficacy of valatinib in combination with imatinib in a phase I/II trial for patients with AML, PMF, and blast phase of chronic myelogenous leukemia. Vatalanib was studied in a phase I clinical trial alone or in combination with cytosine-arabinoside and daunorubicin in patients with myelodysplastic syndromes (MDS) and AML [77]. Sixty-three patients received vatalanib at doses of 500-1000 mg/bid orally. At 1000 mg/bid, dose-limiting toxicities such as lethargy, hypertension, nausea, emesis and anorexia were observed. CR was observed in 5 of 17 evaluable AML patients treated with vatalanib combined with chemotherapy. The authors concluded that vatalanib is generally well tolerated and can be given in combination with chemotherapy in patients with MDS and AML. In a recently study by Barbarroja et al. [78] vatalanib was examined in combination with idarubicin in 4 AML cell lines and 7 AML patients samples. Vatalanib decreased VEGF levels and VEGFR phosphorylation in AML cells, which showed FLT3 internal tandem reduplications/mutations (ITD), raising the question of the actual targeted tyrosine kinase (VEGFR or flt3). In another study, vatalanib was given to 29 patients with PMF at doses of 500 or 750 mg/bid. One patient (3%) achieved CR and 5 (17%) clinical improvement. All together, vatalanib had modest activity in patients with PMF [79].
Cediranib (AZD2171, Recentin®) is a potent inhibitor of both VEGFR-1 and VEGFR-2; it also has activity against c-kit, PDGFR-β, and VEGFR-3 at nanomolar concentrations [80]. In our study, cediranib was well tolerated up to 45 mg/d in patients with a broad range of solid tumors [81]. The most common toxicities included diarrhea, dysphonia, and hypertension. In a phase I study with cediranib in 35 AML patients the most common adverse events were diarrhea, hypertension and fatigue. Six patients experienced an objective response (3 each at 20 and 30 mg). Dose and time-dependent reductions of soluble VEGFR-2 were observed, and there was a correlation between cediranib exposure and plasma VEGF levels [82].
Immunomodulatory drugs (IMiDs)
Thalidomide was originally introduced as sedative and withdrawn in the 1960's due to deleterious side effects. Recently, there is increasing evidence for the efficacy of thalidomide in cancer therapy. Multiple myeloma is one of the first clinical entities for which this could be demonstrated [83,84]. The surprising effects of thalidomide have led to the development of a series of IMiDs with even higher anti-angiogenic potency (Figure 2). It has has been shown that thalidomide has important immunomodulatory effects by decreasing TNF-α synthesis and slectively modulating T cell subsets shifting the T cell population towards T helpers [85]. The interest on thalidomide as an anti-neoplastic agent rose after demonstration of its anti-angiogenic activity in a rabbit model of corneal neovascularization that was induced in response to bFGF [86]. Thalidomide and the newer immunomodulatory drugs (IMiDs) (e.g. lenalidomide) have been shown to significantly decrease the expression of the pro-angiogenic factors VEGF and Interleukin-6 (IL-6) in MM [87]. The newer IMiDs were found to be 2-3 times more potent compared to thalidomide concerning anti-angiogenic activity in various in vivo assays [88]. The anti-angiogenic activity of IMiDs has been shown to be independent of their immunomodulatory effects [89].
Figure 2.
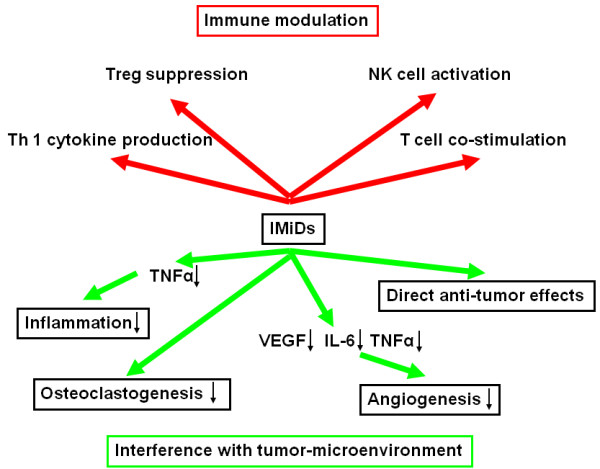
Modes of action of IMiDs. IL-6, interleukin-6; IMiDs, immunomodulatory drugs; NK cells, Natural killer cells; Th1 cells, T helper 1 cells; TNFα, tumor necrosis factor alpha; Treg, regulatory T cells; VEGF, vascular endothelial growth factor.
Thalidomide monotherapy in a phase II trial, in which 84 patients with relapsed and refractory MM received doses ranging from 200 to 800 mg/d, resulted in an overall response rate of 32%. The 2-year event-free survival and overall survival were 20 and 48%, respectively [83,84]. In combination with dexamethasone the response rate was 63% compared to 41% with dexamethasone alone in patients with newly diagnosed MM [90]. Thalidomide was approved for the treatment of newly diagnosed MM.
In patients with AML, thalidomide was examined as mono- and combination therapy. In a phase II study by Thomas et al. [91] thalidomide was analyzed in patients with relapsed or refractory AML previously treated with cytarabine-containing regimens. A total of 16 patients were treated with oral thalidomide 200-800 mg/d. Overall, one patient (6%) achieved CR lasting for 36 months, and two patients had a transient reduction in marrow blasts from 8% and 7% to less than 5% in both cases. There was no correlation between reduction of angiogenesis markers' levels and response. In a phase I/II trial by Steins et al. [92] a dose-escalating trial was performed to study the safety and efficacy of thalidomide in 20 AML patients. Thirteen patients were assessable for both toxicity and response, tolerating a maximum dose of 200-400 mg/d for at least 1 month. Overall, adverse events were fatigue, constipation, rash, and neuropathy (grade 1 to 2 in most patients). In 4 patients, a partial response, defined as reduction of at least 50% of the blast cell infiltration in the bone marrow accompanied by increases of platelet counts and hemoglobin values, was observed. In parallel, MVD significantly decreased in these 5 patients during treatment with thalidomide. A combination therapy of thalidomide and 5-azacytidine, a hypomethylating drug, was examined in 40 patients with MDS and AML [93]. A hematological improvement was observed in 15 of 36 patients (42%), stable disease was observed in 5 of 36 patients (14%), and 10 of 36 patients (28%) had disease progression. Six patients had CR.
In a phase II study with 44 PMF patients thalidomide was examined as monotherapy [94]. Seventeen of 41 evaluable patients (41%) receiving treatment for at least 15 days showed a response. A complette remission (without reversal of bone marrow fibrosis) was achieved in 4 patients (10%), a partial response was achieved in 4 patients (10%), and hematological improvements of anemia, thrombopenia, and/or splenomegaly were observed in 9 patients (21%).
Lenalidomide, a synthetic compound derived by modifying the chemical structure of thalidomide, has also immunomodulatory and anti-angiogenic properties, while showing lower adverse effects rates [95]. In patients with previously treated relapsed/refractory MM, the combination of lenalidomide with dexamethasone increased the response rate from 22.5% to 59.2% compared to dexamethasone alone [96,97]. In 2 phase III trials lenalidomide in combination with dexamethasone, it showed remarkable response rates and better toxicity profile than thalidomide [96,97]. Lenalidomide was approved in combination with dexamethasone for the second-line treatment of MM.
In phase II studies with lenalidomide monotherapy in patients with symptomatic PMF, the overall response rates were 22% for anemia, 33% for splenomegaly, and 50% for thrombocytopenia [98]. In a combination study of lenalidomide with prednisone, 40 patients with PMF were included [99]. Responses were recorded in 12 patients (30%) and are ongoing in 10 (25%). The median time to response was 12 weeks. Three patients (7.5%) had partial response and nine patients (22.5%) had clinical improvement durable for a median of 18 months. Overall response rates were 30% for anemia and 42% for splenomegaly. Interestingly, all eight JAK2-V617F-positive responders experienced a reduction of the baseline mutant allele burden as well.
Proteasome inhibitors
Bortezomib (Velcade®), a boronic acid dipeptide, is a selective, but reversible proteasome inhibitor [100]. It has been approved for clinical use in humans, in particular for treatment MM and mantle cell lymphoma. Beside its direct anti-tumor effects, anti-angiogenic actions of bortezomib have recently been described in vitro and in vivo [100]. In a study by Roccaro et al. [101] the effect of bortezomib on the angiogenic phenotype of MM patient-derived endothelial cells was examined. Bortezomib inhibited the proliferation of endothelial cells and angiogeneis in a dose-dependent manner.
In a phase III study, patients with MM progressing after at least one prior therapy, were randomized to receive single-agent bortezomib or high-dose dexamethasone [102]. Alltogether 669 patients were included. Time to progression was significantly prolonged in the bortezomib treatment arm (median, 6.2 months) compared with the dexamethasone arm (median, 3.5 months). Analysis of overall survival done on the interim database (with 20% of events) showed the superiority of bortezomib for patients. The response rate (complete plus partial response) with bortezomib was also superior to dexamethasone (38% versus 18%). Adverse events on the bortezomib arm were similar to those previously observed in phase II studies; some notable adverse events being asthenia, peripheral neuropathy, thrombocytopenia, and neutropenia. In another phase III study, bortezomib, melphalan, and prednisone (VMP) was examined versus melphalan and prednisone (MP) in previously untreated symptomatic MM patients ineligible for high-dose therapy [103]. VMP resulted in a 35% reduced risk of death compared to MP and prolonged overalls survival. In a phase I/II study by Richardson et al. [104] the combination lenalidomide, bortezomib, and dexamethasone was evaluated in front-line myeloma. The partial response rate was 100% in both the phase II population and overall, with 74% and 67% each achieving very good partial response or better. The combination lenalidomide, bortezomib, and dexamethasone demonstrated favorable tolerability and was highly effective in the treatment of newly diagnosed myeloma. In a phase I study, bortezomib was added to induction chemotherapy in patients with AML [105]. The combination of bortezomib, idarubicin, and cytarabine showed a good safety profile. The recommended dose of bortezomib for phase II studies with idarubicin and cytarabine was 1.5 mg/m2. Overall, 19 patients (61%) achieved complete remission (CR) and three had CR with incomplete platelet recovery.
Conclusions and future directions
Angiogenic and especially VEGF/VEGFR pathways are involved in the pathophysiology of hematological malignancies including multiple myeloma, acute and chronic leukemias, MPN and lymphomas. Although VEGF/VEGFR-related pathways seems to be the most relevant regulators of neoangiogenesis, vasculogenesis and recruitment of endothelial progenitor cells in such instances, but other pathways are important too. Further, VEGF/VEGFR interactions can stimulate proliferation, migration and survival of leukemia/lymphoma cells by autocrinous and paracrinous loops. Novel agents, targeting VEGF, its receptors, and other angiogenic pathways, are in various stages of clinical development and investigation in hematological malignancies. As we know from the the treatment of solid tumors, combination therapies of different anti-angiogenic molecules with chemotherapy or irradiation increases treatment efficacy. Especially, as blocking VEGF activity has been shown to sensitize the vasculature and improve the delivery of cytotoxic drugs to tumor and endothelial cells. However, not all patients treated with anti-angiogenic therapies benefit from this kind of therapy and in most cases, the effect is transient. Therefore, there is an urgent need for biomarkers to identify patients likely to benefit from anti-angiogenic treatments, to select the optimal dose to minimize side effects, and to understand the mechanisms of resistance. Preclinical models suggest multiple mechanisms involved in acquired or primary resistance against anti-angiogenic therapies. Finally, also these "targeted therapies" has side effects profiles which must be considered carefully.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MM and KM selected publications for the review, drafted manuscript. Both authors read and approved the final manuscript.
Contributor Information
Michael Medinger, Email: medingerm@uhbs.ch.
Klaus Mross, Email: mross@tumorbio.uni-freiburg.de.
References
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid, and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu ZP, Hackett NR, Crystal RG, Moore MAS, Hajjar KA, Manova K, Benezra R, Rafii S. Impaired recruitment of bone-marrow derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, Pelletier N, Ferrara N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- Hattori K, Dias S, Heissig B, Hackett NR, Lyden D, Tateno M, Hicklin DJ, Zhu Z, Witte L, Crystal RG, Moore MA, Rafii S. Vascular endothelial growth factor and angiopoietin-1 stimulate postnatal hematopoiesis by recruitment of vasculogenic and hematopoietic stem cells. J Exp Med. 2001;193:1005–1014. doi: 10.1084/jem.193.9.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy WT, Richter L, Frutiger Y, Grogan TM. Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999;59:728–733. [PubMed] [Google Scholar]
- Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, Girardi L, Yurt R, Himel H, Rafii S. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–174. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- Casella I, Feccia T, Chelucci C, Samoggia P, Castelli G, Guerriero R, Parolini I, Petrucci E, Pelosi E, Morsilli O, Gabbianelli M, Testa U, Peschle C. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood. 2003;101:1316–1323. doi: 10.1182/blood-2002-07-2184. [DOI] [PubMed] [Google Scholar]
- Thurston G, Noguera-Troise I, Yancopoulos GD. The delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7:327–331. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- Yan M, Plowman GD. Delta-like 4/Notch signaling and its therapeutic implications. Clin Cancer Res. 2007;13:7243–7246. doi: 10.1158/1078-0432.CCR-07-1393. [DOI] [PubMed] [Google Scholar]
- Diez H, Fischer A, Winkler A, Hu CJ, Hatzopoulos AK, Breier G, Gessler M. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp Cell Res. 2007;313:1–9. doi: 10.1016/j.yexcr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo JA, de Sauvage F, Plowman G, Yan M. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–1087. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, Biswas S, Turley H, Heikamp E, Hainfellner JA, Harris AL. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res. 2007;67:11244–11253. doi: 10.1158/0008-5472.CAN-07-0969. [DOI] [PubMed] [Google Scholar]
- Aguayo A. The role of angiogenesis in the biology and therapy of myelodysplastic syndromes. Curr Hematol Rep. 2004;3:184–191. [PubMed] [Google Scholar]
- Medinger M, Skoda R, Gratwohl A, Theocharides A, Buser A, Heim D, Dirnhofer S, Tichelli A, Tzankov A. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. Br J Haematol. 2009;146:150–157. doi: 10.1111/j.1365-2141.2009.07726.x. [DOI] [PubMed] [Google Scholar]
- Padró T, Ruiz S, Bieker R, Bürger H, Steins M, Kienast J, Büchner T, Berdel WE, Mesters RM. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95:2637–2644. [PubMed] [Google Scholar]
- Pruneri G, Bertolini F, Soligo D, Carboni N, Cortelezzi A, Ferrucci PF, Buffa R, Lambertenghi-Deliliers G, Pezzella F. Angiogenesis in myelodysplastic syndromes. Br J Cancer. 1999;81:1398–1401. doi: 10.1038/sj.bjc.6693515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzankov A, Heiss S, Ebner S, Sterlacci W, Schaefer G, Augustin F, Fiegl M, Dirnhofer S. Angiogenesis in nodal B cell lymphomas: a high throughput study. J Clin Pathol. 2007;60:476–482. doi: 10.1136/jcp.2006.038661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, Dammacco F. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503–508. doi: 10.1111/j.1365-2141.1994.tb08304.x. [DOI] [PubMed] [Google Scholar]
- Loges S, Heil G, Bruweleit M, Schoder V, Butzal M, Fischer U, Gehling UM, Schuch G, Hossfeld DK, Fiedler W. Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol. 2005;23:1109–1117. doi: 10.1200/JCO.2005.05.058. [DOI] [PubMed] [Google Scholar]
- Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, Dammacco F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:30643073. [PubMed] [Google Scholar]
- Majka M, Janowska-Wieczorek A, Ratajczak J, Ehrenman K, Pietrzkowski Z, Kowalska MA, Gewirtz AM, Emerson SG, Ratajczak MZ. Numerous growth factors, cytokines, and chemokines are secreted by human CD34(+) cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood. 2001;97:3075–3085. doi: 10.1182/blood.V97.10.3075. [DOI] [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Fiedler W, Graeven U, Ergün S, Verago S, Kilic N, Stockschläder M, Hossfeld DK. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood. 1997;89:1870–1875. [PubMed] [Google Scholar]
- Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511–521. doi: 10.1172/JCI8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo A, Estey E, Kantarjian H, Mansouri T, Gidel C, Keating M. Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood. 1999;94:3717–3721. [PubMed] [Google Scholar]
- Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66:1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Jundt F, Anagnostopoulos I, Förster R, Mathas S, Stein H, Dörken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002;99:3398–3403. doi: 10.1182/blood.V99.9.3398. [DOI] [PubMed] [Google Scholar]
- Jundt F, Acikgöz O, Kwon SH, Schwarzer R, Anagnostopoulos I, Wiesner B, Mathas S, Hummel M, Stein H, Reichardt HM, Dörken B. Aberrant expression of Notch1 interferes with the B-lymphoid phenotype of neoplastic B cells in classical Hodgkin lymphoma. Leukemia. 2008;22:1587–1594. doi: 10.1038/leu.2008.101. [DOI] [PubMed] [Google Scholar]
- Hubmann R, Schwarzmeier JD, Shehata M, Hilgarth M, Duechler M, Dettke M, Berger R. Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood. 2002;99:3742–3747. doi: 10.1182/blood.V99.10.3742. [DOI] [PubMed] [Google Scholar]
- Duechler M, Shehata M, Schwarzmeier JD, Hoelbl A, Hilgarth M, Hubmann R. Induction of apoptosis by proteasome inhibitors in B-CLL cells is associated with downregulation of CD23 and inactivation of Notch2. Leukemia. 2005;19:260–267. doi: 10.1038/sj.leu.2403592. [DOI] [PubMed] [Google Scholar]
- Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, Bartoli A, Coaccioli S, Screpanti I, Marconi P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856–865. doi: 10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103:3503–3510. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- Jundt F, Pröbsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, Bargou RC, Manz R, Stein H, Dörken B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004;103:3511–3515. doi: 10.1182/blood-2003-07-2254. [DOI] [PubMed] [Google Scholar]
- Vacca A, Dammacco F, Richardson PG, Anderson KC. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66:184–191. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- Sezer O, Niemoller K, Eucker J, Jakob C, Kaufmann O, Zavrski I, Dietel M, Possinger K. Bone marrow microvessel density is a prognostic factor for survival in patients with multiple myeloma. Ann Hematol. 2000;79:574–577. doi: 10.1007/s002770000236. [DOI] [PubMed] [Google Scholar]
- Rajkumar SV, Leong T, Roche PC, Fonseca R, Dispenzieri A, Lacy MQ, Lust JA, Witzig TE, Kyle RA, Gertz MA, Greipp PR. Prognostic value of bone marrow angiogenesis in multiple myeloma. Clin Cancer Res. 2000;6:3111–3116. [PubMed] [Google Scholar]
- Munshi NC, Wilson C. Increased bone marrow microvessel density in newly diagnosed multiple myeloma carries a poor prognosis. Semin Oncol. 2001;28:565–569. doi: 10.1016/S0093-7754(01)90025-9. [DOI] [PubMed] [Google Scholar]
- Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Fonseca R, Greipp PR, Rajkumar SV. Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003;17:2025–2031. doi: 10.1038/sj.leu.2403084. [DOI] [PubMed] [Google Scholar]
- Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, Mitsiades C, Raje N, Richardson P, Anderson KC. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001;98:428–435. doi: 10.1182/blood.V98.2.428. [DOI] [PubMed] [Google Scholar]
- Podar K, Anderson KC. The pathophysiological role of VEGF in hematological malignancies: Therapeutic implications. Blood. 2005;105:138395. doi: 10.1182/blood-2004-07-2909. [DOI] [PubMed] [Google Scholar]
- Rajkumar SV, Mesa RA, Tefferi A. A review of angiogenesis and anti-angiogenic therapy in hematologic malignancies. J Hematother Stem Cell Res. 2002;11:33–47. doi: 10.1089/152581602753448522. [DOI] [PubMed] [Google Scholar]
- Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Kimlinger TK, Greipp PR, Rajkumar SV. Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: evidence favoring loss of marrow angiogenesisinhibitory activity with disease progression. Blood. 2004;104:1159–1165. doi: 10.1182/blood-2003-11-3811. [DOI] [PubMed] [Google Scholar]
- Perez-Atayde AR, Sallan SE, Tedrow U, Connors S, Allred E, Folkman J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol. 1997;150:815–821. [PMC free article] [PubMed] [Google Scholar]
- Yetgin S, Yenicesu I, Cetin M, Tuncer M. Clinical importance of serum vascular endothelial and basic fibroblast growth factors in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2001;42:83–88. doi: 10.3109/10428190109097679. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/S0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Dong X, Han ZC, Yang R. Angiogenesis and antiangiogenic therapy in hematologic malignancies. Crit Rev Oncol/Hematol. 2007;62:105–118. doi: 10.1016/j.critrevonc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Frater JL, Kay NE, Goolsby CL, Crawford SE, Dewald GW, Peterson LC. Dysregulated angiogenesis in B-chronic lymphocytic leukemia: morphologic, immunohistochemical, and flow cytometric evidence. Diagn Pathol. 2008;3:16. doi: 10.1186/1746-1596-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norén-Nyström U, Heyman M, Frisk P, Golovleva I, Sundström C, Porwit A, Roos G, Bergh A, Forestier E. Vascular density in childhood acute lymphoblastic leukaemia correlates to biological factors and outcome. Br J Haematol. 2009;146:521–350. doi: 10.1111/j.1365-2141.2009.07796.x. [DOI] [PubMed] [Google Scholar]
- Aguayo A, Kantarjian HM, Estey EH, Giles FJ, Verstovsek S, Manshouri T. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer. 2002;95:1923–1930. doi: 10.1002/cncr.10900. [DOI] [PubMed] [Google Scholar]
- Hou HA, Ting-Fang Shih T, Liu CY, Chen BB, Tang JL, Yao M, Huang SY, Chou WC, Hsu CY, Tien HF. Changes in MR bone marrow angiogenesis on day 7 after induction chemotherapy can predict outcome of acute myeloid leukemia. Haematologica. 2010. in press . [DOI] [PMC free article] [PubMed]
- Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am J Pathol. 2000;157:15–19. doi: 10.1016/S0002-9440(10)64511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel T, Mazur G, Surowiak P, Wolowiec D, Jelen M, Kuliczkowsky K. Increased expression of vascular endothelial growth factor (VEGF) in bone marrow of patients with myeloproliferative disorders (MPD) Pathol Oncol Res. 2003;9:170–173. doi: 10.1007/BF03033732. [DOI] [PubMed] [Google Scholar]
- Gianelli U, Vener C, Raviele PR, Savi F, Somalvico F, Calori R, Iurlo A, Radaelli F, Fermo E, Bucciarelli P, Bori S, Coggi G, Deliliers GL. VEGF expression correlates with microvessel density in Philadelphia chromosome-negative chronic myeloproliferative disorders. Am J Clin Pathol. 2007;128:966–973. doi: 10.1309/FP0N3LC8MBJUFFA6. [DOI] [PubMed] [Google Scholar]
- Ponzoni M, Savage DG, Ferreri AJ, Pruneri G, Viale G, Servida P, Bertolini F, Orazi A. Chronic idiopathic myelofibrosis: independent prognostic importance of bone marrow microvascular density evaluated by CD105 (endoglin) immunostaining. Mod Pathol. 2004;17:1513–1520. doi: 10.1038/modpathol.3800224. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative isorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A, Skoda RC. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377–1380. doi: 10.1182/blood-2005-11-009605. [DOI] [PubMed] [Google Scholar]
- Alonci A, Allegra A, Bellomo G, Penna G, D'Angelo A, Quartarone E, Musolino C. Evaluation of circulating endothelial cells, VEGF and VEGFR2 serum levels in patients with chronic myeloproliferative diseases. Hematol Oncol. 2008;26:235–239. doi: 10.1002/hon.865. [DOI] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM. TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- Zahiragic L, Schliemann C, Bieker R, Thoennissen NH, Burow K, Kramer C, Zühlsdorf M, Berdel WE, Mesters RM. Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia. 2007;21:1310–1312. doi: 10.1038/sj.leu.2404632. [DOI] [PubMed] [Google Scholar]
- Karp JE, Gojo I, Pili R, Gocke CD, Greer J, Guo C, Qian D, Morris L, Tidwell M, Chen H, Zwiebel J. Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemias: therapy with sequential 1-beta-d-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin Cancer Res. 2004;10:3577–3585. doi: 10.1158/1078-0432.CCR-03-0627. [DOI] [PubMed] [Google Scholar]
- Fiedler W, Mesters R, Tinnefeld H, Loges S, Staib P, Duhrsen U, Flasshove M, Ottmann OG, Jung W, Cavalli F, Kuse R, Thomalla J, Serve H, O'Farrell AM, Jacobs M, Brega NM, Scigalla P, Hossfeld DK, Berdel WE. A phase 2 clinical study of SU5416 in patients with refractory acute myeloid leukemia. Blood. 2003;102:2763–2767. doi: 10.1182/blood-2002-10-2998. [DOI] [PubMed] [Google Scholar]
- Mesters RM, Padró T, Bieker R, Steins M, Kreuter M, Göner M, Kelsey S, Scigalla P, Fiedler W, Büchner T, Berdel WE. Stable remission after administration of the receptor tyrosine kinase inhibitor SU5416 in a patient with refractory acute myeloid leukemia. Blood. 2001;98:241–243. doi: 10.1182/blood.V98.1.241. [DOI] [PubMed] [Google Scholar]
- Giles FJ, Stopeck AT, Silverman LR, Lancet JE, Cooper MA, Hannah AL, Cherrington JM, O'Farrell AM, Yuen HA, Louie SG, Hong W, Cortes JE, Verstovsek S, Albitar M, O'Brien SM, Kantarjian HM, Karp JE. SU5416, a small molecule tyrosine kinase receptor inhibitor, has biologic activity in patients with refractory acute myeloid leukemia or myelodysplastic syndromes. Blood. 2003;102:795–801. doi: 10.1182/blood-2002-10-3023. [DOI] [PubMed] [Google Scholar]
- Drevs J, Müller-Driver R, Wittig C, Fuxius S, Esser N, Hugenschmidt H, Konerding MA, Allegrini PR, Wood J, Hennig J, Unger C, Marmé D. PTK787/ZK 22 a specific vascular endothelial growth factor-receptor tyrosine kinase inhibitor, affects the anatomy of the tumor vascular bed and the functional vascular properties as detected by dynamic enhanced magnetic resonance imaging. Cancer Res. 2584;62:4015–4022. [PubMed] [Google Scholar]
- Mross K, Drevs J, Müller M, Medinger M, Marmé D, Hennig J, Morgan B, Lebwohl D, Masson E, Ho YY, Günther C, Laurent D, Unger C. Phase I clinical and pharmacokinetic study of PTK/ZK, a multiple VEGF receptor inhibitor, in patients with liver metastases from solid tumors. Eur J Cancer. 2005;41:1291–1299. doi: 10.1016/j.ejca.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O'Reilly T, Persohn E, Rösel J, Schnell C, Stover D, Theuer A, Towbin H, Wenger F, Woods-Cook K, Menrad A, Siemeister G, Schirner M, Thierauch KH, Schneider MR, Drevs J, Martiny-Baron G, Totzke F. PTK787/ZK 22 a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2584;60:2178–2189. [PubMed] [Google Scholar]
- Lin B, Podar K, Gupta D, Tai YT, Li S, Weller E, Hideshima T, Lentzsch S, Davies F, Li C, Weisberg E, Schlossman RL, Richardson PG, Griffin JD, Wood J, Munshi NC, Anderson KC. The vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584 inhibits growth and migration of multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2002;62:5019–5026. [PubMed] [Google Scholar]
- Roboz GJ, Giles FJ, List AF, Cortes JE, Carlin R, Kowalski M, Bilic S, Masson E, Rosamilia M, Schuster MW, Laurent D, Feldman EJ. Phase 1 study of PTK787/ZK 22 a small molecule tyrosine kinase receptor inhibitor, for the treatment of acute myeloid leukemia and myelodysplastic syndrome. Leukemia. 2584;20:952–957. doi: 10.1038/sj.leu.2404213. [DOI] [PubMed] [Google Scholar]
- Barbarroja N, Torres LA, Luque MJ, Carretero RM, Valverde-Estepa A, Lopez-Sanchez LM, Rodriguez-Ariza A, Velasco F, Torres A, López-Pedrera C. Additive effect of PTK787/ZK 22 a potent inhibitor of VEGFR phosphorylation, with Idarubicin in the treatment of acute myeloid leukemia. Exp Hematol. 2584;37:679–691. doi: 10.1016/j.exphem.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Giles FJ, List AF, Carroll M, Cortes JE, Valickas J, Chen BL, Masson E, Jacques C, Laurent D, Albitar M, Feldman EJ, Roboz GJ. PTK787/ZK 22 a small molecule tyrosine kinase receptor inhibitor of vascular endothelial growth factor (VEGF), has modest activity in myelofibrosis with myeloid metaplasia. Leuk Res. 2584;31:891–897. doi: 10.1016/j.leukres.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M, Curwen JO, Chester R, Jackson JA, Boffey SJ, Kilburn LL, Barnett S, Richmond GH, Wadsworth PF, Walker M, Bigley AL, Taylor ST, Cooper L, Beck S, Jürgensmeier JM, Ogilvie DJ. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- Drevs J, Siegert P, Medinger M, Mross K, Strecker R, Zirrgiebel U, Harder J, Blum H, Robertson J, Jürgensmeier JM, Puchalski TA, Young H, Saunders O, Unger C. Phase I clinical study of AZD an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2171;20:3045–3054. doi: 10.1200/JCO.2006.07.2066. [DOI] [PubMed] [Google Scholar]
- Fiedler W, Mesters R, Heuser M, Ehninger G, Berdel WE, Zirrgiebel U, Robertson JD, Puchalski TA, Collins B, Jürgensmeier JM, Serve H. An open-label, phase I study of cediranib (RECENTIN) in patients with acute myeloid leukemia. Leuk Res. 2010;34:196–202. doi: 10.1016/j.leukres.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar M, Zeddis J, Barlogie B. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–1571. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- Barlogie B, Desikan R, Eddlemon P, Spencer T, Zeldis J, Munshi N, Badros A, Zangari M, Anaissie E, Epstein J, Shaughnessy J, Ayers D, Spoon D, Tricot G. Extended survival in advanced and refractory multiple myeloma after single-agent thalidomide: identification of prognostic factors in a phase 2 study of 169 patients. Blood. 2001;98:492–494. doi: 10.1182/blood.V98.2.492. [DOI] [PubMed] [Google Scholar]
- Moncada B, Baranda ML, González-Amaro R, Urbina R, Loredo CE. Thalidomide-effect on T cell subsets as a possible mechanism of action. Int J Lepr Other Mycobact Dis. 1985;53:201–205. [PubMed] [Google Scholar]
- D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta D, Treon SP, Shima Y, Hideshima T, Podar K, Tai YT, Lin B, Lentzsch S, Davies FE, Chauhan D, Schlossman RL, Richardson P, Ralph P, Wu L, Payvandi F, Muller G, Stirling DI, Anderson KC. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15:1950–1961. doi: 10.1038/sj.leu.2402295. [DOI] [PubMed] [Google Scholar]
- Teo SK. Properties of thalidomide and its analogues: implications for anticancer therapy. AAPS J. 2005;7:E14–19. doi: 10.1208/aapsj070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge K, Marriott JB, Macdonald CD, Man HW, Chen R, Muller GW, Stirling D, Dalgleish AG. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87:1166–1172. doi: 10.1038/sj.bjc.6600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24:431–436. doi: 10.1200/JCO.2005.03.0221. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Estey E, Giles FJ, Faderl S, Cortes J, Keating M. Single agent thalidomide in patients with relapsed or refractory acute myeloid leukaemia. Br J Haematol. 2003;123:436–441. doi: 10.1046/j.1365-2141.2003.04639.x. [DOI] [PubMed] [Google Scholar]
- Steins MB, Padró T, Bieker R, Ruiz S, Kropff M, Kienast J, Kessler T, Buechner T, Berdel WE, Mesters RM. Efficacy and safety of thalidomide in patients with acute myeloid leukemia. Blood. 2002;99:834–839. doi: 10.1182/blood.V99.3.834. [DOI] [PubMed] [Google Scholar]
- Raza A, Mehdi M, Mumtaz M, Ali F, Lascher S, Galili N. Combination of 5-azacytidine and thalidomide for the treatment of myelodysplastic syndromes and acute myeloid leukemia. Cancer. 2008;113:1596–1604. doi: 10.1002/cncr.23789. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Giles FJ, Albitar M, Cortes JE, Verstovsek S, Faderl S, O'Brien SM, Garcia-Manero G, Keating MJ, Pierce S, Zeldis J, Kantarjian HM. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106:1974–1984. doi: 10.1002/cncr.21827. [DOI] [PubMed] [Google Scholar]
- Kotla V, Goel S, Nischal S, Heuck C, Vivek K, Das B, Verma A. Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2:36. doi: 10.1186/1756-8722-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, San Miguel J, Hellmann A, Facon T, Foa R, Corso A, Masliak Z, Olesnyckyj M, Yu Z, Patin J, Zeldis JB, Knight RD. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–2132. doi: 10.1056/NEJMoa070594. [DOI] [PubMed] [Google Scholar]
- Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, Siegel D, Borrello I, Rajkumar SV, Chanan-Khan AA, Lonial S, Yu Z, Patin J, Olesnyckyj M, Zeldis JB, Knight RD. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133–2142. doi: 10.1056/NEJMoa070596. [DOI] [PubMed] [Google Scholar]
- Tefferi A, Cortes J, Verstovsek S, Mesa RA, Thomas D, Lasho TL, Hogan WJ, Litzow MR, Allred JB, Jones D, Byrne C, Zeldis JB, Ketterling RP, McClure RF, Giles F, Kantarjian HM. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108:1158–1164. doi: 10.1182/blood-2006-02-004572. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A, Kantarjian HM, Manshouri T, Thomas D, Cortes J, Ravandi F, Garcia-Manero G, Ferrajoli A, Bueso-Ramos C, Verstovsek S. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27:4760–4766. doi: 10.1200/JCO.2009.22.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001;7:1419–1428. [PubMed] [Google Scholar]
- Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, Dammacco F, Richardson PG, Anderson KC. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Research. 2006;66:184–191. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- Kane RC, Farrell AT, Sridhara R, Pazdur R. United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clinical Cancer Research. 2006;12:2955–2960. doi: 10.1158/1078-0432.CCR-06-0170. [DOI] [PubMed] [Google Scholar]
- Mateos MV, Richardson PG, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Esseltine DL, Liu K, Cakana A, van de Velde H, San Miguel JF. Bortezomib Plus Melphalan and Prednisone Compared With Melphalan and Prednisone in Previously Untreated Multiple Myeloma: Updated Follow-Up and Impact of Subsequent Therapy in the Phase III VISTA Trial. J Clin Oncol. 2010;28:2259–2266. doi: 10.1200/JCO.2009.26.0638. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Weller E, Lonial S, Jakubowiak AJ, Jagannath S, Raje NS, Avigan DE, Xie W, Ghobrial IM, Schlossman RL, Mazumder A, Munshi NC, Vesole DH, Joyce R, Kaufman JL, Doss D, Warren DL, Lunde LE, Kaster S, Delaney C, Hideshima T, Mitsiades CS, Knight R, Esseltine DL, Anderson KC. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010. in press . [DOI] [PMC free article] [PubMed]
- Attar EC, De Angelo DJ, Supko JG, D'Amato F, Zahrieh D, Sirulnik A, Wadleigh M, Ballen KK, McAfee S, Miller KB, Levine J, Galinsky I, Trehu EG, Schenkein D, Neuberg D, Stone RM, Amrein PC. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin Cancer Res. 2008;14:1446–1454. doi: 10.1158/1078-0432.CCR-07-4626. [DOI] [PubMed] [Google Scholar]