

Abstract
Listeria monocytogenes infection induces a strong inflammatory response characterized by the production of IL-12 and IFN-γ and protective immunity against this pathogen is dependent on CD8+ T cells (CTL). Recent studies have suggested that these inflammatory cytokines affect the rate of memory CD8+ T cell generation as well as the number of short lived effector cells generated. The role of the closely related cytokine, IL-23, in this response has not been examined. We hypothesized that IL-12 and IL-23 produced by dendritic cells collectively enhance the generation and function of memory cells. To test this hypothesis, we employed a DC vaccination approach. Mice lacking IL-12 and IL-23 were vaccinated with wild-type (WT), IL-12−/−, or IL-12/23−/− DC and protection to Lm was monitored. Mice vaccinated with WT and IL-12−/− DC were resistant to lethal challenge with Lm. Surprisingly, mice vaccinated with IL-12/23−/− DC exhibited significantly reduced protection when challenged. Protection correlated with the relative size of the memory pools generated. In summary, these data indicate that IL-23 can partially compensate for the lack of IL-12 in the generation protective immunity against Lm.
1. Introduction
Cytokines play very important roles in shaping the magnitude and type of immune response elicited by infection (1, 2). These soluble factors can augment or suppress T cell activation at key points during immune responses (3-6). As a result of their potent effects, the contribution of these molecules to the generation of memory cells is being extensively investigated for their therapeutic potential (2, 7-12).
Members of the interleukin-12 (IL-12) family are potent regulators of immune responses, as reviewed in (13, 14). This family is composed of three cytokines-IL-12, IL-23, and IL-27. IL-12 augments and sustains Th1 immune responses which are very important in the eradication of intracellular pathogens as well as tumors (15-18). In contrast, IL-23 supports Th17 mediated immune responses which are important in the control of extracellular pathogens as well as the induction of autoimmunity (19-23). IL-27 promotes Th1 responses, inhibits Th2 responses, and decreases the proliferative capacity of melonomas (24-29).
The roles of IL-12 in CD8+ T cell activation have been studied in several systems and are well documented (4, 7, 30-32). These include inducing IFN-γ production, augmenting proliferation, and increasing the survival of these lymphocytes (4, 30, 31, 33). Recently, it has been demonstrated that systemic inflammation can reduce the number of memory CD8+ T cells generated and slow the rate of memory development (11, 34-36). IL-12 has been specifically implicated as a key inflammatory mediator in the development of memory CD8+ T cells (34, 36). For example, a recent study found that the generation of memory CD8+ T cells was enhanced while the number of effector cells was diminished in Lm-infected IL-12 deficient mice (p35−/−) compared to WT mice (36). Likewise, expression of the IL-12 receptor on CD8+ T cells was found to be required for this regulation. CD8+ T cells lacking expression of the high affinity chain of the IL-12 receptor (IL-12Rβ2−/−) formed a larger memory pool than WT T cells following immunization of with recombinant Lm-OVA (36). Other studies have demonstrated that IL-12 augmented the generation of short-lived effector cells (SLEC- KLRG1 high, IL-7Rα low) during primary responses; however, it did not have a substantial effect on memory precursor effector cell generation (MPEC- KLRG1 low, IL-7Rα high) (7, 37). Therefore, the importance of IL-12 in the generation of memory CD8+ T cells remains controversial.
While IL-12 is known to promote Th1 responses, IL-23 has emerged as a key regulator of Th17 responses. In the absence of Th1 or Th2 polarizing cytokines, IL-23 supports a Th17-type immune response, characterized by the production of IL-17 (38-40). This cytokine response augments the recruitment of phagocytes and lymphocytes to inflammatory foci (38). In addition, IL-23 can support Th1 responses by augmenting IFN-γ production and their proliferation (38-42). Although IL-23’s role in CD4+ T cell activation is now well documented, its impact on CD8+ T cell activation and memory generation has not been as thoroughly studied. IL-12 and IL-23 share a common subunit, p40, and a common receptor chain, β1, which has complicated the study of the specific roles of these two cytokines. Several early studies focused on IL-12 utilized mice that lacked p40, which had the unintended effect of targeting both cytokines (43). Thus, the specific roles of these two cytokines in the generation of CD8 T cell memory have not been thoroughly addressed. Furthermore, it been not been determined if these cytokines provided by DC are sufficient to prime protective memory or if other cellular sources of the cytokines are required.
Dendritic cell (DC) vaccination has been quite successful in murine models at eliciting protective immunity against Lm (7, 44, 45) and holds promise in clinical trials against tumors as well (46). Unlike direct infection with live Lm, which has been shown to be a potent inducer of inflammation, DC vaccination effectively induces immunity presumably in the absence of systemic inflammation (45). Therefore, we used a DC vaccination approach to determine if IL-12 or IL-23 produced by DC impacted the generation or function of memory CD8+ T cells in vivo.
We infected DC with a recombinant strain of Listeria monocytogenes (Lm-OVA) as a stimulus for DC maturation and a source of model antigen (OVA). We then vaccinated IL-12p40−/− mice (mice lacking IL-12 and IL-23) with Lm-OVA infected WT, IL-12−/−, or IL-12/23−/− dendritic cells (DC) and measured protection against Lm-OVA challenge. We found that mice vaccinated with Lm-OVA-infected IL-12−/−, or IL-12/23−/− DC were more susceptible to Lm-OVA challenge compared to mice vaccinated with similarly infected WT DC. Mice vaccinated with DC lacking IL-12 or IL-12/23 had higher bacterial burdens in the spleens and livers when challenged and exhibited severe morbidity when compared with mice vaccinated with WT DC. We found a correlation between the number of OVA-specific memory CD8+ T cells and resistance to Lm-OVA challenge; however, we did not observe compromised secondary CD8+ T cell responses to sub-lethal infection, regardless of the DC population used as vaccine. Our studies demonstrate that when these cytokines are provided exclusively by DC, both IL-12 and IL-23 contribute to protective immunity against Lm, yet in the absence of IL-12, IL-23 can serve a compensatory function in support of robust CTL responses. Thus, this study provides important insights as to the roles of IL-12 and IL-23 in the overall immunogenicity of DC vaccination.
2. Materials and methods
2.1. Listeria strains
Listeria monocytogenes strains 10403s (WT), was obtained from Dr. Daniel Portnoy (Univ. of California, Berkeley, CA) and Lm-OVA strain, was obtained from Dr. Hao Shen (University of Pennsylvania). For experiments, bacteria grown to stationary phase at 30°C in brain–heart infusion broth (BHI) were washed twice and resuspended in RPMI medium at a concentration to achieve the desired moi when added to DC monolayers. The moi was confirmed by plating dilutions of the inoculum on BHI agar and enumerating cfu after 24 h at 37°C.
2.2. Mice
C57BL/6 (WT), IL-12p35−/−BL/6-IL12atm/Jm, IL-12p40−/−BL/6-IL12btm/Jm and OT-1 TCR transgenic mice specific for OVA(257-264) presented by H-2Kb were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained and bred in the animal facility at Wake Forest University School of Medicine.
2.3. Surface and Intracellular Cytokine Staining
The activation state of OVA-specific CD8+ T cells (OT-1) was determined using MHC class I tetramers specific for OVA257-264 labeled with APC which were prepared according to published protocols (47) in combination with various markers of T cell activation. These markers included CD44 PE (clone IM7), CD62L PE (clone MEL-14), CD69 PE (clone H1.2F3) all from BD, San Diego, CA, and KLRG1 PE (clone 2F1), from Abcam, Cambridge MA. In order to determine OVA-specific CD8+ T cell function ex vivo or after restimulation with OVA peptide (SIINFEKL) for 5 hours, T cells were stained using CD8 Per-CP (clone 53-6.7), IFN-γ PE (clone XMG1.2), or TNF-α APC (clone MP6-XT22) all from BD. In order to address cytotoxic potential, T cells were stained using CD8 Per-CP (clone 53-6.7), Perforin PE (clone δG9) all from BD, and Granzyme B APC (clone GB11) from Caltag Laboratories, Burlingame CA. Synthetic OVA peptide, residues 257-264 (SIINFEKL), was synthesized at Wake Forest University School of Medicine Peptide Synthesis facility.
2.4. Dendritic Cell Propagation
Bone marrow derived DC were generated as previously described (3). Bone marrow was removed from the tibias and femurs of 8- to 10-week-old WT (C57BL/6), IL-12p35−/−, or IL-12p40−/− mice. Red blood cells were lysed, and the progenitor cells (5 × 105/ mL) were resuspended and plated in RPMI 1640 containing 10% FCS supplemented with 10 ng/mL GM-CSF (generated from a recombinant baculovirus expression system). Dendritic cells were cultured for 6 days at 37° in 5% CO2 and given fresh medium and cytokine on days 2 and 4. DC used for the described experiments were between 90-95% CD11c+ and expressed low levels of CD40, CD80, and CD86 prior to infection.
2.5. Infection of Dendritic Cells
Dendritic cells were seeded at 5 × 105/ well in a 48 well plate and infected with Lm-OVA (H. Shen, University of Pennsylvania) at a multiplicity of infection (MOI) of 1. Four hours post infection penicillin/streptomycin was added at 10 μg/ml. Twenty-four hours post-infection, mice were vaccinated with 2.5 × 105 DC/ mouse intravenously. Parallel samples of DC were stained for CD80, CD86, and CD40 to ensure that each DC population expressed similar levels of costimulatory molecules (data not shown). At the MOI used for these studies each DC population exhibited a 4-5 fold increase of CD86, a 1.5 fold increase in CD80, and a 2 fold increase in CD40 expression compared to the levels found on untreated DC (data not shown). A portion of the Lm-OVA infected DC that were treated with antibiotics were lysed and plated on BHI agar plates overnight at 37°C to check for viable bacteria. No colonies were detected, indicating that no viable bacteria were transferred with the DC used for vaccination (data not shown).
2.5. Adoptive Transfer of OT-1
OT-1 TCR transgenic CD8+ T cells specific for ovalbumin257-264-H-2Kb were adoptively transferred i.v. into mice 2 days prior to DC vaccination at 105 OT-1/ mouse. Prior to transfer, cells were stained for their surface expression of CD69, CD62L, and CD44. Staining for these markers indicated that > 97% of these cells exhibited a naïve phenotype (CD69 and CD44 low/CD62L high).
2.6. DC Vaccination
Mice were vaccinated using a modification of a previously published protocol (44). Briefly, Lm-OVA infected DC were seeded at a concentration of 2.5 × 105 DC/ 500 μl in serum free media. Mice were administered the 500 μl of DC in serum-free RPMI media intravenously via the lateral tail vein. To compare DC vaccination with direct infection, control mice were infected with Lm-OVA (H. Shen, Univ. Pennsylvania) in PBS at a concentration 3 × 103/mouse.
2.7. Bacterial Enumeration ex vivo
To assess the numbers of bacteria colonizing the liver and spleen, portions of each organ were removed and weighed. The organs were then homogenized and lysed in 5 ml of sterile water while vortexing. Serial dilutions were made in 96-well plates and plated on BHI agar. The total number of colonies per gram of liver or spleen was calculated after incubating overnight at 37°C.
2.8. Survival Analysis
p40−/− mice were challenged with 5 LD50 Listeria-OVA forty days after being vaccinated with either Lm-OVA infected WT DC, p35−/− DC, or p40−/− DC. Mice were sacrificed when they exhibited the following signs of morbidity: ruffled fur, hunched posture, labored breathing, unresponsiveness to auditory or tactile stimulation, and severe weight loss. The weights of the mice were recorded prior to challenge with Lm-OVA, and every day thereafter for two weeks. The mice were monitored 3 times daily in accordance with the guidelines outlined by the Animal Care and Use Committee (ACUC) of Wake Forest University School of Medicine.
2.9. T cell Enumeration
The total number of cells isolated from the spleens and livers of vaccinated mice were determined based on counts of trypan blue-excluding cells. The total number of OVA-specific or functional T cells (tagged by tetramer or cytokine-specific antibody) isolated from each organ was determined by multiplying the percentage of tagged T cells (determined via FACs analysis) by the total number of cells recovered. Numbers of tetramer positive cells correlated with the number of functional cells detected by ICS in all cases.
3. Results
3.1. Mice Vaccinated with IL-12/23 Deficient DC were Susceptible to Lm Challenge
To determine if IL-12 or IL-23 produced by DC was required for the generation of protective immunity against L. monocytogenes, IL-12/23 deficient mice (p40−/−) were vaccinated with Lm-OVA-infected DC generated from either C57BL/6 (WT DC), IL-12 deficient (p35−/− DC), or IL-12/IL-23 deficient mice (p40−/− DC). Forty days after vaccination, mice were challenged with 5 LD50 Lm-OVA and protection was monitored for two weeks. Mice were euthanized if they exhibited severe morbidity. We observed that mice vaccinated with WT DC were completely protected from lethal challenge with Lm-OVA and exhibited very mild illness, including slightly ruffled fur and only moderate weight loss (Fig. 1A and 1B). Mice immunized with p35−/− DC were also quite resistant to lethal challenge, with 80% of the vaccinated mice surviving the infection. However, the weight loss in mice immunized with p35−/− DC was more marked than that observed in mice vaccinated with WT DC (Fig. 1B). Yet, by one week post challenge the surviving mice had fully recovered and did not exhibit any signs of morbidity. In contrast, the p40−/− DC immunized mice failed to control the challenge infection, and only 40% of these mice survived for the duration of the study (Fig. 1A). The morbidity in the surviving mice in this group was also more pronounced, indicated by severe and prolonged weight loss (Fig. 1B). As a positive control, p40−/− mice were directly vaccinated with Lm-OVA which has been shown to elicit protective immunity (48). When challenged, 100% of these mice survived the infection (data not shown).
Fig. 1.
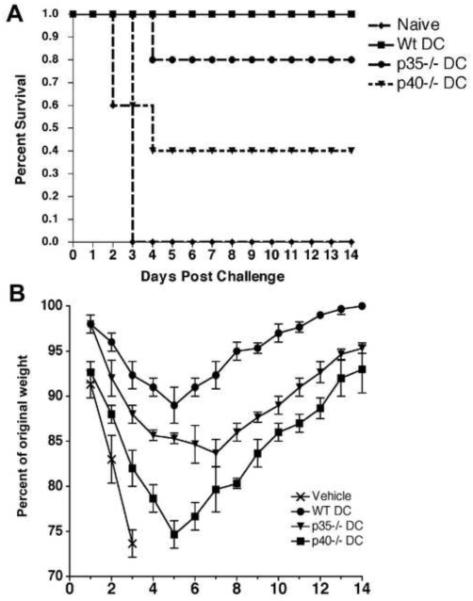
Survival of lethal Lm challenge is augmented by IL-12 and IL-23 delivered by DC during vaccination. p40−/− mice were immunized with WT, p35−/−, or p40−/− DC. Forty days post vaccination mice were challenged with a lethal dose of Lm-OVA. Survival (A) and weight loss (B) were monitored over two weeks in the challenge mice (N=5/group). Statistical analyses were performed using a Student’s t-Test with the WT DC vaccinated group serving as the positive control.
3.2. DC Vaccination with IL-12p40−/− DC Induced Organ-Specific Reduction in Secondary Effector CD8+ T cells Following Challenge
It has been established by several groups that CD8+ T cell mediated immunity is critical for protective immunity against Lm (49, 50), but it is not clear what specific roles IL-12 and IL-23 produced by DC play in initiating this response. Thus, we next addressed whether the decreased protection we observed in mice vaccinated with DC lacking IL-12 and IL-23 correlated with reduced secondary CD8+ T cell responses upon challenge. CD8+ T cell function in the spleens and livers of DC-vaccinated mice was analyzed ex vivo on day 3 post challenge with a sub-lethal dose of Lm-OVA (Day 43).
Direct ex vivo analysis of CD8+ T cells from the liver indicated that the number of OVA-specific CD8+ T cells in the livers of mice vaccinated with p40−/− DC was decreased over 5-fold on day 3 post-challenge with Lm, compared to mice vaccinated with WT or p35−/− DC (Fig. 2A). In contrast, the number of splenic OVA-specific CD8+ T cells in mice vaccinated with WT, p35−/−, or p40−/− DC on day 3 post challenge was not significantly different (Fig. 2B and 2D). Further, we found that the number of splenic CD8+ T cells producing IFN-γ was not significantly different between any of the DC vaccinated groups directly ex vivo (data not shown) or upon restimulation with OVA peptide (Fig. 2C and 2E). The mean fluorescence intensities (MFI) of IFN-γ and TNF-α of the T cells also was not different indicating that the amount of cytokine produced on a per cell basis was similar (data not shown). We also observed no significant differences in the number of OVA-specific CD8+ T cells expressing Granzyme B or perforin (data not shown). Taken together, these observations indicate that in the absence of both IL-12 and IL-23 during priming, secondary CD8+ T cell responses were decreased in nonlymphoid organs such as the liver upon Lm challenge but that in the absence of IL-12 alone, IL-23 played a compensatory role. Intriguingly, there were no differences in CD8+ T cell responses in the spleen, regardless of cytokines expressed by the DC.
Fig. 2.
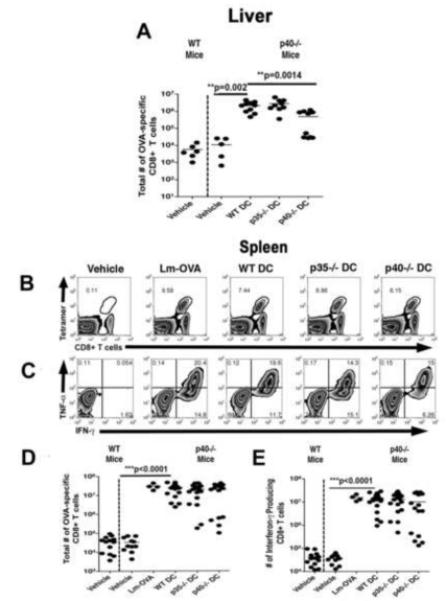
DC-produced IL-12 and IL-23 augment secondary CD8+ T cell responses in the liver of challenge mice. Mice vaccinated as described were challenged with 1 LD50 of Lm-OVA forty days post immunization. CD8+ T cell responses were determined in the spleen and liver on day 3 post challenge. The number of OVA-specific CD8+ T cells in the liver (A) and spleen (B and D), and the function of these T cells isolated from the spleen (C and E) were determined via FACs analysis after surface and intracellular cytokine staining. Statistical analyses were performed using a Student’s t-Test with the WT DC vaccinated group serving as the positive control.
3.3. Vaccination with DC lacking IL-12 and IL-23 Promoted Only Weak Bacterial Clearance from the Liver and Spleen Upon Sub-Lethal Challenge
In addition to monitoring CD8+ T cell responses in vaccinated mice challenged with a sub-lethal dose of Lm, we sought to determine if these responses correlated with bacterial clearance. Mice were vaccinated as previously described and challenged forty days later with 1 LD50 of Listeria. Bacterial loads were assessed in the liver and spleen 3 days post challenge. In general, liver colonization was observed more frequently in the DC vaccinated groups than colonization of the spleen (Fig. 3A and B). Again, the positive control mice, (p40−/− mice immunized with Lm-OVA at 3 × 103 CFU/ mouse) were highly resistant to this challenge dose as evidenced by the lack of detectable bacteria in the liver (Fig. 3A), in agreement with previously published data (32). Vaccination with WT DC infected with Lm-OVA also conferred resistance to this challenge (Fig. 3). We found that 85% of the p40−/− mice immunized with WT DC had no detectable Lm-OVA in their livers, and all had cleared bacteria from the spleen by day 3 post challenge (Fig. 3B and 3D). Additionally, in the few mice from this group that were still infected, the bacterial loads were significantly lower than those observed in challenged naïve WT mice (Fig. 3A).
Fig. 3.
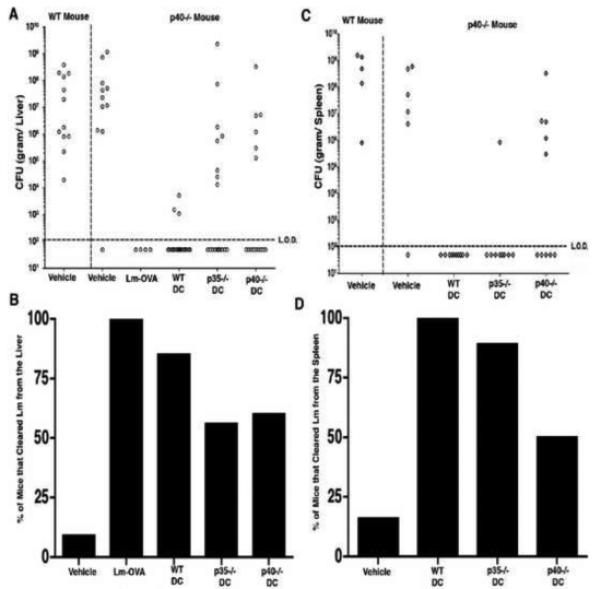
IL-12 and IL-23-enhanced resistance to Lm which is also evident at lower challenge doses. p40−/− mice were immunized with WT, p35−/−, or p40−/− DC. Forty days post vaccination mice were challenged with 1 LD50 of Lm-OVA. Lm-OVA colonization of the liver (A) and the spleen (C) were determined on day 3 post-challenge. The percentage of mice that had cleared Lm-OVA from the liver (B) and spleen (D) was determined by the following formula: (# of mice with no detectable bacteria/# of mice with detectable bacteria) x 100.
Vaccination of the p40−/− mice with Lm-OVA infected p35−/− DC led to a slight increase in susceptibility to Lm-OVA challenge, most notably in the liver (Fig. 3A). We observed that 56% of mice vaccinated with p35−/− DC contained no detectable bacteria in their livers on day 3 post challenge (Fig. 3B). However, the mice in this group that had detectable colonies in this organ had high burdens that were comparable to those observed in naïve animals (Fig. 3A). Interestingly, this group of mice had more efficient clearance of bacteria from the spleen than in the liver. In fact, we found that 89% of the p35−/− DC vaccinated mice had no recoverable bacteria in the spleen (Fig. 3D). These data suggest that in the absence of IL-12, IL-23 can partially compensate by stimulating T cell responses that more readily localize to non-lymphoid organs.
Strikingly, p40−/− DC vaccination provided little protection to challenge as evidenced by substantial colonization of both the spleen and liver. We found that 40% of the vaccinated mice had a heavy bacterial burden in the liver (Fig. 3B) and 50% demonstrated colonization of the spleen (Fig. 3D), similar to the naïve WT mice (Fig. 3A and 3B). These data indicate that DC-derived IL-12 and IL-23 augment protective immunity against Lm which is evident even at lower challenge doses.
3.4. IL-12 and IL-23 Produced by DC Increase the Generation of Memory CD8+ T cells
Since we observed that IL-12 and IL-23 increased protective immunity against Lm, and augmented secondary CD8+ T cell responses in the liver by day 3 post challenge, we next wanted to determine how these cytokines affected the generation of memory CD8+ T cells. To measure this effect, mice were vaccinated with either WT, p35−/−, or p40−/− DC and their T cell responses were analyzed forty days after immunization. We chose this time point to determine the functional capacity of memory CD8+ T cells prior to challenge with Lm, which could potentially explain the differences we observed in our challenge studies (survival and bacterial burden).
Vaccination with WT and p35−/− DC elicited large numbers of OVA-specific CD8+ T cells in both the spleen and liver at day 40 post-challenge. However, immunization with p40−/− DC generated substantially fewer memory CD8+ T cells in both organs (more than ten-fold fewer in the liver and over three-fold less in the spleen) compared to that generated by WT DC vaccination (Fig. 4A and 4B). In addition, the frequency of T cells producing IFN-γ and TNF-α ex vivo was significantly lower in p40−/− DC vaccinated mice than in WT DC and p35−/− DC-vaccinated mice. We observed almost a three-fold decrease in the number of CD8+ T cells that produced IFN-γ in response to OVA peptide stimulation compared to vaccination with WT DC (Fig. 4C and 4D). Further examination of the memory T cells revealed that IL-12 and IL-23 did not affect the surface expression of CD44, CD62L, or KLRG1 (data not shown) suggesting that there were no striking phenotypic differences in the T cell populations elicited, only in the number of these cells generated.
Fig. 4.
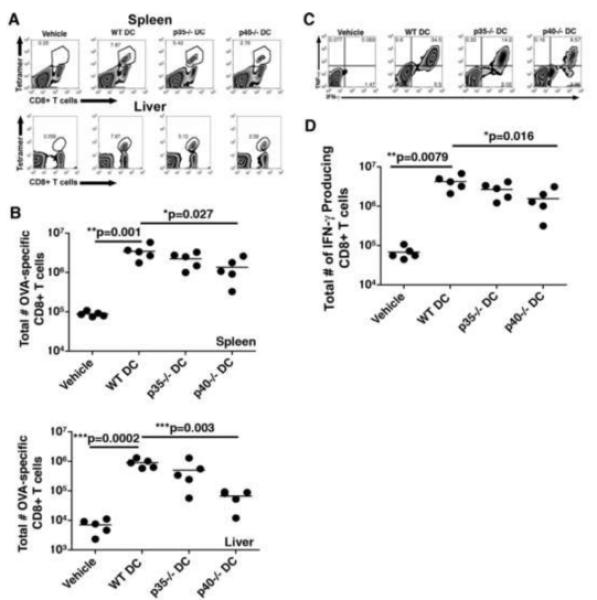
Memory CD8+ T cell development is compromised in the absence of IL-12 and IL-23. Mice were vaccinated as described and forty days post-immunization CD8+ T cell responses were assessed in the spleen and liver. The number of OVA-specific CD8+ T cells in the spleen and liver (A and B), and the function of these T cells isolated from the spleen (C and D) were determined via FACs analysis after surface and intracellular cytokine staining. Statistical analyses were performed using a Student’s t-Test with the WT DC vaccinated group serving as the positive control.
3.5. IL-12 and IL-23 Produced by DC Do not Affect CD8+ T cell Numbers or IFN-γ Production During the Primary T cell Response
Studies using the Lm model system have revealed that immunization strategies using heat-killed (HKLM) or LLO(−) Lm results in sub-optimal primary CD8+ T cell responses characterized by decreased numbers of CTL and diminished IFN-γ production compared with WT Lm immunization which is known to generate protective immunity (51-54). Surprisingly, the same defects observed in primary CD8+ T cell responses elicited by HKLM and LLO- Lm were also observed in secondary CD8+ T cell responses, suggesting that if primary CD8+ T cell responses do not reach a certain activation threshold this may result in the suboptimal generation of memory CD8+ T cells and ultimately their inability to confer protective immunity (53). Further examination of the innate immune responses in vivo revealed that diminished DC maturation (costimulatory molecule upregulation and cytokine production) induced by HKLM or LLO- Lm may account in part for the decrease in T cell response and resulting lack of protection (52). Since IL-12 and IL-23 are produced by Lm-infected mature DC, and are known to be important modulators of T cell responses, we next wanted to determine if primary responses were compromised in the absence of DC-derived IL-12 and IL-23 delivered during vaccination. In order address this question, we monitored T cell responses (total number of OVA-specific CD8+ T cells, and IFN-γ production) at the peak of the T cell response (day 7) after vaccination of the p40−/− mice with WT, p35−/−, or p40−/− DC. We observed that expansion of CD8+ T cells was not affected by the absence of IL-12 and IL-23 during priming, indicated by similar total numbers of OVA-specific cells (mean = 8 × 106/mouse) on day 7 in all DC vaccination strategies in the spleen (Fig. 5C). Surprisingly, the numbers of IFN-γ producing, OVA-specific CD8+ T cells were not affected by the presence or absence of DC-produced IL-12 or IL-23 (Fig. 5A and 5B). Although IL-12 has been shown by several investigators to augment IFN-γ production and proliferation of CD8+ T cells, we postulate that IL-12 and IL-23 produced exclusively by DC do not affect the expansion phase of the T cell response. Instead, these cytokines appear to affect a later stage of the immune response, ultimately impacting the number of memory cells maintained.
Fig. 5.
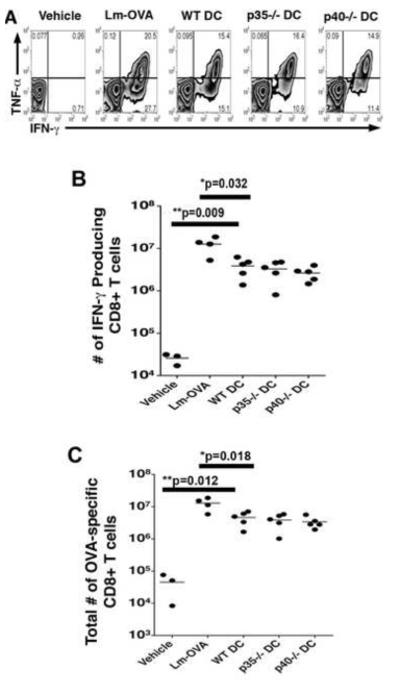
Primary CD8+ T cell responses are not augmented by the presence of DC-produced IL-12 and IL-23. Mice were vaccinated as described and 7 days post-immunization CD8+ T cell responses were determined in the spleen. The number of OVA-specific CD8+ T cells (C), and the function of these T cells isolated from the spleen (A and B) were determined via FACs analysis after surface and intracellular cytokine staining. Statistical analyses were performed using a Student’s t-Test with the WT DC vaccinated group serving as the positive control.
4. Discussion
The members of the Interleukin-12 cytokine family, specifically IL-12 and IL-23, are known to impact T cell responses in distinct ways, reviewed in (13, 14). Dendritic cells, thought to be critical for priming of T cell responses, are also known to produce these cytokines in response upon infection with bacteria (our paper and another on Il-23 from DC). However, it remains to be determined if these cytokines provided exclusively by DC are sufficient to regulate the number and functional profile of the primary, secondary, or memory T cell response. Here, we report that IL-12 and IL-23 produced exclusively by DC are key to the development of protective T cell responses against Listeria monocytogenes. Intriguingly, our results reveal that although the cells producing IL-12 or IL-23 are present only during the initiation of the primary response (in the form of DC vaccination) these cytokines have a profound effect on the secondary and memory responses as well. Furthermore, in the absence of IL-12, IL-23 can play a compensatory role, enhancing the localization of Lm-specific T cells to non-lymphoid organs such as the liver.
Most pathogens contain multiple TLR agonists, and during infection with virulent pathogens, multiple inflammatory cytokines and chemokines are often produced. If the pathogen is a potent inducer of inflammation and elicits a broad cytokine response (IFN-γ, IFN α/β, TNF-α, and/or IL-12/23), the redundant activities in this broad response could mask the individual activities of each cytokine (55). Therefore, the role of specific cytokines such as IL-12 vs. IL-23 may be more readily discerned under conditions of less extensive inflammation (such as immunization with attenuated pathogens or DC vaccination) (45). These types of studies are particularly important for the continued improvement of vaccines, since they are designed to generate the desired T cell response without extensive inflammation.
There is mounting evidence to suggest that high level inflammatory cytokine production (IL-12 and IFN-γ in particular) during primary responses can delay the generation of memory CD8+ T cells (2, 7, 8, 36, 37, 56). In conditions under which inflammation is attenuated including live Lm infection followed by antibiotic treatment, DC vaccination, and in IL-12 deficient (p35−/−) mice, T cells with the characteristics of memory cells are detectable at earlier timepoints or at higher frequency following infection or immunization (11, 35-37, 45, 57, 58). Another related study has demonstrated that high level IL-12 production during primary responses increases the number of short-lived effectors cells (SLEC) that develop during this phase of T cell activation (34). More recent studies however, have demonstrated that IL-12 does not alter the generation of memory precursor effector cells (MPEC) (7, 37). In addition, several reports have observed that IL-12 enhances the generation and function (increased secondary expansion upon antigen reencounter) of memory CD8+ T cells (12, 59, 60). Together, these studies highlight the importance of IL-12 in memory cell generation, but the nature of the effect may be modulated based on the relative intensity of the overall inflammatory response. The data from our study, along with previously published work, suggests that IL-12 and IL-23 produced by dendritic cells augment protective CD8+ T cell responses when inflammation is limiting and the only source of the cytokine is the DC. However, if inflammation is robust (i.e. infection with virulent pathogens) other cytokines may compensate for the lack of IL-12 and IL-23 in the generation of CD8+ T cell mediated protective immunity.
A major difference between our study and the previous studies examining the role of IL-12 and IL-23 on CD8 T cell memory, was the experimental approach. In the current study we vaccinated mice lacking IL-12 and IL-23 (p40−/−) with DC either expressing or lacking these cytokines. Thus, naïve CD8+ T cells could only receive these cytokines from the DC we vaccinated with or not at all. Using this system, we were able to determine how a transient exposure to IL-12 and/or IL-23 effected memory T generation, secondary T cell responses, and protective immunity in vivo. Specifically, we have concluded that both IL-12 and IL-23 are important in the generation of memory CD8+ T cells, under conditions where inflammation is not robust. Our results revealed that in the absence of IL-12, IL-23 could augment memory CD8+ T cell responses, indicating a previously unrecognized compensatory role for this cytokine. However, if neither cytokine was present (in p40−/− DC vaccination), the mice were significantly less protected upon challenge than those vaccinated with p35−/− or WT DC.
In addition to augmenting the number of memory CD8+ T cells, IL-23 enhanced secondary responses in the liver upon challenge (Fig. 3). Interestingly, the increased number of memory CD8+ T cells observed in the presence of IL-23 was not the result of enhanced T cell expansion during the primary response (Fig. 5). Therefore, we hypothesize that IL-23 may enhance the survival of T cells during contraction, increase the survival of memory cells, or increase the rate of homeostatic proliferation of these cells resulting in the larger memory pool. It especially intriguing that we observed a striking effect of IL-23 on the number of secondary effector cells in the liver than in the spleen upon challenge (Fig. 4). These data suggest that IL-23 may impact the homing of T cells to lymphoid vs. non-lymphoid organs. Support for this notion is provided by a recent study demonstrating that IL-23 is required for the production of IL-17 and recruitment of PMN to the liver upon Lm infection (61). Likewise, a recent study focused on the chemokines CCL19 and CCL21, indicates that the capacity of T cells to migrate in an EAE model is dependent not on these CCR7 ligands, but CCR7-dependent induction of IL-23 (62). Thus, we are only beginning to appreciate the role of IL-23 in determining the migratory fate of T cells.
Our study taken together with current literature, collectively support a model in which IL-12 and IL-23 serve as rheostats, translating the severity of an infection and influencing the resultant level of response (2, 7, 8, 11, 32, 34, 36, 56, 59, 60, 63). If IL-12/23 is abundant, memory generation is delayed in favor of the generation of SLECs with the intent of resolving the infection. However, if these cytokines are present in low amounts, SLEC formation is reduced, and more cells are committed to the generation of memory CD8+ T cells. We have identified a previously unrecognized role for IL-23 in increasing CD8+ T cell mediated immunity particularly in the absence of IL-12. Thus, these cytokines remain important targets for consideration in the design of vaccines aimed at generating protective CD8+ T cell responses.
Acknowledgements
This research was supported by the NIH Research Project Grant Program (R01) Grant #AI057770-01A1 to E.M.H. Additional support was provided by the NIH Predoctoral Fellowship Award for Minority Students (F31) Grant #AI73245-01A1 to C.J.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Geginat J, Sallusto F, Lanzavecchia A. J Exp Med. 2001;194:1711–9. doi: 10.1084/jem.194.12.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harty JT, Badovinac VP. Nat Rev Immunol. 2008;8:107–19. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- 3.Brzoza KL, Rockel AB, Hiltbold EM. J Immunol. 2004;173:2641–51. doi: 10.4049/jimmunol.173.4.2641. [DOI] [PubMed] [Google Scholar]
- 4.Chang J, Cho JH, Lee SW, Choi SY, Ha SJ, Sung YC. J Immunol. 2004;172:2818–26. doi: 10.4049/jimmunol.172.5.2818. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, Hunter CA, Kastelein RA, Cua DJ. J Clin Invest. 2006;116:1317–26. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dercamp C, Chemin K, Caux C, Trinchieri G, Vicari AP. Cancer Res. 2005;65:8479–86. doi: 10.1158/0008-5472.CAN-05-1319. [DOI] [PubMed] [Google Scholar]
- 7.Cui W, Joshi NS, Jiang A, Kaech SM. Vaccine. 2009;27:2177–87. doi: 10.1016/j.vaccine.2009.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joshi NS, Kaech SM. J Immunol. 2008;180:1309–15. doi: 10.4049/jimmunol.180.3.1309. [DOI] [PubMed] [Google Scholar]
- 9.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL. Nat Immunol. 2005;6:1236–44. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 10.Messingham KA, Badovinac VP, Jabbari A, Harty JT. J Immunol. 2007;179:2457–66. doi: 10.4049/jimmunol.179.4.2457. [DOI] [PubMed] [Google Scholar]
- 11.Badovinac VP, Harty JT. J Immunol. 2007;179:53–63. doi: 10.4049/jimmunol.179.1.53. [DOI] [PubMed] [Google Scholar]
- 12.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. J Immunol. 2009;182:2786–94. doi: 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trinchieri G. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 14.Trinchieri G, Pflanz S, Kastelein RA. Immunity. 2003;19:641–4. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Science. 1993;260:547–9. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 16.Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. J Exp Med. 1993;177:1199–204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sypek JP, Chung CL, Mayor SE, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. J Exp Med. 1993;177:1797–802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeh HJ, 3rd, Hurd S, Storkus WJ, Lotze MT. J Immunother Emphasis Tumor Immunol. 1993;14:155–61. doi: 10.1097/00002371-199308000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. Immunol Rev. 2008;226:132–46. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abraham C, Cho JH. Annu Rev Med. 2008 [Google Scholar]
- 21.Overwijk WW, de Visser KE, Tirion FH, de Jong LA, Pols TW, van der Velden YU, van den Boorn JG, Keller AM, Buurman WA, Theoret MR, Blom B, Restifo NP, Kruisbeek AM, Kastelein RA, Haanen JB. J Immunol. 2006;176:5213–22. doi: 10.4049/jimmunol.176.9.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Quinn DB, Palmer MT, Lee YK, Weaver CT. Adv Immunol. 2008;99:115–63. doi: 10.1016/S0065-2776(08)00605-6. [DOI] [PubMed] [Google Scholar]
- 23.Abraham C, Cho J. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.20894. [DOI] [PubMed] [Google Scholar]
- 24.Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, Chen Y, Tato CM, McClanahan TK, de Waal Malefyt R, Hunter CA, Cua DJ, Kastelein RA. J Immunol. 2009;182:5748–56. doi: 10.4049/jimmunol.0801162. [DOI] [PubMed] [Google Scholar]
- 25.Shinozaki Y, Wang S, Miyazaki Y, Miyazaki K, Yamada H, Yoshikai Y, Hara H, Yoshida H. Int J Cancer. 2009;124:1372–8. doi: 10.1002/ijc.24107. [DOI] [PubMed] [Google Scholar]
- 26.Yoshimoto T, Morishima N, Mizoguchi I, Shimizu M, Nagai H, Oniki S, Oka M, Nishigori C, Mizuguchi J. J Immunol. 2008;180:6527–35. doi: 10.4049/jimmunol.180.10.6527. [DOI] [PubMed] [Google Scholar]
- 27.Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Expert Opin Biol Ther. 2007;7:1705–21. doi: 10.1517/14712598.7.11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oniki S, Nagai H, Horikawa T, Furukawa J, Belladonna ML, Yoshimoto T, Hara I, Nishigori C. Cancer Res. 2006;66:6395–404. doi: 10.1158/0008-5472.CAN-05-4087. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu M, Shimamura M, Owaki T, Asakawa M, Fujita K, Kudo M, Iwakura Y, Takeda Y, Luster AD, Mizuguchi J, Yoshimoto T. J Immunol. 2006;176:7317–24. doi: 10.4049/jimmunol.176.12.7317. [DOI] [PubMed] [Google Scholar]
- 30.Curtsinger JM, Lins DC, Mescher MF. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valenzuela J, Schmidt C, Mescher M. J Immunol. 2002;169:6842–9. doi: 10.4049/jimmunol.169.12.6842. [DOI] [PubMed] [Google Scholar]
- 32.Orgun NN, Mathis MA, Wilson CB, Way SS. J Immunol. 2008;180:4109–15. doi: 10.4049/jimmunol.180.6.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kieper WC, Prlic M, Schmidt CS, Mescher MF, Jameson SC. J Immunol. 2001;166:5515–21. doi: 10.4049/jimmunol.166.9.5515. [DOI] [PubMed] [Google Scholar]
- 34.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Immunity. 2007;27:281–95. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haring JS, Badovinac VP, Harty JT. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Pearce EL, Shen H. J Immunol. 2007;179:2074–81. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
- 37.Wilson DC, Matthews S, Yap GS. J Immunol. 2008;180:5935–45. doi: 10.4049/jimmunol.180.9.5935. [DOI] [PubMed] [Google Scholar]
- 38.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 39.Harrington LE, Mangan PR, Weaver CT. Curr Opin Immunol. 2006;18:349–56. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 40.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Immunity. 2006;24:677–88. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, Guglani L, Alcorn JF, Strawbridge H, Park SM, Onishi R, Nyugen N, Walter MJ, Pociask D, Randall TD, Gaffen SL, Iwakura Y, Kolls JK, Khader SA. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. J Immunol. 2005;175:788–95. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 43.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 44.Hamilton SE, Harty JT. J Immunol. 2002;169:4936–44. doi: 10.4049/jimmunol.169.9.4936. [DOI] [PubMed] [Google Scholar]
- 45.Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. Nat Med. 2005;11:748–56. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- 46.Palucka AK, Ueno H, Fay J, Banchereau J. J Immunother. 2008;31:793–805. doi: 10.1097/CJI.0b013e31818403bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Immunity. 1998;8:177–87. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 48.Krawczyk CM, Shen H, Pearce EJ. Infect Immun. 2007;75:3556–60. doi: 10.1128/IAI.00086-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ladel CH, Flesch IE, Arnoldi J, Kaufmann SH. J Immunol. 1994;153:3116–22. [PubMed] [Google Scholar]
- 50.Seaman MS, Perarnau B, Lindahl KF, Lemonnier FA, Forman J. J Immunol. 1999;162:5429–36. [PubMed] [Google Scholar]
- 51.Bouwer HG, Gibbins BL, Jones S, Hinrichs DJ. Infect Immun. 1994;62:1039–45. doi: 10.1128/iai.62.3.1039-1045.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muraille E, Giannino R, Guirnalda P, Leiner I, Jung S, Pamer EG, Lauvau G. Eur J Immunol. 2005;35:1463–71. doi: 10.1002/eji.200526024. [DOI] [PubMed] [Google Scholar]
- 53.Lauvau G, Vijh S, Kong P, Horng T, Kerksiek K, Serbina N, Tuma RA, Pamer EG. Science. 2001;294:1735–9. doi: 10.1126/science.1064571. [DOI] [PubMed] [Google Scholar]
- 54.Barry RA, Bouwer HG, Portnoy DA, Hinrichs DJ. Infect Immun. 1992;60:1625–32. doi: 10.1128/iai.60.4.1625-1632.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kapsenberg ML. Nat Rev Immunol. 2003;3:984–93. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 56.Hand TW, Kaech SM. Immunol Res. 2008 doi: 10.1007/s12026-008-8027-z. [DOI] [PubMed] [Google Scholar]
- 57.Badovinac VP, Harty JT. Immunol Rev. 2006;211:67–80. doi: 10.1111/j.0105-2896.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 58.Pearce EL, Shen H. Immunol Rev. 2006;211:197–202. doi: 10.1111/j.0105-2896.2006.00399.x. [DOI] [PubMed] [Google Scholar]
- 59.Lee JB, Lee KA, Chang J. Int Immunol. 2007;19:1039–48. doi: 10.1093/intimm/dxm072. [DOI] [PubMed] [Google Scholar]
- 60.Ye Z, Xu S, Moyana T, Yang J, Xiang J. Cell Mol Immunol. 2008;5:147–52. doi: 10.1038/cmi.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meeks KD, Sieve AN, Kolls JK, Ghilardi N, Berg RE. J Immunol. 2009;183:8026–34. doi: 10.4049/jimmunol.0901588. [DOI] [PubMed] [Google Scholar]
- 62.Kuwabara T, Ishikawa F, Yasuda T, Aritomi K, Nakano H, Tanaka Y, Okada Y, Lipp M, Kakiuchi T. J Immunol. 2009;183:2513–21. doi: 10.4049/jimmunol.0800729. [DOI] [PubMed] [Google Scholar]
- 63.Bashyam H. J Exp Med. 2007 [Google Scholar]