

Abstract
The ability of δ ribozyme to catalyze the cleavage of an 11-mer RNA substrate was examined under both single- and multiple-turnover conditions. In both cases only small differences in the kinetic parameters were observed in the presence of either magnesium or calcium as cofactor. Under multiple-turnover conditions, the catalytic efficiency of the ribozyme (kcat/KM) was higher at 37 °C than at 56 °C. The cleavage reaction seems to be limited by the product release step at 37 °C and by the chemical cleavage step at 56 °C. We observed substrate inhibition at high concentrations of the 11-mer substrate. Cleavage rate constants were determined with a structural derivative characterized by an ultrastable L4 tetraloop. The kinetic parameters (kcat and KM) and dissociation constant (Kd) were almost identical for both ribozymes, suggesting that the stability of the L4 loop has a negligible impact on the catalytic activities of the examined ribozymes. Various cleavage inhibition and gel-shift assays with analogues, substrate, and both active and inactive ribozymes were performed. The 2′-hydroxyl group adjacent to the scissile phosphate was shown to be involved in binding with the ribozyme, while the essential cytosine residue of the J4/2 junction was shown to contribute to substrate association. We clearly show that substrate binding to the δ ribozyme is not restricted to the formation of a helix located downstream of the cleavage site. Using these results, we postulate a kinetic pathway involving a conformational transition step essential for the formation of the active ribozyme/substrate complex.
Self-cleaving sequences are present in both the genomic and antigenomic RNA of hepatitis δ virus (HDV).1 These sequences, known as δ self-catalytic domains, are required for the processing of multimeric strands of both polarities into monomers during rolling circle replication of the virus (for reviews see refs 1–3). Trans-acting δ ribozyme systems have been generated by separating the RNA molecule at various positions according to different secondary structure predictions (4, 5). Thus far, the proposed pseudoknot secondary structure model best fits the experimental data for both the antigenomic and genomic RNAs (2). This model includes four stems (P1–P4), two internal loops (L3 and L4), and three single-stranded junctions identified in terms of the linked stems (J1/2, J1/4, and J4/2) (see Figure 1).
Figure 1.

Secondary structure and sequence of δ ribozyme RzP1.1. The numbering of the nucleotides is from 5′ to 3′ for both the substrate and the ribozyme molecules. Paired regions are numbered P1–P4, while joining regions are named according to the paired elements they link. The structure is shown according to the pseudoknot model (5). A dotted line represents the sequence removed in the trans-acting ribozyme as compared to the self-cleaving RNA strand. The arrow indicates the substrate cleavage site. The SphI and SmaI restriction sites used for modification of the ribozyme are indicated. In the inset the ultrastable L4 loop found in ribozyme RzP1.1/4.1 is shown
Trans-acting δ ribozymes of either polarity have been designed and used almost exclusively to study the impact of mutations on cleavage activity (2). In general, trans-acting ribozymes were shown to cleave at a slower rate than the corresponding self-cleaving RNA strand. However, these rates were comparable to those of similar intermolecular constructs of both the hammerhead and hairpin ribozymes. Characterization of various versions of the δ ribozymes, regardless of the variability of each version, has allowed the determination of composite kinetic parameters (6–8). Genomic and antigenomic HDV self-catalytic domains have been divided into substrate and enzyme molecules but have not yet been analyzed as extensively as similar hammerhead and hairpin ribozymes. To learn more about the reaction pathway of δ ribozyme cleavage, we focused on the kinetic analysis of a trans-acting form of the antigenomic δ ribozyme, which was divided into substrate and enzyme at the J1/2 junction of the pseudoknot model. Two versions of this ribozyme, which differs from the wild-type form by a shortened P4 stem and an extension of stem P2, were generated (see Figure 1). These versions differ by their L4 loops, which are composed of either an ultrastable tetraloop (UUCG, in RzP1.1/4.1) or its reverse form (GCUU, in RzP1.1), which is known for its relative instability (9). Although the modification of the L4 loop and the extension of the P2 stem have been shown to result in an increased cleavage activity of both cis- and trans-acting δ ribozymes, no detailed kinetic studies have been performed (6, 7, 10). In this paper, the kinetic parameters for ribozymes with modified L4 loops were determined at both 37 and 56 °C under both multiple- and single-turnover conditions. Since the δ ribozyme has the unique property of using calcium as efficiently as magnesium as cofactor, we determined the kinetic parameters in the presence of each of these ions. Finally, we investigated the binding process using analogues, substrate, and both active and inactive ribozymes.
EXPERIMENTAL PROCEDURES
Plasmids Carrying δ Ribozymes
Cloning of the transacting ribozyme sequence was achieved through the annealing of two pairs of complementary and overlapping oligonucleotides representing the entire length of the ribozyme (57 nt; Figure 1), and subsequent ligation of the resulting double-stranded DNA fragment into pUC19 to create a plasmid harboring the δ ribozyme (referred to as pRzP1.1; for details of construction see ref 11). The minigene was designed so as to have unique SphI and SmaI restriction sites (see Figure 1). The sequence of the T7 RNA promoter was included at the 5′ end of the ribozyme so as to permit in vitro transcription. Mutant ribozymes were constructed by replacing the SphI–SmaI fragment of pRzP1.1 by an oligonucleotide duplex containing the desired altered sequence (pRzP1.1/P4.1 or pRzP1.1/P4.1/J4-2.1, namely, the A47 mutant). The sequences of all engineered ribozymes were confirmed by DNA sequencing. Plasmids containing wild-type and mutant ribozymes were digested with SmaI, purified by both phenol and chloroform extractions, and precipitated for further use as templates in in vitro transcription reactions.
RNA Synthesis
In vitro transcription reactions were performed according to standard procedures with T7 RNA polymerase (12). Unlabeled ribozyme was synthesized by in vitro runoff transcription on SmaI-linearized pUC19 construct (10 μg). Unlabeled and internally labeled substrate RNA ([α-32P] GTP) were synthesized from partial oligodeoxynucleotides duplexes flanked on the 5′ end by the T7 promoter sequence. Briefly, deoxyoligonucleotides (500 pmol) containing the substrate and the T7 promoter sequence were denatured by heating at 95 °C for 5 min in a 20 μL mixture containing 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, and 50 mM KCl2 and allowed to cool slowly to 37 °C. The in vitro transcription reaction was carried out with the partial duplex as template. All transcripts were passed through Sephadex G-50 spun columns and purified on 20% poly-acrylamide gels (PAGE) with 50 mM Tris–borate (pH 8.3)/1 mM EDTA/7 M urea running buffer as previously described (13). The reaction products were visualized either by ultraviolet shadowing over a fluorescent thin-layer chromatography plate or by autoradiography. The bands corresponding to the correct sizes for the ribozymes and substrates were cut out. These transcripts were eluted overnight at 4 °C in a solution containing 0.1% SDS and 0.5 M ammonium acetate and were then precipitated by adding 0.1 volume of 3 M sodium acetate, pH 5.2, and 2.2 volumes of ethanol. The amounts of purified transcripts were quantified by spectrophotometry at 260 nm.
Synthesis and Purification of RNA and RNA/DNA Mixed Polymer
RNA and RNA–DNA mixed polymers were synthesized on an automated oligonucleotide synthesizer (Keck Biotechnology Resource Laboratory, Yale University), and deprotected according to previously described procedures (14). These polymers were purified by 20% PAGE. Major bands were excised and eluted as described above.
End-Labeling of RNA
Synthetic oligoribonucleotide (1 pmol) was end-labeled in a mixture containing 1.6 pmol of [γ-32P]ATP, 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 50 mM KCl, and 3 units of T4 polynucleotide kinase at 37 °C for 30 min. The excess [γ-32P]ATP was removed by applying the reaction mixture onto a spin column packed with a G-50 Sephadex gel matrix. The concentration of labeled transcripts was adjusted to 0.05 pmol/μL. The 3′ product (7-mer) was 3′-end-labeled with [α-32P]Cp in the presence of T4 RNA ligase according to the procedure of the manufacturer (Pharmacia).
Kinetic Analysis under Single-Turnover Conditions
Single-turnover conditions as described by Hertel et al. (15) were used with some modifications. Briefly, various concentrations of ribozyme, ranging from 5 nM to 4 μM, were mixed with trace amounts of substrate (final concentration <2 nM) in a final volume of 18 μL and subjected to denaturation by heating at 95 °C for 2 min. The mixtures were quickly placed on ice for 2 min and then equilibrated to the reaction temperature for 5 min prior to initiation of the reaction. To initiate cleavage, 2 μL of a solution containing 500 mM Tris-HCl, pH 7.9, and 100 mM either MgCl2 or CaCl2 was added to the mixtures. The cleavage reactions were followed until the end point of cleavage was reached. The reactions were sampled at various time points by removing 2 μL aliquots and adding them to 8 μL of ice-cold formamide/dye mixture (95% formamide, 10 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol). These samples were analyzed by 20% PAGE, and the radioactive bands were quantified with a PhosphorImager (Molecular Dynamics). The fractions of substrate cleaved were determined, and the rate of cleavage (kobs) was obtained from fitting the data to the equation At = A0(1 − e−kt), where At is the percentage of cleavage at time t, A0 is the maximum percentage of cleavage (or end point of cleavage), and k is the rate constant (kobs). Each rate constant was calculated from at least two measurements. The values of kobs obtained were then plotted as a function of concentrations of ribozyme for the determination of the kinetic parameters: apparent kcat and KM. Values obtained from independent experiments varied less than 15%.
Cleavage Reactions under Multiple-Turnover Conditions
The catalytic parameters KM and kcat were determined under conditions of excess substrate in 20 μL reactions. Substrate (concentrations ranging from 50 nM to 10 μM) and ribozyme (50 nM) were denatured together in the presence of 50 mM Tris-HCl (pH 7.9) and a trace amount (<2 nM) of internally labeled substrate. The mixture was then preincubated at the testing temperature for 5 min. The reaction was initiated by the addition of bivalent metal ions to a final concentration of 10 mM. The reactions were stopped by removing aliquots (1.5 μL) at regular time intervals and adding them to 10 μL of ice-cold formamide/dye mixture. Reaction products were analyzed on 20% PAGE gels, and the fraction of substrate cleaved at each time point was determined by radioanalytic scanning with a PhosphorImager. Initial reaction velocities were calculated over a time span that was within the linear range (<1 turnover), and Lineweaver–Burk plots were used to calculate KM and kcat. Measurements of the kinetics of inhibition were performed with either the 3′ end product, the 5′ end product, or the chemically synthesized SdC4 analogue at 37 °C under the conditions described above. To avoid substrate inhibition phenomenon, three different concentrations (50, 100, and 250 nM) of either 3′ end product or SdC4 analogue were tested as inhibitor in the presence of various substrate concentration varying from 50 to 750 nM. Either in the absence of inhibitor or in the presence of the 5′ end product (with several concentration ranging from 250 nM to 20 μM), the substrate concentration was ranging from 50 nM to 1 μM. These experiments were performed at least in duplicate and average KI values are reported.
Determination of Equilibrium Dissociation Constants
The equilibrium dissociation constants were determined by a slight modification of the method described previously (11). Various ribozyme concentrations (usually ranging from 1 nM to 1 μM) were individually incubated with trace amounts of either the substrate analogue or the 3′ product until equilibrium was reached (1 h) and were then loaded onto nondenaturing gels. Quantification of bound and free substrates was performed with a PhosphorImager as described earlier.
RESULTS
Pre-Steady-State Conditions
Ribozyme RzP1.1 possessing the GCUU L4 loop was used in the initial kinetic determinations under pre-steady-state conditions ([Rz] ≫ [S]). The experiments were performed at both 37 and 56 °C in the presence of either magnesium or calcium as cofactor (Figure 2). As an example of a typical autoradiogram obtained in these experiments, that for the incubation of 5 pmol of ribozyme with 0.1 pmol of substrate at 56 °C in the presence of 10 mM MgCl2 is shown in Figure 2A. In the present case, the reaction reached up to 95% of cleaved substrate. In general, we observed that up to 85–95% of substrate was cleaved within the incubation time, suggesting that most, if not all, of the bound ribozymes were in a catalytically active form. The small fraction of uncleaved substrate may correspond either to ribozymes folded into inactive conformation that could not bind the substrate into a productive fashion or, more probably, to an equilibrium between the active and inactive ribozyme/substrate complexes that is dependent on the occurrence of a conformational transition (see Discussion). By use of trace amounts of labeled substrate, pseudo-first-order rate constants for cleavage (kobs) were determined with increasing enzyme concentrations so as to obtain the apparent kcat and KM values (see Figure 2B and Table 1). The apparent kcat values from the various test conditions were consistent; for example, at 37 °C in the presence either of magnesium or calcium the kcat values obtained were 0.32 min−1 and 0.41 min−1, respectively. The apparent KM values with either calcium or magnesium were identical at 37 °C (10.3 and 9.9 nM, respectively). In contrast, the apparent values of KM were slightly different at 56 °C, 330 nM and 570 nM in the presence of either magnesium or calcium, respectively (see Table 1). Thus, the apparent KM are 30–60-fold larger at 56 °C than at 37 °C. Considering that the kcat values were almost identical at 37 °C and 56 °C, the variation of the apparent KM suggests the occurrence of a temperature-dependent step during the formation of the active ternary complex. The kcat and KM values (0.32 min−1 and 9.9 nM) estimated at 37 °C in the presence of magnesium as cofactor are almost identical to those recently reported for the same ribozyme in an independent experiment (0.34 min−1 and 17 nM; 11). In this experiment the apparent kcat and KM values were also determined for a structural derivative characterized by a stabilized UUCG L4 loop (RzP1.1/4.1) to be 0.23 min−1 and 9.3 nM, respectively, with magnesium as cofactor and 0.4 min−1 and 12.7 nM, respectively, with calcium as cofactor. Thus, both ribozyme versions (RzP1.1 and RzP1.1/4.1) have identical kinetic parameters, suggesting that the stability of the L4–P4 hairpin has a negligible impact on the ability of these ribozymes to mediate cleavage at physiological temperatures.
Figure 2.

Activity assays under pre-steady-state conditions. (A) Example of a 20% PAGE autoradiogram of a cleavage assay at 56°C in the presence of 10 mM CaCl2. Indicated at the top are the times (in minutes) at which aliquots were removed from the reaction mixture. S, XC, and BPB are substrate, xylene cyanol, and bromophenol blue, respectively. (B) Plots of kobs versus ribozyme concentration in the presence of either 10 mM MgCl2 (○) or CaCl2 (□) at 37 °C. Kinetic parameters were determinated by means of nonlinear curve-fitting based on pseudo-first-order analysis under different conditions.
Table 1.
Summary of Pre-Steady-State and Steady-State Kinetic Parametersa
| temperature (°C) | cofactor | pre-steady-state |
steady-state |
||||
|---|---|---|---|---|---|---|---|
| kcat (min−1) | KM (nM) | kcat/KM (min−1·M−1) | kcat (min−1) | KM (nM) | kcat/KM (min−1·M−1) | ||
| 37 | Mg2+ | 0.32 ± 0.03 | 9.9 ± 1.0 | 3.0 × 107 | 0.69 ± 0.03 | 510 ± 22 | 1.4 × 106 |
| 37 | Ca2+ | 0.41 ± 0.05 | 10.3 ± 0.9 | 4.0 × 107 | 0.71 ± 0.05 | 262 ± 19 | 2.7 × 106 |
| 56 | Mg2+ | 1.2 ± 0.1 | 330 ± 36 | 3.6 × 106 | 1.1 ± 0.1 | 1300 ± 140 | 8.5 × 105 |
| 56 | Ca2+ | 1.0 ± 0.1 | 570 ± 59 | 1.8 × 106 | 1.3 ± 0.1 | 2200 ± 200 | 5.9 × 105 |
See Experimental Procedures section for experimental details.
Steady-State Conditions
The cleavage activity of ribozyme RZP1.1 with the GCUU L4 loop was also determined under steady-state conditions ([S] ≫ [Rz]) at 37 °C with 10 mM magnesium as cofactor (Figure 3A). Time course experiments were carried out with various concentrations of substrate, and the ratio of product to enzyme concentrations was plotted. This experiment was aimed at determining whether the product release step becomes limiting (16), as the 3′ reaction product is still able to form a complex with the ribozyme through Watson–Crick base pairing. We observed a burst of rate as previously reported (6, 10). The more products accumulated, the slower the turnover was. The rate constant was estimated to be 0.5 min−1 during the first turnover, and then it decreased progressively during subsequent turnovers until it became negligible, reaching 0.05 min−1 during the fourth turnover (Figure 3B). This decrease in rate was observed at the different substrate concentrations tested at 37 °C with either calcium or magnesium as cofactor (data not shown). In contrast, we did not observe any decrease in rate when the experiments were performed at 56 °C (data not shown). For example, in the presence of a 100-fold excess of substrate over ribozyme we observed 35 turnovers that occurred with a similar rate constant. Thus, we concluded that at 56 °C the enzyme–product dissociation step is no longer rate-limiting.
Figure 3.

Activity assays under steady-state conditions. (A) Monitoring of ribozyme cleavage with time. Internally labeled (less than 1 nM) and unlabeled substrate (500 nM) were cleaved by 50 nM ribozyme in the presence of 10 mM MgCl2 at 37 °C. The concentration of product over that of ribozyme ([P]/[E]) represents the number of turnovers. (B) Lineweaver–Burk plots of kinetic experiments performed at both 37 °C (○) and 56 °C (□) in the presence of 10 mM magnesium. The amount of ribozyme was fixed at 50 nM, preannealed with varying concentrations of substrate (S). Initial velocities (vi) were calculated over a time span that was within the linear range (<1 turnover).
To establish the Michaelis–Menten parameters of this ribozyme, we considered only the percentage of cleaved substrate during the first turnover. Under these conditions the initial velocity increased with increasing substrate concentration up to 1 μM at 37 °C and 4 μM at 56 °C (Figure 3B). Beyond these substrate concentrations the initial velocities decreased. This profile is characteristic of substrate inhibition (16); hence, we believe that in high concentrations the substrate interacts with a region of the ribozyme different from the recognition domain, and this interaction results in inactivation of the ribozyme. At 56 °C this interaction would be less stable, and consequently a higher substrate concentration is required to observe it. Similar results were also observed with calcium as cofactor (data not shown).
Extrapolation of the linear portion of the Lineweaver–Burk plots yielded Michaelis–Menten parameters under various conditions (see Table 1). At 37 °C, the KM was slightly higher in the presence of magnesium as compared to calcium as cofactor, 510 nM to 262 nM, respectively. Inversely, at 56 °C the KM was slightly higher (2-fold) in the presence of calcium as compared to magnesium. The KM values seem to depend on temperature as, for example, in the presence of magnesium the apparent KMs at 37 and 56 °C are 510 and 1300 nM, respectively. In the presence of calcium as cofactor, however, the apparent KM increases 8-fold over the same temperature range (262 and 2200 nM, respectively). In contrast, the estimated kcat values ranged from 0.71 min−1 to 1.3 min−1 regardless of the temperature and the metal ion cofactor (see Table 1). Overall, the apparent second-order rate constants (i.e., kcat/KM) appeared relatively similar for both cofactors at both temperatures, and the turnover of the ribozyme is slightly faster at 37 °C than at 56 °C. The experiments were repeated four times at 37 °C and twice at 56 °C, with reproducible results being obtained, ensuring that the reported differences are real.
Temperature Sensitivity of Ribozyme Cleavage
The cleavage activities of the two L4 loop versions of δ ribozyme were estimated at different temperatures under multiple-turnover conditions. The values of kobs were estimated at several temperatures between 23 and 65 °C, and an Arrhenius plot was drawn (Figure 4). The optimal temperatures of the ribozymes harboring the GCUU and the UUCG L4 loops (RzP1.1 and RzP1.1/P4.1, respectively) were estimated to be ~41 °C and 45 °C, respectively. Thus, the presence of the ultrastable tetraloop in RzP1.1/P4.1 seems to increase slightly the resistance to nonphysiological temperatures. These estimated optimal temperatures are relatively low compared to those of hammerhead ribozymes (55 °C; 17). Using the linear portion of the curve, we determined activation energies of 17.5 and 18.6 kcal·mol−1 for the UUCG and GCUU L4 loop ribozymes, respectively.
Figure 4.
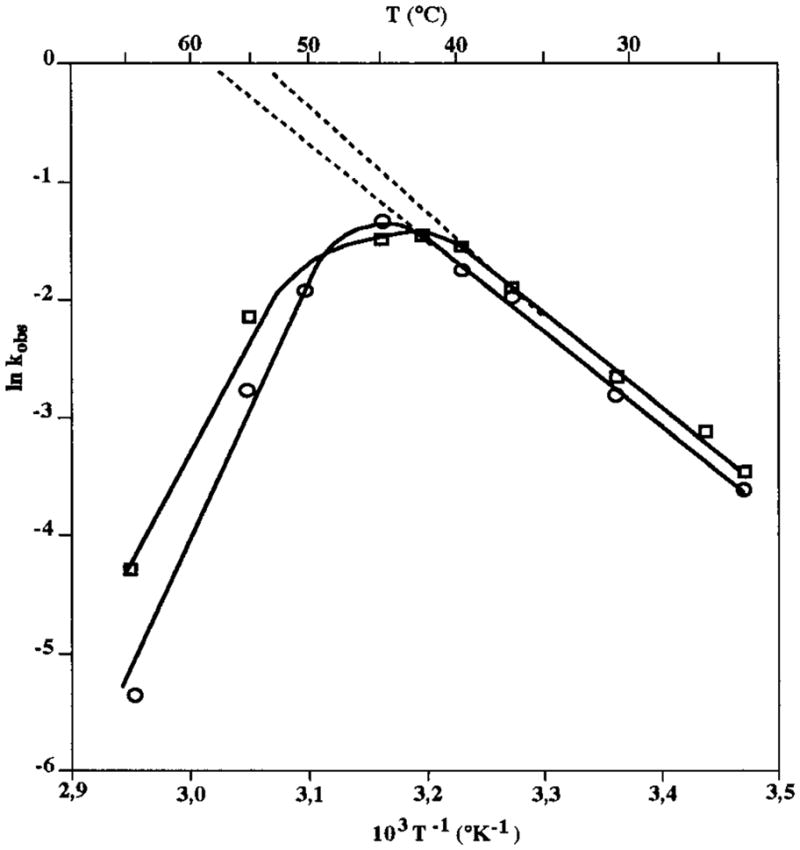
Temperature dependence of the rate constant (kobs). Reactions were performed under steady-state conditions with 250 nM substrate being incubated with 50 nM ribozyme at the desired temperature. Rate constants were determined and plotted as a function of the temperature in Celsius (°C) as well as 1/temperature in kelvins (K). (□, ○) Ribozymes with either the GCUU (RzP1.1) or the UUCG (RzP1.1/4.1) ultrastable L4-loop, respectively.
Inhibition Assays
Inhibition assays were performed at 37 °C with three different chemically synthesized analogues: (i) an 11 nt RNA identical to the substrate except for the presence of a deoxyribose residue at position 4 (the cleavage site), namely, SdC4; (ii) a 7 nt RNA corresponding to the 3′ product (the substrate portion base-paired to the ribozyme); and (iii) a 4 nt RNA corresponding to the 5′ product. Substrates with a deoxyribonucleotide at the cleavage site, as well as the 7-mer 3′ product, had previously been shown to act as analogues (18–20). Under steady-state conditions, a 250 nM concentration of either the SdC4 analogue or the 3′ product was used in order to minimize substrate inhibition phenomena. All experiments lead to the conclusion that both the 3′ end product and the SdC4 analogue are competitive inhibitors (e.g., Figure 5). The estimated inhibition constants (KI) of the SdC4 analogue and the 3′ product were 321 nM (± 24 nM) and 712 nM (± 73 nM), respectively. The KI value of SdC4 analogue is in the same range as the apparent KM estimated for the substrate under similar conditions, indicating that the binding of both molecules appears to be similar. The slightly higher apparent KI of the 3′ product may suggest that the interaction between the substrate and the δ ribozyme is not restricted to the base-paired domain (the P1 stem). Indeed, the 5′ portion of the substrate would probably contribute to the substrate–ribozyme interaction; however, no inhibition was detected when the 5′ product analogue was tested as inhibitor (Figure 5). Higher concentrations (ranging from 250 nM up to 20 μM) of the 5′ product were tested, but no inhibition was detected (data not shown).
Figure 5.

Lineweaver–Burk plots of inhibition assays. Kinetics were performed either in the absence (○), or in the presence (□) of 250 nM SdC4 analogue, the 3′ product (△) or the 5′ product (×).
Binding Shift Assays
To learn from both the substrate and product binding to δ ribozyme, we performed gel shift assays under conditions similar to those of cleavage assays. Trace amounts of end-labeled analogue (SdC4 or the 3′ product) were incubated at 37 °C with various concentrations of ribozyme, and the mixture was analyzed on native polyacrylamide gels. Figure 6 shows a binding curve of assays performed with the SdC4 analogue and the ribozyme (RzP1.1) in the presence of either magnesium or calcium. The dissociation constants (Kd) were estimated to be 30 and 52 nM in the presence of either magnesium or calcium, respectively. Thus, calcium and magnesium promote the formation of the substrate–analogue complex in a similar manner. If this difference in Kd is significant, it would suggest that the substrate–ribozyme association is weaker in the presence of calcium as compared to magnesium. Similarly, a Kd of 28 nM was determined for the SdC4 analogue and the RzP1.1/4.1 ribozyme with the ultrastable L4 loop (Table 2). From an independent experiment, we recently reported a Kd of 31 nM for the same complex (11). Thus, modification of the L4 loop did not alter the binding of the SdC4 analogue to the ribozymes. Using the ultrastable L4 loop version of the δ ribozyme, we observed a Kd of 122 nM for the 3′ product 3′-end-labeled with [α-32P]Cp, which is an uncleavable analogue like SdC4 (Table 2). The binding of the 3′ product was slightly weaker than that of the SdC4 analogue. However, the maximum bound complex (e.g., capacity) was estimated at 14% with the 3′ product 3′-end-labeled compared to 80% with the SdC4 analogue. These results suggest that the 3′ product/ribozyme complex is weaker. When the 3′ product was 5′-end-labeled, the Kd and the maximum bound complex were estimated to be 42 nM and 14%, supporting the conclusion that the product/ribozyme complex is weaker (data not shown).
Figure 6.

Example of a binding shift assay between the SdC4 analogue and the active ribozyme. Different concentrations of ribozyme were individually mixed with trace amounts of end-labeled substrate (<1 nM), incubated at 37 °C for 1 h, and analyzed on a nondenaturing polyacrylamide gel. Fractions of bound and free substrate were determined, and Kd, which is the equilibrium dissociation constant, was obtained from fitting the data to the equation % bound substrate = [Rz]/Kd + [Rz], where [Rz] is the concentration of ribozyme. The fraction of substrate bound in the presence of either 10 mM MgCl2 (□) or CaCl2 (○) is indicated.
Table 2.
Equilibrium Dissociation Constantsa
| analogue | substrate | active ribozyme | inactive ribozyme (A47 mutant) |
|---|---|---|---|
| uncleavable | SdC4 | 28 (80) | 191 (50) |
| 3′ product | 122 (14) | 276 (6) | |
| cleavable | all RNA | 25 (70) |
See Experimental Procedures section for experimental details; Kd units are nanomolar; values in parentheses are % maximum bound complex.
Binding shift assays in the presence of magnesium were also performed with a mutated ribozyme that harbors an adenosine at position 47 (RzP1.1/P4.1/J4-2.1). The mutation of the essential cytosine for an adenosine at position 47 yields a δ ribozyme completely inactive at both 37 and 56 °C (data not shown). The A47 mutant bound the SdC4 analogue with a Kd of 191 nM (Table 2). This dissociation constant is 7-fold weaker than that of the active ribozyme, suggesting that C47 may play either a direct or an indirect role in the binding of the substrate. We also observed that the A47 mutant ribozyme bound the 3′ product with a Kd of 276 nM, supporting the notion that the cytosine at position 47 of the active ribozyme contributes to the ribozyme/substrate association. The capacity of the inactive ribozyme to bind the 3′ product was limited (6%), suggesting that this complex was relatively weak. The inactive A47 mutant was used to evaluate the binding of the substrate, which is a cleavable molecule as compared to the uncleavable analogue. The A47 mutant ribozyme binds the RNA substrate with a Kd of 25 nM (Table 2). The only difference between the SdC4 analogue and the RNA substrate is the 2′-hydroxyl group adjacent to the scissile phosphate. Therefore, the 8-fold stronger binding of the RNA substrate as compared to the SdC4 binding indicates that the 2′-hydroxyl group adjacent to the scissile phosphate is involved in substrate binding to the ribozyme.
DISCUSSION
The Two δ Ribozyme Versions
In this study we characterized trans-acting δ ribozymes under both pre-steady-state and steady-state conditions. Previously, it has been reported that a large proportion of δ ribozymes would fold into inactive conformations (7). We postulated that the sequence of the P4–L4 stem–loop, which has an accessory function in catalytic core assembly (5), may be responsible for these misfoldings. Consequently, we constructed two identical δ ribozymes carrying either an unstable GCUU loop (RzP1.1) or an ultrastable UUCG loop (RzP1.1/4.1) (Figure 1). Both ribozymes have a P2 stem extended by three G–C pairs in order to stabilize the stem and ensure efficient transcript synthesis. Unexpectedly, their kinetic parameters (kcat and KM) and dissociation constant (Kd) were virtually identical. These results suggest that the stability of the L4 loop appears to have a negligible impact on the ability of the ribozyme to mediate binding and cleavage at 37 °C. However, the results of cleavage assays performed at different temperatures showed that the version with the ultrastable tetraloop seems to have a somewhat higher resistance to nonphysiological temperatures. Previously, it has been demonstrated that a temperature of 45–55 °C could enhance δ ribozyme turnover as compared to 37 °C (21). We suspected that the version with the ultrastable loop should have more resistance to higher temperatures. Since the designed P4 stem was sufficient to provide stablity to the ribozyme, and the ultrastable L4 loop did not affect the optimal temperaturature of the ribozyme, we propose that the inactive formation of these ribozymes is a consequence of the misfolding of another region of the catalytic RNA; for example, the three Watson–Crick base pairs of the P3 stem.
Most of the ribozymes characterized in this study adopted active conformations, unlike other versions of the δ ribozyme (7). At 37 °C under steady-state conditions, we observed a burst that occurred near one unit of ribozyme concentration. This observation supports the notion that most of the ribozyme was active. Furthermore, the burst yielding a slower rate of product formation indicates a change in the rate-limiting step from the cleavage step in the first turnover to the product release step in subsequent turnovers. Slow product release was also suggested as one possible cause of low turnover for other versions of the δ ribozyme (10, 18). In contrast, we observed that at 56 °C the cleavage step remains the limiting step for all turnovers.
In addition, substrate inhibition occurred in steady-state conditions at concentrations greater than 1 and 4 μM at 37 and 56 °C, respectively. These results suggest that at high substrate concentrations there is probably a greater amount of the inactive form of the ribozyme due to an undesirable interaction. Sequence analysis revealed some potential regions of the substrate and the ribozyme that could be responsible for this interaction. Apart from the P1 domain, the L3 loop harbors the sequence C9UCCUC14, which might pair to the substrate G1GGCGGG7 sequence. The helix formation between the substrate and L3 loop might prevent the folding of the ribozyme required for catalysis. At 56 °C, this putative interaction might be less stable, and therefore a larger portion of substrate is required in order to obtain a similar inhibition.
Magnesium versus Calcium as Cofactor
The apparent kcat values determined in the presence either of magnesium or calcium were identical under similar conditions, suggesting that both ions support a similar chemical cleavage. In contrast, the estimated KM and Kd values were slightly different (e.g., <2-fold). For example, under steady-state conditions, we determined values of KM of 510 and 262 nM at 37 °C, and 1.3 and 2.2 μM at 56 °C, in the presence of magnesium and calcium, respectively. Kd values of 28 nM with magnesium and 52 nM with calcium were obtained by binding shift assays. This difference is insignificant, although it is possible that these two metal ions might interact differently with the active ribozyme/substrate complex. A previous report of Puttaraju et al. (21) stated that calcium enhanced reaction turnovers as compared to magnesium, while Suh et al. (22) show that both calcium and magnesium support similar cleavage activity. Our results with calcium agree with those of Suh et al. (22), leading us to suggest that the efficiency of cleavage with either magnesium or calcium depends on the ribozyme sequence. From independent experiments, we observed that magnesium and calcium gave slightly different cleavage patterns due to metal ion-induced cleavage when one of these ribozymes was subjected to identical tests with either calcium or magensium (D. A. Lafontaine, S. Mercure, and J. P. Perreault, unpublished data). This indicates that δ ribozyme binds calcium and magnesium with some minor differences that can cause small variations in the binding affinity, resulting in slightly different KM and Kd values.
To learn how the metal ion cofactors are involved in δ ribozyme catalysis, we determined the nature of the rearrangement of the ribozyme conformation due to the addition of the substrate and magnesium. Assuming that upon addition of the metal ion to the ribozyme both the substrate and the substrate/ribozyme complex incorporate magnesium ions prior to activity, we used various conditions including mixing magnesium with the ribozyme prior to the addition of substrate, and vice versa, to initiate catalytic cleavage reactions (data not shown). However, we could not detect any significant effect of the various initiation methods on the cleavage reactions, suggesting that the addition of metal ions into the ribozyme/substrate complex is so rapid that it could not be distinguished by this method. Similar experiments were performed with a genomic-derived version of δ ribozyme (8). In that study, it was shown that the cleavage rate could be maximized by a preincubation of substrate and ribozyme together, and then initiation of the reaction by the subsequent addition of the magnesium. The same report suggested that the magnesium ions do not interact with the pro-R oxygen directly but are essential to the formation of the active complex of the ribozyme and its substrate (8). Our results also support this suggestion, since both calcium and magnesium gave similar values of kcat (for the chemical step) and KM and Kd (for the binding process).
Kinetic Pathway
To analyze our experimental data, we considered a kinetic pathway that required a conformational transition step for formation of the active ternary complex:
where E, S, P1, and P2 are respectively the ribozyme, the substrate, the 5′ end product, and the 3′ end product. Since all kinetic analyses were performed in the presence of saturating concentrations of either magnesium or calcium (10 mM), metal ion binding was not considered. This working hypothesis is supported by the observation that in the presence of active ions (like Mg2+ and Ca2+) the ribozyme/substrate complex goes through a conformational transition that alters the patterns of metal ion probing (D. A. Lafontaine, S. Mercure, and J.-P. Perreault, unpublished data). In contrast, inactive metal ions (like Cd2+ and Zn2+) do not support this conformational transition.
Steady-state results show that the rate-limiting step is temperature-dependent. At 37 °C it is the product release step, while at 56 °C it is the chemical step. Since the kcat values were estimated by using only data from the first turnover, they can be compared to the kcat estimated from pre-steady-state experiments designed to measure the rate of reaction of the conversion of ES to EP (kchem) and are unaffected by the rate of complex association. The fact that the kcat values under both single- and multiple-turnover conditions are similar suggests that under these two conditions the rate-limiting step is the rate of the chemical cleavage. The kcat values in the present work are in agreement with the values ranging between 0.1 to 1.0 min−1 reported previously for other versions for the δ ribozyme (6, 10, 18).
Under pre-steady-state conditions the apparent Michaelis–Menten constant (KM), which is the concentration of E at half-maximum rate, represents the apparent dissociation constant of the ES′ complex. On the other hand, the apparent KM determined under steady-state conditions is a measure of the ribozyme bound in any form to the substrate (ES and ES′). Similar values of apparent KM and Kd under steady-state or pre-steady-state conditions implies that the majority of E-bound molecules are capable of going through the reaction steps without the rate-limiting conformational changes within the complex (16). At 37 and 56 °C, we estimated the apparent KM values to be 9.9 nM and 330 nM under pre-steady-state conditions and 510 nM and 1300 nM under steady-state conditions, respectively. These data are in ageement with those reported for other versions of δ ribozymes studied at 37 °C (6, 10, 18). The differences in KM between the pre-steady-state and steady-state experiments indicate that the ES complex is undergoing a slow internal conformational rearrangement yielding the ES′ complex. We believe that under pre-steady-state conditions the conformational transition is bypassed and the reaction goes directly to ES′ complex formation prior to initiation (e.g., addition of magnesium). The fact that the single-turnover KM (10 nM) and binding shift Kd (25–28 nM) are similar suggest that the majority of E molecules that bind substrate are capable of catalyzing the reaction at 37 °C. In contrast, the pre-steady-state KM values were significantly higher at 56 °C (>33-fold), near the steady-state KM, suggesting that a large proportion of the ES dissociates rather than goes through the reaction steps. Also, one possible explanation for the higher apparent KM under steady-state conditions, which is similar to the KI of the competitive SdC4 analogue, would be that the ES complex is slowly transformed into the active ES′ complex. Overall, the kcat/KM values (106–107 min−1·M−1) are comparable to those of other ribozymes, such as the ~ 107 min−1·M−11 reported for the group I intron (23). In this latter system the reaction is also limited by substrate association under single-turnover conditions and byproduct release under multiple-turnover conditions.
Both the 3′ product and the SdC4 analogues give relatively similar Kd values in gel shift assays with the active ribozyme (122 and 28 nM, respectively) and with the inactive A47 mutant ribozyme (276 and 190 nM, respectively). The similarity of the equilibrium dissociation constants was expected because the base-pairing regions of both analogues are identical. However, the maximum bound complex (the capacity to bind) was significantly reduced with the 3′ product as compared to the SdC4 analogue with both the active and inactive ribozymes (6-fold lower). One hypothesis to explain this decrease is that the 3′-product does not pass through the conformational transition that the SdC4 analogue does. Therefore, the conformation transition contributes to stabilizing the substrate/ribozyme complex, resulting in a higher proportion of bound complex, without decreasing the Kd value. As a consequence, we propose that the 5′ portion of the substrate might have an important contribution to the mechanism; at least one interaction in the ribozyme/substrate complex involves a chemical group of the substrate that is located upstream of the scissile phosphate (in the 5′ portion). These gel shift assays also showed a 7-fold difference in Kd in the binding of both the SdC4 analogue and the 3′ product to the active ribozyme as compared to the inactive ribozyme. This difference indicates that the cytosine residue at position 47 contributes to the binding mechanism. Cross-linking experiments and tridimensional δ ribozyme models suggest that residue C47 is in close proximity to the scissile phosphate (19, 20, 24, 25). Therefore, it appears reasonable to hypothesize that C47 can interact with a chemical group near the cleavage site. We also observed a difference in the binding of the SdC4 analogue and the RNA substrate to the A47 mutant ribozyme (8-fold stronger), suggesting that the 2′-hydroxyl group adjacent to the scissile phosphate is involved in the binding with the ribozyme. Thus, the results reported here suggest that the ribozyme C47, the 2′-hydroxyl adjacent to the cleavage site, as well as another chemical group in the substrate 5′ portion, may directly or indirectly contribute to the binding mechanism. Both the substrate recognition and binding processes of the δ ribozyme are not restricted exclusively by the formation of a helix composed of either six or seven Watson–Crick base pairs (e.g., P1 stem), but rather the 5′ portion of the substrate also plays a major role in both processes, and as a consequence it cannot be ignored in further studies.
Footnotes
Abbreviations: HDV, hepatitis δ virus; PAGE, polyacrylamide gel electrophoresis; Rz, ribozyme; S, substrate.
This work was supported by a Medical Research Council (MRC, Canada) grant and by a scientific team grant from Fonds pour la Formation de Chercheurs et l’Aide à la Recherche (FCAR, Québec). D.L. holds a predoctoral fellowship from Fonds pour la Recherche en Santé au Québec (FRSQ). S.M. and S.A. hold postdoctoral fellowships from the Canadian Liver Foundation and the Natural Science and Engineering Research Council (NSERC, Canada), respectively. J.-P.P. is an MRC scholar.
References
- 1.Lazinski DW, Taylor J. RNA. 1995;1:225–233. [PMC free article] [PubMed] [Google Scholar]
- 2.Been MD, Wickham GS. Eur J Biochem. 1997;247:741–753. doi: 10.1111/j.1432-1033.1997.00741.x. [DOI] [PubMed] [Google Scholar]
- 3.Mercure S, Lafontaine D, Roy G, Perreault JP. Médecine/Science. 1997;13:662–669. [Google Scholar]
- 4.Branch AD, Robertson HD. Proc Natl Acad Sci USA. 1991;88:10163–10167. doi: 10.1073/pnas.88.22.10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Been MD. Trends Biochem Sci. 1994;19:251–256. doi: 10.1016/0968-0004(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 6.Been MD, Perrotta AT, Rosenstein SP. Biochemistry. 1992;31:11843–11852. doi: 10.1021/bi00162a024. [DOI] [PubMed] [Google Scholar]
- 7.Kawakami J, Yuda K, Suh YA, Kumar PK, Nishikawa F, Maeda H, Taira K, Ohtsuka E, Nishikawa S. FEBS Lett. 1996;394:132–136. doi: 10.1016/0014-5793(96)00941-6. [DOI] [PubMed] [Google Scholar]
- 8.Fauzi H, Kawakami J, Nishikawa F, Nishikawa S. Nucleic Acids Res. 1997;25:3124–3130. doi: 10.1093/nar/25.15.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varani G. Annu Rev Biophys Biomol Struct. 1995;24:379–404. doi: 10.1146/annurev.bb.24.060195.002115. [DOI] [PubMed] [Google Scholar]
- 10.Lee BS, Lai YC, Ping YH, Huang ZS, Lin JY, Wu HN. Biochemistry. 1996;35:12303–12312. doi: 10.1021/bi961219m. [DOI] [PubMed] [Google Scholar]
- 11.Ananvoranich S, Perreault JP. J Biol Chem. 1998;273:13182–13188. doi: 10.1074/jbc.273.21.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milligan JF, Uhlenbeck OC. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 13.Beaudry D, Bussière F, Lareau F, Lessard C, Perreault JP. Nucleic Acids Res. 1995;23:745–752. doi: 10.1093/nar/23.5.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perreault JP, Altman S. J Mol Biol. 1992;226:399–409. doi: 10.1016/0022-2836(92)90955-j. [DOI] [PubMed] [Google Scholar]
- 15.Hertel KJ, Herschlag D, Uhlenbeck OC. EMBO J. 1996;15:3751–3757. [PMC free article] [PubMed] [Google Scholar]
- 16.Fersht AR. Enzyme Structure and Mechanism. 2. WH Freeman; New York: 1985. pp. 121–154. [Google Scholar]
- 17.Ulhenbeck OC. Nature. 1987;328:596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 18.Perrotta AT, Been MD. Biochemistry. 1992;31:16–21. doi: 10.1021/bi00116a004. [DOI] [PubMed] [Google Scholar]
- 19.Bravo C, Lescure F, Laugaa P, Fourrey JL, Favre A. Nucleic Acids Res. 1996;24:1351–1359. doi: 10.1093/nar/24.7.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenstein SP, Been MD. Biochemistry. 1996;35:11403–11413. doi: 10.1021/bi9609984. [DOI] [PubMed] [Google Scholar]
- 21.Puttaraju M, Perrotta AT, Been MD. Nucleic Acids Res. 1993;21:4253–4258. doi: 10.1093/nar/21.18.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suh YA, Kumar PKR, Taira K, Nishikawa S. Nucleic Acids Res. 1993;21:3277–3280. doi: 10.1093/nar/21.14.3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herschlag D, Cech TR. Biochemistry. 1990;29:10172–10180. doi: 10.1021/bi00496a004. [DOI] [PubMed] [Google Scholar]
- 24.Tanner NK, Schaff S, Thill G, Petit-Koskas E, Crain-Denoyelle AM, Westhof E. Curr Biol. 1994;4:488–498. doi: 10.1016/s0960-9822(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 25.Branch AD, Polaskova JA. Nucleic Acids Res. 1995;23:4180–4189. doi: 10.1093/nar/23.20.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]