

Abstract
The MA protein from HIV-1 is a small, multifunctional protein responsible for regulating various stages of the viral replication cycle. To achieve its diverse tasks MA interacts with host cell proteins and it has been reported that one of these is the ubiquitous calcium -sensing calmodulin (CaM) which is up-regulated upon HIV-1 infection. The nature of the CaM-MA interaction has been the subject of structural studies using peptides based on the MA sequence that have led to conflicting conclusions. The results presented here show that CaM binds intact MA with 1:1 stoichiometry in a Ca2+-dependent manner and that the complex adopts a highly extended conformation in solution as revealed by small-angle X-ray scattering. Alterations in tryptophan fluorescence suggest that the two tryptophans at the N-terminus of MA mediate the CaM interaction. Major chemical shift changes occur in the NMR spectrum of MA upon complex formation, while chemical shift changes in the CaM spectrum are quite modest and are assigned to residues within the target-protein binding hydrophobic clefts of CaM. The NMR data indicate that CaM binds MA via its N-and C-terminal lobes and induces a dramatic conformational change involving a significant loss of secondary and tertiary structure within MA. Circular dichroism experiments suggest that MA looses ~20% of its α-helical content upon CaM binding. Thus CaM binding is expected to impact upon the accessibility of interaction sites within MA that are involved in its various functions.
INTRODUCTION
Human immunodeficiency virus 1 (HIV-1) infection results in elevated levels of expression of the intracellular calcium sensor calmodulin (CaM) in the host cell1. Furthermore, it has been reported that CaM forms a complex with the HIV-1 Gag polyprotein and with its multifunctional N-terminal subunit, MA (also known as p17)2 that is proteolytically released by HIV-1 protease during the late stages of infection and viral maturation3; 4; 5. Gag is a single 55 kDa polypeptide chain capable of directing virion assembly and budding in the absence of other HIV-1 components (reviewed by Wills and Craven6). Its MA subunit forms the matrix layer around the core of the HIV-1 virus7; 8. Two predominant species of MA are important in vivo; the cleaved form within the mature virion and the host cytoplasm during viral infection, and the Gag-associated form found during the late stages of viral replication and export. MA is involved in directing Gag to the plasma membrane9; 10, mediated via its N-terminal myristoyl group11, and in recruiting additional proteins for viral assembly12. In addition to its structural functions in the mature virus, free MA plays diverse roles during the early stages of viral replication including the regulation of early post-entry steps and inclusion in the viral pre-integration complex13; 14.
The structure of MA has been solved by NMR15; 16 and crystallography17, and consists of a largely α-helical globular N-terminal ‘head’ with a flexible C-terminal ‘tail’. The crystal structure contains six MA molecules of similar conformation in the asymmetric unit arranged in trimers, while NMR data indicate MA is monomeric in solution. The NMR and crystal structures of the monomers are similar; with the major differences being in the flexible tail and the helix connecting it to the globular head.
While it has been reported that CaM and MA interact, the nature of the interaction is not known and its role in HIV-1 infection is not understood2. CaM is the ubiquitous intracellular calcium sensor that regulates a wide range of physiological functions through its ability to bind many different proteins such that its binding is both specific and yet diverse (reviewed by Crivici and Ikura18). CaM is relatively small (148 residues), highly conserved amongst eukaryotes19 and is described as having a ‘dumbbell’ shape with two globular lobes connected by an extended α-helix20 that is known to be flexible in solution21; 22. Each lobe consists of a pair of ‘EF-hand’ motifs23 that comprise a cup-shaped domain with two Ca2+-binding loops forming the base of the cup and four α-helices the sides. Hydrophobic residues lining the base of the cup form a cleft that is opened upon Ca2+-binding24; 25, thus exposing hydrophobic residues that mediate target protein recognition and binding26
The specificity of CaM binding to target proteins is generally conferred by the presence of large hydrophobic ‘anchor’ residues within a target sequence, spaced to bind at each of the hydrophobic clefts of CaM. Electrostatic interactions stabilize binding and govern the orientation of the target binding site (reviewed by Vetter and LeClerc27). The flexibility of the interconnecting helix of CaM enables the target-binding clefts in each lobe to be oriented in order to accommodate the wide variety of stereochemistries found amongst the diverse CaM target proteins28. The ‘classical’ Ca2+-dependent binding mode for CaM is illustrated by the complex it forms with the α-helical peptides from smooth and skeletal muscle myosin light chain kinase (MLCK)26; 29. In this mode, two large hydrophobic anchor residues in MLCK, spaced 12 residues apart, each bind to one of the CaM hydrophobic clefts to form a hydrophobic ‘tunnel’ as the CaM dumbbell collapses in order to encompass the peptide. The collapse is facilitated by an unwinding of the central helix, which acts as a flexible hinge, and the orientation of the peptide is determined by basic residues in the N-terminus of the peptide that form salt bridges to the C-terminal lobe of CaM. Numerous variations from this arrangement have been documented and include those complexes in which CaM has an extended rather than collapsed conformation30 or binds a target peptide in the opposite orientation31, as a dimer32, or in the absence of Ca2+ 33; 34; 35.
CaM-peptide studies have implicated the N-terminal ~50 residue sequence of MA in CaM binding. Sequence segments consisting of residues 11–25 and 31–46 of MA each contain a single tryptophan (W16 and W36), both of which are in the globular domain with W16 sequestered within the hydrophobic core and W36 partially exposed to solvent (Figure 1). Tryptophan fluorescence spectroscopy shows that these peptides bind to Ca2+-CaM with nanomolar affinities2. Residues 11–46 of MA constitute the two perpendicularly oriented α-helices connected by a basic loop region, and it has been proposed that they form a nearly contiguous CaM-binding surface 15. The peptide studies to date have not unambiguously resolved the question of the stoichiometry of the putative Ca2+-CaM-MA complex2. Previous small angle X-ray scattering (SAXS) studies led to a proposed novel arrangement for the interaction that does not involve the canonical target-recognition hydrophobic clefts of CaM, and instead depends upon electrostatic interactions with CaM’s interconnecting helix.
Figure 1. CaM-binding sites on the MA structure.
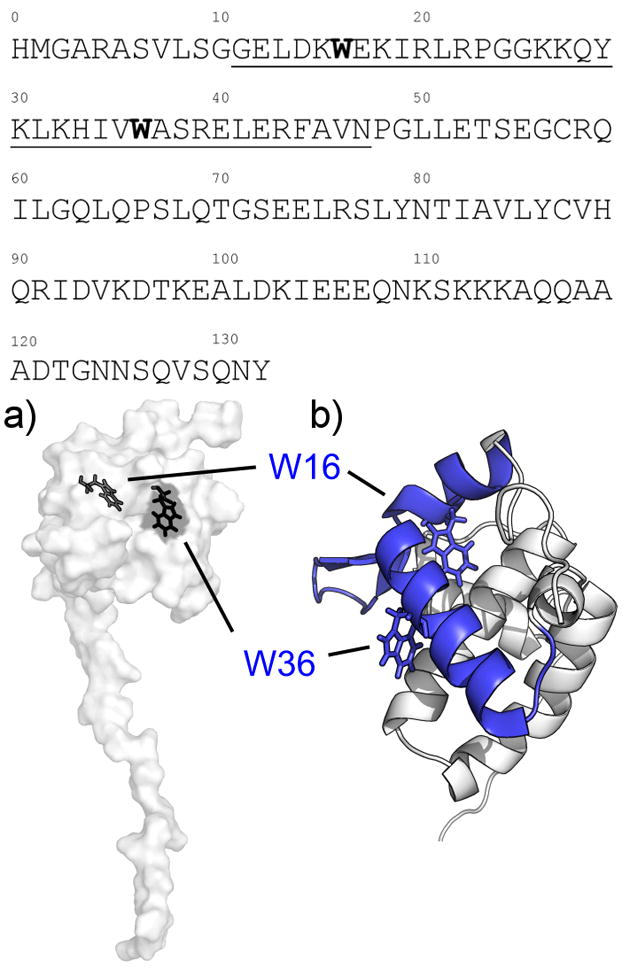
(Top) Amino acid sequence for the MA construct used in this experiment, depicting the proposed CaM-binding sites (underlined) and the tryptophan residues located within (bold). a) Surface depiction of the MA NMR structure (PDB: 2HMX) showing the tryptophan residues (black) located within the N-terminal lobe of MA, with W16 completely buried within the hydrophobic core while W36 is partially exposed to solvent. b) Closeup view of the N-terminal globular domain of MA and the CaM-binding site (blue) in cartoon form. The site is composed of two short α-helices connected by a basic loop region.
We report here a set of biophysical studies that focus on the interaction between CaM and full-length MA in order to definitively establish the binding stoichiometry and the nature of the interaction. We find that CaM binds MA in a 1:1 stoichiometry, the binding is Ca2+-dependent and is weakened with increasing concentrations of NaCl. Unexpectedly, the native structure of MA is dramatically disrupted upon complex formation, and we discuss the implications of this result for MA function.
RESULTS AND DISCUSSION
Both apo and Ca2+-CaM bind MA with affinities that vary inversely with ionic strength
To qualitatively assess the interaction between CaM and MA ‘pulldown’ assays were performed using covalently-linked CaM agarose beads and free MA in solutions containing increasing concentrations of NaCl in the presence and absence of Ca2+. The results show that MA binds to CaM in a Ca2+-dependent manner and that the affinity decreases with increasing ionic strength (Figure 2). No observable interaction was detected between apo-CaM and MA in solutions containing more than 50 mM NaCl, while for Ca2+-CaM 500 mM NaCl was required to abolish MA binding.
Figure 2. Effect of Ca2+ and NaCl on the interaction between CaM and MA monitored by SDS-PAGE pulldown assays.
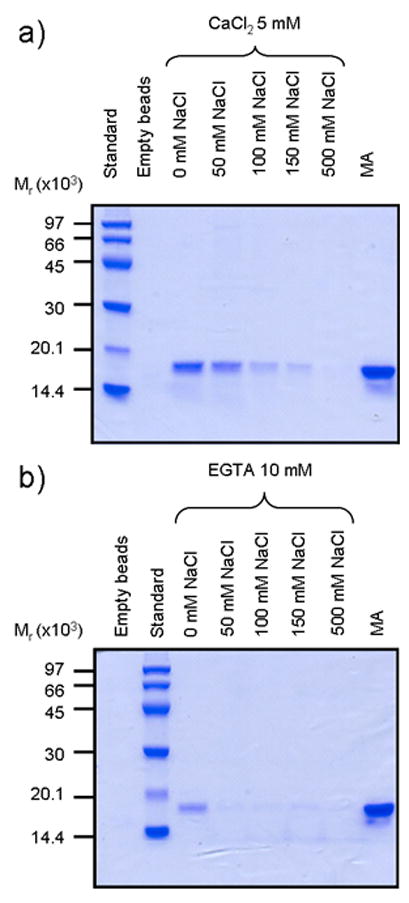
Molecular mass marker sizes (Mr) are shown along the left edge. a) Interactions between CaM and MA in high calcium buffer. Lane 1: molecular mass standards; lane 2: CaM-agarose beads only; lanes 3–7: CaM-agarose + MA in 0, 50, 100, 150, or 500 mM NaCl; lane 8: MA alone. b) Interactions between CaM and MA in low calcium buffer. Lane 1: CaM-agarose beads only; lane 2: molecular mass standards; lanes 3–7: CaM-agarose + MA in 0, 50, 100, 150, or 500 mM NaCl; lane 8: MA only.
Tryptophan fluorescence spectroscopy was used to probe the interaction between CaM and MA. As CaM lacks tryptophan residues and MA contains only the two tryptophans located within the putative CaM binding sequences (W16 and W36) tryptophan fluorescence is a useful probe for monitoring binding.,. From these data, equilibrium binding constants between CaM and MA were quantified. Addition of CaM to MA results in a blue-shifted tryptophan emission peak with λmax changing from ~350 nm to ~340 nm in the raw spectra (Figure 3a) and is accompanied by an increase in the maximum emission intensity by up to a factor of 1.8 for MA complexed with Ca2+-CaM in the absence of NaCl. While both apo and Ca2+-CaM are capable of binding MA in the absence of NaCl, their dissociation constants differ significantly with Keq of 170 ± 0.06 nM for the complex of MA and Ca2+-CaM and Keq of 0–4 mM for apo-CaM (Table 1). The Ca2+-CaM affinity for MA decreased by more than 2 orders of magnitude (Keq = 40 ± 20 μM, Figure 3b) at 150 mM NaCl and was further reduced at 500 mM NaCl (Keq = 0–1 mM, Figure 3c). No interaction between apo-CaM and MA could be detected at NaCl concentrations in excess of 150 mM.
Figure 3. Dependence of the affinity between Ca2+-CaM and MA as a function of NaCl concentration.

a) Tryptophan fluorescence emission spectra for the titration of Ca2+-CaM and MA in 0 mM NaCl demonstrating the shift in peak emission wavelength from MA alone ( | ) to saturated Ca2+-CaM-MA (solid line). b) Change in fluorescence intensity (ΔF/F0) from MA at 334 nm upon binding Ca2+-CaM as a function of [NaCl]: 0 mM NaCl (■); 100 mM (●); 150 mM (▲); 300 mM (▼); and 500 mM (◆). Each data point is the average of measurements and the error bars correspond to ± 1 standard deviation. c) CaM-MA Keq vs. NaCl concentration in the presence of Ca2+ (■), and in the absence of Ca2+ (with EGTA chelator, □).
Table 1. Fit parameters and calculated equilibrium constants for tryptophan fluorescence titrations of CaM and MA.
The first column indicates the presence (Ca2+) or absence (EGTA) of calcium and the second column shows the concentration of NaCl in the sample (0–500 mM). The equilibrium constant Keq for each curve was calculated from fits to the tryptophan fluorescence data for CaM-MA in the program ORIGIN, with the fit quality described by χ2 and R2.
| Ca2+/EGTA | [NaCl] (mM) | Keq (M) | χ2 | R2 |
|---|---|---|---|---|
| Ca2+ | 0 | 1.71 × 10−7 ± 6.34 × 10−8 | 0.00137 | 0.99752 |
| Ca2+ | 100 | 5.01 × 10−6 ± 7.18 × 10−7 | 0.00045 | 0.99842 |
| Ca2+ | 150 | 4.34 × 10−5 ± 2.45 × 10−5 | 0.00035 | 0.98819 |
| Ca2+ | 300 | 1.06 × 10−4 ± 1.18 × 10−4 | 0.00009 | 0.98728 |
| Ca2+ | 500 | 2.80 × 10−4 ± 1.13 × 10−3 | 0.00004 | 0.97219 |
| EGTA | 0 | 2.80 × 10−4 ± 3.71 × 10−3 | 0.00002 | 0.98306 |
| EGTA | 150 | Below measurement limit | - | - |
Combined, the pulldown and tryptophan fluorescence investigations indicate that the stability of the CaM-MA is sensitive to ionic strength, suggesting that electrostatic interactions between the proteins are important for complex formation. The tryptophan fluorescence data indicate that the solvent accessibility of W16 and W36 in the putative CaM-binding sequence of MA is altered implying that the tryptophan chemical environments in MA become more hydrophobic upon CaM-binding. As W16 is buried within the MA core and W36 is partially solvent-exposed when observed in high resolution structures15; 17, our results support the idea that that these MA residues form contacts with the CaM hydrophobic clefts and become more sequestered from solvent. The addition of calcium, which binds at the base of the EF-hand clefts of calmodulin to expose the hydrophobic binding pockets within the N- and C-terminal lobes, strengthens the interaction between both proteins.
Ca2+-CaM and MA forms an extended 1:1 complex in solution
In the absence of NaCl, Ca2+-CaM-MA eluted as a single peak in size exclusion chromatography with a molecular mass, Mr, of 29900 (± 3 %, Supplemental Figure) consistent with the formation of a 1:1 complex between CaM (Mr 16690 ± 5 % from MALLS, theoretical Mr 16706) and MA (Mr 12980 ± 5 %, theoretical Mr 14979). In 100 mM NaCl the complex eluted as two peaks with values for Mr of 20470 (± 8 %) and 14950 (± 15 %) in similar positions to CaM (Mr 16380 ± 9 %) and MA (Mr 13850 ± 4 %) analyzed under the same conditions. We concluded that low salt conditions are important in stabilizing the CaM-MA complex.
Small angle X-ray scattering (I(q) vs. q, Figure 5) was used to determine the shapes of Ca2+-CaM, MA, and the Ca2+-CaM-MA complex in solution and the derived structural parameters are summarized in Table 2. Guinier plots for data on each component and the complex (Figure 4a inset) are linear as expected for monodisperse solutions. The probable distribution of vector lengths between scattering centers, P(r) vs. r, for Ca2+-CaM, MA, and Ca2+-CaM-MA (Figure 4b) yield values for the forward scattering intensity (I(0)) and associated molecular mass ( Mr), radius of gyration (Rg), and maximum linear dimension (Dmax) which agree well with previously reported parameters from SAXS data for Ca2+-CaM21 and with calculated average parameters derived from the NMR ensemble for MA (PDB entry 2HMX, 20 structures, using the program CRYSOL36). The Mr values also agree with the experimental masses from MALLS and calculated masses based on the protein amino acid sequences (from PROTPARAM37). Overall, these results indicate that both Ca2+-CaM and MA exist as monodisperse monomers under these solution conditions and confirm that the Ca2+-CaM-MA complex has 1:1 stoichiometry. The complex has an Rg value that is ~50% larger than Ca2+-CaM or MA alone and the P(r) function indicates a relatively elongated structure with a maximum linear dimension that is ~60% greater than either of the components.
Figure 5. 15N-1H-HSQC spectra for 15N-CaM bound to MA and 15N-MA bound to Ca2+-CaM.
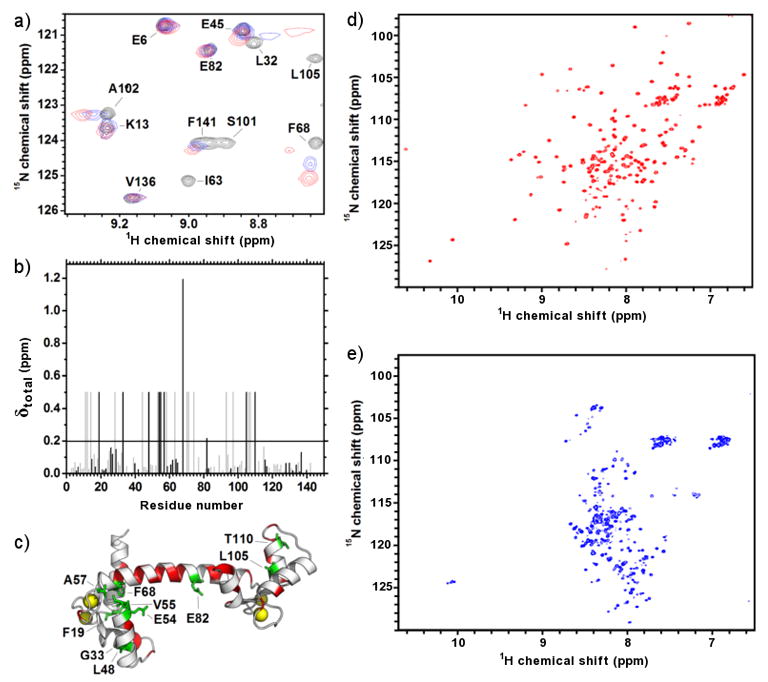
a) Focused region of the HSQC spectra showing the titration of 15N-CaM (black) with unlabelled MA (0.4:1 molar ratio, blue; 1:1 molar ratio, red). b) Amide chemical shift perturbations for 15N-CaM bound to MA indicating unambiguously assigned residues (black) and those which are tentatively assigned (grey). The significant threshold for perturbation (average perturbation + 1 standard deviation) is indicated by a horizontal line. Amide resonances that disappear during complex formation are set to an arbitrary value of 0.5. c) Annotated structure (pdb: 1CLL) of Ca2+-CaM showing unambiguously assigned and perturbed residues (green) and unassigned residues (red). Bound calcium atoms are shown as yellow spheres. The proposed interaction surface of CaM-MA from previous SAXS studies41 (inset i, orange) and the interaction surface between in Ca2+-CaM-MLCK26 (inset ii, black) is on the same Ca2+-CaM structure for comparison. d) HSQC spectra for 15N-MA in the absence of CaM showing well dispersed peaks. e) HSQC spectra for 15N-MA in complex with unlabelled CaM demonstrating perturbations in most of the amide resonances.
Table 2. Structural parameters for the Ca2+-CaM-MA complex and its components determined from the scattering data (I(q) versus q).
The Rg, Dmax, and I(0) values were obtained from P(r) analysis, c (in g.mL−1) was determined directly from UV spectroscopy for CaM and MA and calculated from component concentrations for the complex. The molecular mass Mr was calculated by the method described by Orthaber et al.49, and the expected masses derived from amino acid sequences are given in parentheses next to the experimentally derived values, with Δ% denoting the percentage difference between experimental and expected Mr. The error in the I(0)-derived Mr values arise from uncertainties in protein concentration and partial specific volume. Previously determined values for Rg and Dmax are given in parentheses where available in the literature (Ca2+-CaM) or estimated from high-resolution structures (MA).
| Sample | ΔρM2 (1020cm2.g−2) | I(0)/c (cm−1.mL.g−1) | Mr | Δ% | Rg (Å) | Dmax (Å) |
|---|---|---|---|---|---|---|
| Ca2+-CaM | 4.84 | 13 | 16280 (16706) | −2 | 20.9 (21.321) | 70 (6321) |
| MA | 4.68 | 12 | 16000 (14979) | 7 | 19.9 (20.4) | 70 (80) |
| Ca2+-CaM-MA | 4.77 | 29 | 37320 (31685) | 18 | 30.5 | 110 |
Figure 4. SAXS analysis of Ca2+-CaM, MA, and Ca2+-CaM-MA.

a) I(q) vs. q plots of SAXS data for the complex (black ■, 9.7 mg.mL−1), Ca2+-CaM (red ▲, 5.0 mg.mL−1), and MA (blue ●, 8.0 mg.mL−1). The plots for Ca2+-CaM and MA have been arbitrarily shifted on the vertical axis for clarity. Superimposed lines indicate the P(r) fit to the data. (Inset) Guinier plots (data points) and linear fits (lines) to the SAXS data for the complex (black ■), Ca2+-CaM (red ▲), and MA (blue ●) to qRg < 1.3. The plots for Ca2+-CaM and MA have been arbitrarily shifted on the vertical axis for clarity. All curves are linear as expected for monodisperse samples. Propagated errors based on counting statistics were smaller than symbol size in low q region (< 0.15 Å−1) and indicated by the scatter around the fitted lines at higher q. b) SAXS P(r) profiles for the complex (black ■, Dmax = 110 Å), Ca2+-CaM (red ▲, Dmax = 70 Å), MA (blue ●, Dmax = 60 Å). c) Ca2+-CaM-MA envelope (grey), (inset) different orientations of Ca2+-CaM-MA (grey), d) free Ca2+-CaM with superimposed structure (left, red, PDB entry: 1CLL) plus scale bar and free MA with superimposed structure (right, blue, PDB entry: 2HMX, NMR ensemble models 1–10 out of 20). Structures are drawn to relative scale. MA contains two unstructured regions in the NMR ensemble model (rendered as backbone ribbons): an N-terminal domain around residues 1–10 and the C-terminal ‘tail’ around residues 106–132 which is unstructured according to the NMR data; the shape restoration suggests that the unstructured C-terminal tail is more compact than indicated by the NMR coordinate files.
Ab initio shape restoration models derived from the SAXS data for CaM and MA are consistent with their known structures (Figure 4d). The CaM envelope shows evidence for the expected dumbbell shape and is slightly foreshortened compared to the crystal structure38 as observed in previous SAXS experiments21 and attributed to the flexibility of the central helix. The MA envelope depicts two globular lobes with the larger having the expected dimensions for the MA ‘head’ domain and the smaller lobe which would accommodate the disordered C-terminus of the NMR structure15 suggesting the C-terminus is somewhat compact. The highly elongated shape of complex has a total volume consistent with that expected for a 1:1 complex (~73,000 Å3 with overall dimensions ~110 Å × 50 Å × 40 Å, Figure 6c) showing two aspherical lobes.
Figure 6. Far-UV CD spectra tracking changes in secondary structure for CaM-MA.
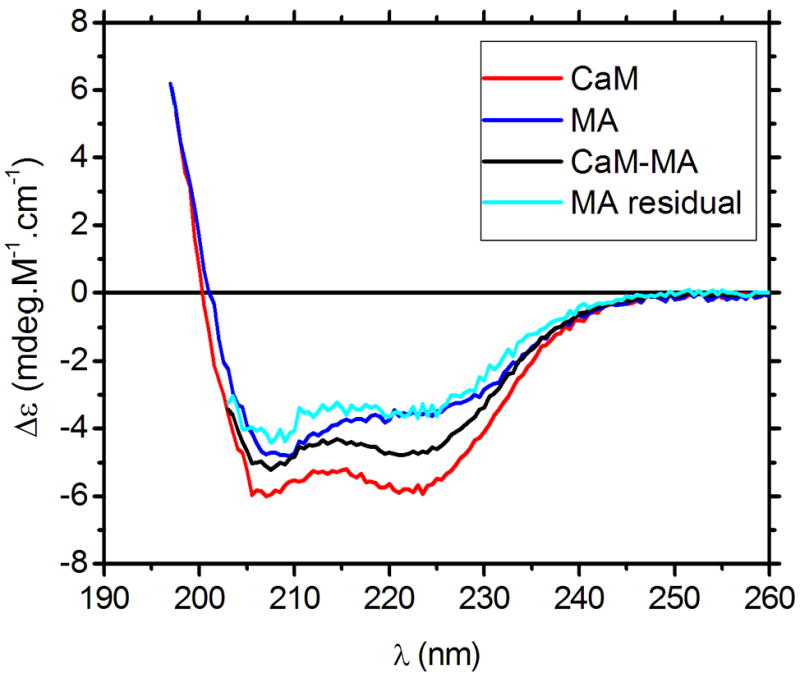
Spectra were recorded for Ca2+-CaM (red line; 8 μM), MA (blue line; 8 μM), and Ca2+-CaM-MA (black line, 8 μM) at 25 °C. The MA residual spectrum (cyan line) was calculated from subtraction of the Ca2+-CaM spectrum from the Ca2+-CaM-MA spectrum, and displays significant differences from the MA spectrum.
MA interacts with the hydrophobic binding clefts of Ca2+-CaM
NMR experiments were performed in order to identify specific interactions between the two proteins with the view that these might aid in modeling the CaM and MA component structures into the SAXS-derived envelope for the complex. 1H-15N-HSQC spectra were acquired for 15N-labeled Ca2+-CaM titrated with sequential additions of MA to a final molar ratio of 1.4:1 (Figure 5a). The amide resonances for 15N- Ca2+-CaM were obtained from previously published data39 and over 70% of the CaM sequence was assigned based on the published chemical shifts. Amide signals that disappeared or were significantly perturbed (see methods) over the course of the titration experiment (Figure 5b) were considered to be either part of the MA binding interface, or involved in binding-induced conformational changes. Most of the assigned resonances in the 15N-Ca2+-CaM spectra gradually moved from their original positions in the free 15N- Ca2+-CaM spectrum to their final positions in MA-saturated CaM, allowing unambiguous tracking of their chemical shifts during the titration. No further changes were observed in the spectrum following the addition of more than 1 molar equivalent of MA (data not shown), indicating a specific interaction between Ca2+-CaM and MA.
Of the ten unambiguously assigned 15N-Ca2+-CaM residues that have significantly perturbed amide resonances upon binding MA (Figure 5c), four are known to interact with the archetypal CaM binding partner myosin light chain kinase peptide (MLCK)26; 29 or form part of the hydrophobic target-recognition clefts20. Another four residues are adjacent to a residue previously identified to interact with MLCK, while two lie within three residues distance in the amino acid sequence. An additional fifteen 15N-Ca2+-CaM amide resonances that are also significantly perturbed upon MA binding, but have similar chemical shifts to other signals in the 15N-HSQC spectrum were able to be tentatively assigned. Overall, these results are consistent with significant interactions between the canonical hydrophobic target-binding clefts of CaM and that recognition and binding of MA thus shares similarities to previously identified CaM complexes26; 28; 29; 30; 31; 32.
Izumi and colleagues36 have previously generated a model for Ca2+-CaM-MA based on SAXS experiments on CaM bound to a peptide corresponding to residues 11–47 of MA and proposed that a group of residues near or within the central helix of calmodulin (E47, D50, E54, D56, E67, R74, D78, E82 and E139) form direct contacts with MA via electrostatic interactions in a novel interaction. Our NMR data show that two of the residues in this binding scheme have both unambiguous assignments and perturbed amide resonances when MA binds to Ca2+-CaM (E54 and E82), while R74 is perturbed but is ambiguously assigned. Four CaM residues in this scheme are ambiguously assigned in our NMR data have amide resonances that are not significantly altered upon MA binding (E47, D50, D56, and E67) while two cannot be assigned (D78 and E139). Of the three residues listed above with perturbed chemical shifts, two (R74 and E82) are located within the flexible central helix that typically deforms upon calmodulin binding to other target proteins (CaM residues 73–8328), and all lie within two residues of a residue sitting in the canonical CaM target-binding cleft. Most of the residue signals that were unambiguously assigned and perturbed in our NMR analysis were not explicitly predicted to shift in the MA(11–47) model by Izumi and colleagues (8 out of 10 residues: F19, G33, L48, V55, A57, F68, L105, and T110). Indeed, our NMR data show binding-induced perturbations in three amide chemical shifts from residues in the CaM N-terminal lobe (F19, V55, and F68) which direct their side chains into the cleft around the base of the CaM hydrophobic pocket, as well as two residues (G33 and L48) located on the distal side of the same pocket. Although a similar number of chemical shift perturbations were expected from residues in the C-terminal lobe, relatively few were actually observed to change upon MA binding, comprising only two (L105 and T110) out of thirteen chemical shifts unambiguously assigned to residues in that lobe. The lack of perturbed chemical shifts in the C-terminal lobe may be due to the relative scarcity of assignments on our NMR experiment for residues in this region that have previously been identified to form contacts with target molecules (F92, V108, M109, F141, M144, and M14524; 41). It is also possible that the CaM-MA interaction is principally mediated by the N-terminal lobe, and we note that the proposed model by Izumi and colleagues also shows a disproportionate number of MA contacts from that lobe.
MA undergoes major conformational changes upon binding
A series of 1H-15N-HSQC spectra for 15N-MA with sequential additions of unlabeled Ca2+-CaM to a final molar ratio of 1.4:1 were also acquired. Over 80 % of the amide resonances for 15N-MA and two indole resonances were assigned from published data40. The 1H-15N-HSQC spectrum for MA contains well-dispersed resonances (Figure 5d) that underwent major chemical shift perturbations over the course of the titration such that only a few signals remained in their original positions upon complex saturation (Figure 5e). A general migration of many 15N-MA resonances to the region of the spectrum normally associated with unstructured proteins or α-helical structure occurred during the titration. No intermediate signals were observed between the free and Ca2+-CaM-bound chemical shifts for amides in 15N-MA, leading to difficulties in tracking their positions over the course of the titration. These observations indicate that a major conformational change occurs in MA upon forming a complex with Ca2+-CaM. No further changes were observed in the spectrum following the addition of more than 1 molar equivalent of Ca2+-CaM (data not shown) consistent with a 1:1 specific interaction.
Only eleven unambiguously assigned amide resonances remained in similar positions in both free and bound MA plus an additional eighteen resonances that were tentatively assigned. The corresponding residues encompass every class of amino acid except those which are acidic and are spaced at semi-regular intervals along the protein backbone. Interestingly, signals for the indole ring nitrogens of W16 and W36 (the two tryptophan residues in the putative MA CaM-binding sequence) disappeared in bound-MA despite their corresponding backbone amide resonances appearing relatively unaffected. As these residues are likely anchor residues for binding to the hydrophobic clefts of CaM, it is possible that the interaction between Ca2+-CaM and MA involves the indole side chains of W16 and W36 without affecting the corresponding amide, or that the secondary structure in that part of MA is similar in both the free and bound forms. The sheer number of chemical shift changes limits our ability to identify key residues at the interface. However, the data indicate that virtually every part of the MA structure is perturbed upon binding CaM.
Formation of Ca2+-CaM-MA causes a significant loss of α-helical structure
To determine the extent of CaM-induced structural changes in MA due to a loss of secondary vs. tertiary structure, far-UV circular dichroism (CD) experiments were performed on Ca2+-CaM, MA, and the 1:1 complex (Figure 6). The secondary structure content calculated from each spectrum (Table 3) confirm that Ca2+-CaM and MA are predominantly α-helical, while an equimolar mixture of the two proteins contains significantly less α-helix than expected based on the mass-weighted average of the values from each uncomplexed component (Table 3). No significant deviations were observed for β-sheet content and the loss of α-helical structure was accompanied by a correspondingly larger amount of undefined content. From the NMR data, we inferred that the majority of the structural changes caused by Ca2+-CaM-MA formation occur in MA rather than in Ca2+-CaM. We therefore generated an approximate ‘MA residual’ spectrum by subtracting the Ca2+-CaM signal from that of the complex. Although the MA and MA residual spectra are visually similar, the MA residual spectrum contains less α-helix and more undefined structure, while β-sheet and turn content is essentially unchanged. These results suggest that MA retains most of its α-helical secondary structure and the dramatic loss of dispersion in the NMR spectrum of MA upon complex formation has a significant contribution from a loss of tertiary contacts.
Table 3. Secondary structure content of Ca2+-CaM, MA, and their complex derived from circular dichroism data.
Values for secondary structure content are the average results from CDpro and the quoted errors are the standard deviations between predictions calculated by three different algorithms. The stated values for helix and sheet are the summed from the amounts of ‘regular’ and ‘distorted’ versions of each structure calculated by CDpro. For comparison, the secondary structure content expected for an unaltered CaM-MA complex and the ‘MA residual’ spectrum generated from subtracting the Ca2+-CaM spectrum from the Ca2+-CaM-MA spectrum is also given below.
| Spectrum | Helix (%) | Sheet (%) | Turn (%) | Undefined (%) |
|---|---|---|---|---|
| Ca2+-CaM | 60 ± 5 | 6 ± 3 | 13 ± 6 | 21 ± 4 |
| MA | 53 ± 3 | 8 ± 1 | 17 ± 1 | 22 ± 5 |
| Ca2+-CaM-MA | 49 ± 2 | 6 ± 3 | 14 ± 1 | 32 ± 4 |
| Expected complex | 57 ± 4 | 69 ± 2 | 14 ± 4 | 22 ± 4 |
| MA residual | 41 ± 2 | 8 ± 1 | 15 ± 2 | 37 ± 2 |
CONCLUSIONS
The results of our experiments show that CaM binds to intact MA in a calcium dependent manner to form an elongated 1:1 complex in solution and that MA undergoes significant conformational change on binding Ca2+-CaM. Izumi and colleagues41 have previously generated a model for the Ca2+-CaM-MA interaction using MA peptides corresponding to residues 11–47 and proposed that a group of charged residues in CaM on the N-terminal lobe and within the central helix form direct electrostatic contacts with MA. They proposed a novel interaction that excluded the involvement of CaM’s hydrophobic clefts that are typically responsible for CaM-target binding, and rather suggested that CaM could interact with MA without the involvement of the large hydrophobic residues internal to the MA core that would require a conformational change in the MA structure. While our results regarding the sensitivity of the CaM-MA interaction to ionic strength support the fact that electrostatic interactions are important, our combined NMR, fluorescence, and CD data show that Ca2+-CaM binding to intact MA does ultimately involve the hydrophobic clefts in the two lobes of CaM as well as the tryptophans within the MA core and thus induces structural changes that disruption both tertiary and secondary structure in MA.
The MA protein is involved in a diverse number of functions throughout the HIV-1 replication cycle. It forms the matrix of the mature virion, is the only known Gag product of the pre integration complex14; 42, it interacts with phosphatidylinositol 4,5 bisphosphate within plasma membranes40 (plasma membrane targeting) and has an N-terminally linked myristoyl switch for membrane anchoring11. As well as these functions, MA has a purported nuclear localization signal43; 44 encoded within its sequence and can bind RNA45 and DNA46. The basic N-terminal region of MA appears crucial for many of these processes and our results show that the binding of CaM to this same region has significant structural consequences for the rest of the protein. The introduction of a layer of ‘structural plasticity’ within MA induced by Ca2+-CaM binding could affect many of the viral and cellular events MA has integrated into, but at present the precise details regarding the CaM-MA interaction ‘in vivo’ remain poorly understood in part because of the lack of understanding to date of the conditions under which a stable interaction occurs.
Materials and Methods
Protein expression and purification
Calmodulin purification
Plasmid DNA encoding the Xenopus laevis calmodulin gene (pET3a-CaM; provided by M. Ikura, U. of Toronto) was transformed into Escherichia coli BL21 (DE3) grown in lysogeny broth47 (LB) media supplemented with 100 μg.mL−1 ampicillin. The cells were grown at 37 °C and CaM expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and harvested 4 h later by centrifugation (6000 × g, 10 min). Pelleted cells were resuspended in CaM-lysis buffer (20 mM tris(hydroxymethyl)aminomethane (TRIS) pH 7.4, 1 mM ethylene diamine tetra acetic acid (EDTA)) containing Complete™ protease inhibitor tablets (Roche), and small quantities (~ 1 mg) of lyophilized ribonuclease A (RNase A) and deoxyribonuclease A (DNase A; Sigma) and lysed by three freeze-thaw cycles of liquid N2 and 80 °C water before extracting the soluble fraction by centrifugation (50000 × g, 1 h). This fraction was mixed with concentrated CaCl2 to a final concentration of 10 mM then passed over a packed matrix of phenyl-sepharose 6B fast flow beads (GE Healthcare Life Sciences) pre-equilibrated with wash buffer (20 mM TRIS pH 7.4, 10 mM CaCl2). The beads and bound proteins were washed several times with fresh wash buffer and wash buffer supplemented with 500 mM NaCl before CaM was eluted in 50 mM TRIS, 150 mM NaCl, 1 mM ethylene glycol tetra acetic acid (EGTA). Protein-containing fractions were detected using the Bradford assay48 and subsequently pooled and further purified by Superdex 75 size exclusion chromatography (SEC; in 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) pH 7.0, 100 mM NaCl, 5 mM CaCl2, and 2 mM tris(2-carboxyethyl)phosphine (TCEP)). CaM stocks were concentrated to ~ 20 mg.mL−1 by vacuum centrifugation and dialyzed against SEC buffer supplemented with 0.1 % (w/v) NaN3, before storage at 4 °C. The purity of the final CaM stocks was assessed using SDS-PAGE with Coomassie Blue staining and the identity of the purified protein confirmed by MALDI mass spectrometry. CaM concentrations were determined by UV absorbance using a molar extinction coefficient at 276 nm (ε276nm) of 3030 M−1 cm−1, calculated from the primary sequence of the protein with the program PROTPARAM37.
HIV-1 MA purification
Plasmid encoding the HIV-1 MA gene (pET16b-MA; provided by W. Sundquist, U. of Utah) was transformed into E. coli BL21 (DE3) Rosetta2 cells (Merck) grown in LB supplemented with 100 μg.mL−1 ampicillin and 33 μg.mL−1 chloramphenicol. This construct produces a version of MA with a His-tag and Factor Xa protease site incorporated into its N-terminus. Protein overexpression was performed in an identical manner to CaM except that the cells were left to express MA after induction for 16 h at 15 °C. Purification of MA was accomplished by the method described in Massiah et al.15 with modifications. Harvested cells were resuspended in MA-lysis buffer (30 mM imidazole pH 7.0, 500 mM NaCl, 5 mM β-mercaptoethanol (β-ME)) supplemented with protease inhibitor tablets, RNase A and DNase A, before lysis by three freeze-thaw cycles in liquid N2 and 25 °C water. Soluble lysates were recovered by centrifugation (50000 × g, 1 h) and passed over a nickel-nitrilotriacetic acid (Ni-NTA, Qiagen) agarose column pre-equilibrated with MA-lysis buffer without supplements. The column was washed with 5–10 column volumes of buffer A (75 mM imidazole, 500 mM NaCl, 5 mM β-ME) and buffer B (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 6.5, 50 mM NaCl, 2.5 mM β-ME) to remove loosely-bound protein. MA was eluted from the Ni-NTA beads with 250 mM imidazole pH 7.0, 100 mM NaCl, 5 mM β-ME. Fractions containing MA were pooled and dialyzed against buffer B containing 10 μg.mL−1 Factor Xa (Sigma) for 16 h. This procedure removes the His-tag from MA and leaves a residual histidine residue at the N-terminus. The cleaved protein mixture was passed over a 50:50 mix of Ni-NTA agarose and benzamidine agarose to remove any remaining His-tagged protein and Factor Xa. Further purification of MA was performed using SEC as described for CaM and the purity was assessed by SDS-PAGE and MALDI-MS. Sample concentrations were determined by UV spectrometry using a ε280nm of 17085 M−1 cm−1 calculated from PROTPARAM.
CaM-agarose pulldown assays
To qualitatively assess the effect of NaCl and calcium on the interaction between MA and CaM, concentrated MA (in SEC buffer) was added to aliquots of CaM-agarose beads (containing CaM covalently attached to an agarose matrix; Sigma) to a final concentration of 5 μM. The beads were suspended in buffers containing 50 mM MOPS pH 7.0 and 1 mM TCEP, supplemented with 0, 50, 100, 150, or 500 mM NaCl in combination with either 5 mM CaCl2 or 10 mM EGTA. The mixtures were incubated for 10 min at 25 °C before the CaM-agarose beads were pelleted by centrifugation at 17000 × g for 1 min and the soluble fraction discarded. Unbound MA was rinsed from the beads using three centrifugation/resuspension cycles in the corresponding assay buffer. Reducing SDS-PAGE loading buffer (50 mM TRIS pH 6.8, 2 % sodium dodecyl sulfate (SDS), 10 % glycerol, 4 % β-ME) was then added to the beads and bound protein content was analyzed by SDS-PAGE with Coomassie Blue staining. Samples of pure MA and clean CaM-agarose were included as controls. Special care was taken throughout this procedure to ensure minimal loss of beads during washing and that bead mass and buffer volumes were equivalent across all samples within handling error.
Tryptophan fluorescence
Fluorescence spectra from protein samples and their corresponding buffer solutions were obtained at 25 °C using a Varian Eclipse fluorescence spectrophotometer with the following settings: λexcitation = 295 nm; λemission = 300 – 500 nm; slit widths = 5 nm; scan rate = 120 nm.min−1; data interval = 1 nm. Aliquots of both MA (5 μM) and CaM (227 μM) were individually dialyzed against either high calcium buffer (50 mM MOPS pH 7.0, 5 mM CaCl2, 2 mM TCEP) containing 0, 50, 100, 150, or 500 mM NaCl or low calcium buffer (50 mM MOPS pH 7.0, 10 mM EGTA, 2 mM TCEP) containing 0 or 150 mM NaCl. To determine the binding affinity between apo/Ca2+-CaM and MA, concentrated CaM samples were titrated into the respective MA samples (5 μM) and left to equilibrate for 10 min at each titration step, prior to recording each emission spectra in triplicate and averaging to produce the final spectra. The total volume change after all titrations was limited to < 5 % of the MA sample volume. All sample spectra were corrected for solvent effects by subtracting the emission spectra of buffers across the full emission range. The peak tryptophan fluorescence signal from fully complexed CaM-MA occurred at 334 nm and all data analyses are performed at this wavelength. Control spectra using CaM alone confirmed that the intrinsic fluorescence caused by CaM’s tyrosine residues contributed less than 5 % of the emission signal and consequently this contribution has been discounted in the data analysis. Changes in emission intensity from MA caused by addition of CaM were fitted in the program package ORIGIN with the equations Keq = ([CaM][MA])/[MAB] and ΔF = (FMAB − FMA)/FMA*[MAB]/[MAT], where: Keq is the equilibrium constant; [CaM], [MA], and [MAB] are the concentrations of CaM, MA, and MA bound to CaM respectively; ΔF is the fluorescence change due to the addition of CaM expressed as a fraction of the signal from MA alone; FMA and FMAB are the fluorescence contributions from free MA ([MA]) and bound MA respectively, and [MAT] is the total concentration of MA in the sample ([MAT] = [MA] + [MAB]).
Multi-angle laser light scattering
Multi-angle laser light scattering (MALLS) data were obtained for purified MA, CaM, and a 1:1 molar mix of these two components dialyzed in 50 mM MOPS pH 7.0, 50 mM MOPS pH 7.0, 5 mM CaCl2, 2 mM TCEP containing either 0 or 100 mM NaCl. The MALLS system consisted of a Superdex 200 10/30 gel filtration column driven by an ÄKTA FPLC platform, feeding into a Wyatt Technology miniDAWN light scattering unit and an Optilab DSP refractometer. The system was equilibrated with 5 column volumes of the same buffer as the samples prior to each experiment. To determine the relative molecular mass (Mr) of each species, the system was calibrated to an absolute scale using the intrinsic Rayleigh scattering of toluene. A uniform refractive index to concentration gradient (dn/dc) of 0.19 mL.g−1 was assumed for all proteins. The calibration was verified by analyzing a sample of bovine serum albumin on the system immediately prior to the samples of interest.
Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) experiments were performed on samples of MA (8.0 mg.mL−1), CaM (5.0 mg.mL−1), and a 1:1 molar mixture of CaM and MA (9.7 mg.mL−1) dialyzed against 50 mM MOPS pH 7.0, 5 mM CaCl2, 2 mM TCEP. The dialysate was retained for solvent blank measurements. A lysozyme standard was prepared by dissolving and dialyzing the lyophilized protein (USB Corporation) in 150 mM NaCl, 40 mM sodium acetate (pH 3.8), to a final concentration of ~ 20 mg.mL−1, which was serially diluted to produce a concentration series used to verify Mr calculations for the proteins on interest. These lysozyme samples gave values for Mr within 2 % of the expected values (Mr 14630 vs. expected Mr 14295) when analyzed by the method of Orthaber49 and water-calibrated absolute scaling as described below.
SAXS data were acquired using an Anton Paar SAXSess with line-collimation and CCD detector as described in Jeffries et al.50. The initial stages of data reduction and analysis were performed as described previously51. Briefly, the instrument slit and integration lengths were set to 10 mm and all data were collected using the CCD camera, except for beam intensity profiles which were recorded on image plates. Samples were exposed for 30–120 min at 20 °C in 10 s increments and averaged in blocks of 15 or 30 min to improve counting statistics and monitor for potential radiation damage to the sample. Data reduction to I(q) vs. q (where q = (4πsinθ)/λ, 2θ is the scattering angle, and λ = 1.54 Å is the wavelength of the radiation) and subtraction of the solvent scattering intensity were performed with the program SAXSquant1D (Anton Paar, Austria). All data were placed on an absolute scale using the scattering of water as a standard49. Guinier analysis52 was performed in the program PRIMUS53, while P(r) analysis was performed using GIFT54 or GNOM55 both of which include corrections for the data smearing due to slit geometry of the instrument. GIFT and GNOM calculate the indirect Fourier transformation of the data that represents the probable pair-wise distribution P(r) of vector lengths within the scattering particle, from which the forward scattering intensity at zero scattering angle (I0), radius of gyration (Rg), and maximum dimension (Dmax) can be derived. For comparison, Rg and Dmax values from previous SAXS experiments were obtained for Ca2+-CaM21, while a theoretical Rg for MA was calculated using the program CRYSOL36 from the full NMR structure ensemble in PDB entry 2HMX, from which an estimate for Dmax was also derived using the program PYMOL56. Theoretical values for the scattering length density (ρ) of CaM and MA and the components in the supporting SAXS buffer were calculated with the programs MULCh57. The partial specific volume (v) of both proteins were calculated from their primary amino acid sequence using NucProt58. The calculated ρ and v and experimentally determined I0 and protein concentration values were used to calculate the molecular masses of the scattering particles in each sample as described in Orthaber et al.49. We expect that concentration measurements for CaM are less accurate than those for MA due to the low extinction coefficient for that protein (CaM: ε276nm = 3030 M−1cm−1, MA: ε280nm = 17085 M−1cm−1).
Ab initio shape restoration calculations for the Ca2+-CaM-MA complex were performed using the output files from GNOM as input to the program DAMMIF59. Ten independent DAMMIF calculations were calculated for each sample, and these were averaged with the program DAMAVER60 to generate a consensus envelope for each species. The mean normalized spatial discrepancy within each set of models is less than 0.7 which indicates a high degree of similarity between models.
NMR spectroscopy
CaM and MA isotopically labeled with 15N (denoted 15N-CaM and 15N-MA respectively) were expressed and purified as described above with LB replaced by M9 minimal media containing 15N-NH4Cl as the sole nitrogen source. 15N-CaM and MA were dialyzed into 10 mM KCl (pH 6.3), 5 mM CaCl2, 1 mM TCEP, while 15N-MA and CaM were dialyzed into 10 mM sodium phosphate (pH 5.5), 5 mM CaCl2, 1 mM TCEP. At this pH and concentration the calcium phosphate remained soluble. Samples were concentrated to 0.1–0.5 mM by filtration through Vivaspin™ 20 3500 MWCO concentrator units. All 1H-15N-HSQC spectra were recorded on a Bruker Avance 600 spectrometer equipped with a cryoprobe. NMR data were processed in the program XWINNMR/TOPSPIN (Bruker) and analyzed with SPARKY (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Multiple spectra for 15N-CaM and 15N-MA were collected at temperatures between 298 K (25 °C) and 320 K (47 °C) in order to quantify any temperature effects. Unlabeled MA and CaM at concentrations in excess of 1 mM were titrated into their corresponding 15N-labeled binding partners (0.1–0.3 mM) to a final molar ratio of 1.4:1 in 0.2:1 molar intervals. Each titration sample was equilibrated for a minimum of 10 min per addition prior to each spectra measurement at 298 K.
Previously published NMR 1H-15N chemical shift assignments for CaM39 (BMRB: 547) and MA40 (BMRB: 7250) were used to annotate our 1H15N-HSQC spectra for 15N-CaM and 15N-MA, allowing 75% (CaM) and 90% (MA) of the amide resonances to be assigned as well as those for the indole nitrogens of W16 and W36 in MA. Weighted combined 1H and 15N resonance perturbations (Δδtot) for each assigned residue were calculated using the equation Δδtot = [(δHNWHN)2+ (δNWN)2]1/2, where δi and Wi are the resonance perturbation and weight factor of nucleus i respectively, WHN = 1, and WN = 0.154 61. Amide resonances that were perturbed by more than the experimental average + 1 standard deviation were considered to be significant, and include those that disappear from the spectra over the course of the titrations.
Circular dichroism spectroscopy
Circular dichroism (CD) spectra for CaM, MA, and an equimolar mixture of the two components were recorded at 25 °C in 1 mm pathlength cuvettes using a Neslab RTE-111 temperature–controlled Jasco J-720 spectropolarimeter calibrated with a camphor-10-sulfonic acid standard (0.6 g.mL−1) immediately prior to the experiment. The protein samples (8 μM) were dialyzed in 10 mM sodium phosphate (pH 5.5), 100 μM CaCl2, and 1 mM TCEP for 16 h prior to the experiment. CD spectra were measured in the wavelength range 260–185 nm while monitoring the high tension (HT) voltage of the sample. Data corresponding to HT values greater than 600 V were considered unreliable and discarded. The final spectra were the average of three scans and baseline corrected by subtracting the spectral contribution from buffer alone. Estimates of secondary structure were made using the program suite CDpro62.
Supplementary Material
Acknowledgments
We thank Prof. Mitsuhiko Ikura (University of Toronto, Canada) and Prof. Wes Sundquist (University of Utah, USA) for the gifts of pET3a-CaM and pET16b-MA respectively, Dr Ben Crossett (Australian Proteome Analysis Facility, Sydney, Australia) for mass spectrometry and Dr David Langley (University of Sydney, Sydney, Australia) for reading and commenting on the manuscript. We also thank Mercedes Ballesteros (University of Sydney, Sydney, Australia) for assistance in the production of 15N-labelled proteins for the NMR experiments and Prof. Joel Mackay for helpful suggestions with respect to the tryptophan fluorescence and NMR experiments.
ABBREVIATIONS
- HIV-1
human immunodeficiency virus 1
Footnotes
This project was supported by a U.S. National Institutes of Health P50 Award (GM082545) and an Australian Research Council Discovery Project (DP0770321) to J.T.. J.Y.H.C holds an Australian Nuclear Science and Technology Organization Scholarship.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Radding W, Pan ZQ, Hunter E, Johnston P, Williams JP, McDonald JM. Expression of HIV-1 envelope glycoprotein alters cellular calmodulin. Biochem Biophys Res Commun. 1996;218:192–7. doi: 10.1006/bbrc.1996.0034. [DOI] [PubMed] [Google Scholar]
- 2.Radding W, Williams JP, McKenna MA, Tummala R, Hunter E, Tytler EM, McDonald JM. Calmodulin and HIV type 1: interactions with Gag and Gag products. AIDS Res Hum Retroviruses. 2000;16:1519–25. doi: 10.1089/088922200750006047. [DOI] [PubMed] [Google Scholar]
- 3.Mervis RJ, Ahmad N, Lillehoj EP, Raum MG, Salazar FH, Chan HW, Venkatesan S. The gag gene products of human immunodeficiency virus type 1: alignment within the gag open reading frame, identification of posttranslational modifications, and evidence for alternative gag precursors. J Virol. 1988;62:3993–4002. doi: 10.1128/jvi.62.11.3993-4002.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan AH, Swanstrom R. Human immunodeficiency virus type 1 Gag proteins are processed in two cellular compartments. Proc Natl Acad Sci U S A. 1991;88:4528–32. doi: 10.1073/pnas.88.10.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiegers K, Rutter G, Kottler H, Tessmer U, Hohenberg H, Krausslich HG. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J Virol. 1998;72:2846–54. doi: 10.1128/jvi.72.4.2846-2854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wills JW, Craven RC. Form, function, and use of retroviral gag proteins. AIDS. 1991;5:639–54. doi: 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Gelderblom HR, Hausmann EH, Ozel M, Pauli G, Koch MA. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology. 1987;156:171–6. doi: 10.1016/0042-6822(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 8.Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R, Krausslich HG, Boulanger P, Fuller SD. Organization of immature human immunodeficiency virus type 1. J Virol. 2001;75:759–71. doi: 10.1128/JVI.75.2.759-771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan X, Yu X, Lee TH, Essex M. Mutations in the N-terminal region of human immunodeficiency virus type 1 matrix protein block intracellular transport of the Gag precursor. J Virol. 1993;67:6387–94. doi: 10.1128/jvi.67.11.6387-6394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Parent LJ, Wills JW, Resh MD. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol. 1994;68:2556–69. doi: 10.1128/jvi.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A. 1990;87:523–7. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu X, Yuan X, Matsuda Z, Lee TH, Essex M. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J Virol. 1992;66:4966–71. doi: 10.1128/jvi.66.8.4966-4971.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiernan RE, Ono A, Englund G, Freed EO. Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J Virol. 1998;72:4116–26. doi: 10.1128/jvi.72.5.4116-4126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller MD, Farnet CM, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71:5382–90. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 16.Matthews S, Barlow P, Boyd J, Barton G, Russell R, Mills H, Cunningham M, Meyers N, Burns N, Clark N, et al. Structural similarity between the p17 matrix protein of HIV-1 and interferon-gamma. Nature. 1994;370:666–8. doi: 10.1038/370666a0. [DOI] [PubMed] [Google Scholar]
- 17.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci U S A. 1996;93:3099–104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crivici A, Ikura M. Molecular and structural basis of target recognition by calmodulin. Annu Rev Biophys Biomol Struct. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- 19.Watterson DM, Iverson DB, Van Eldik LJ. Spinach calmodulin: isolation, characterization, and comparison with vertebrate calmodulins. Biochemistry. 1980;19:5762–8. doi: 10.1021/bi00566a015. [DOI] [PubMed] [Google Scholar]
- 20.Babu YS, Bugg CE, Cook WJ. Structure of calmodulin refined at 2.2 A resolution. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 21.Heidorn DB, Trewhella J. Comparison of the crystal and solution structures of calmodulin and troponin C. Biochemistry. 1988;27:909–15. doi: 10.1021/bi00403a011. [DOI] [PubMed] [Google Scholar]
- 22.Barbato G, Ikura M, Kay LE, Pastor RW, Bax A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 1992;31:5269–78. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- 23.Kretsinger RH, Nockolds CE. Carp muscle calcium-binding protein. II. Structure determination and general description. J Biol Chem. 1973;248:3313–26. [PubMed] [Google Scholar]
- 24.Vigil D, Gallagher SC, Trewhella J, Garcia AE. Functional dynamics of the hydrophobic cleft in the N-domain of calmodulin. Biophys J. 2001;80:2082–92. doi: 10.1016/S0006-3495(01)76182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuboniwa H, Tjandra N, Grzesiek S, Ren H, Klee CB, Bax A. Solution structure of calcium-free calmodulin. Nat Struct Biol. 1995;2:768–76. doi: 10.1038/nsb0995-768. [DOI] [PubMed] [Google Scholar]
- 26.Meador WE, Means AR, Quiocho FA. Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science. 1992;257:1251–5. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 27.Vetter SW, Leclerc E. Novel aspects of calmodulin target recognition and activation. Eur J Biochem. 2003;270:404–14. doi: 10.1046/j.1432-1033.2003.03414.x. [DOI] [PubMed] [Google Scholar]
- 28.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993;262:1718–21. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 29.Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–8. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 30.Juranic N, Atanasova E, Filoteo AG, Macura S, Prendergast FG, Penniston JT, Strehler EE. Calmodulin wraps around its binding domain in the plasma membrane Ca2+ pump anchored by a novel 18-1 motif. J Biol Chem. 2009 doi: 10.1074/jbc.M109.060491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osawa M, Tokumitsu H, Swindells MB, Kurihara H, Orita M, Shibanuma T, Furuya T, Ikura M. A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase kinase. Nat Struct Biol. 1999;6:819–24. doi: 10.1038/12271. [DOI] [PubMed] [Google Scholar]
- 32.Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–4. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- 33.Tsvetkov PO, Protasevich II, Gilli R, Lafitte D, Lobachov VM, Haiech J, Briand C, Makarov AA. Apocalmodulin binds to the myosin light chain kinase calmodulin target site. J Biol Chem. 1999;274:18161–4. doi: 10.1074/jbc.274.26.18161. [DOI] [PubMed] [Google Scholar]
- 34.Schumacher MA, Crum M, Miller MC. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure. 2004;12:849–60. doi: 10.1016/j.str.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 35.Houdusse A, Gaucher JF, Krementsova E, Mui S, Trybus KM, Cohen C. Crystal structure of apo-calmodulin bound to the first two IQ motifs of myosin V reveals essential recognition features. Proc Natl Acad Sci U S A. 2006;103:19326–31. doi: 10.1073/pnas.0609436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svergun D, Barberato C, Koch MHJ. CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography. 1995;28:768–773. [Google Scholar]
- 37.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 38.Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. Calmodulin structure refined at 1.7 A resolution. J Mol Biol. 1992;228:1177–92. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 39.Ikura M, Kay LE, Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–67. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 40.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103:11364–9. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izumi Y, Watanabe H, Watanabe N, Aoyama A, Jinbo Y, Hayashi N. Solution X-ray scattering reveals a novel structure of calmodulin complexed with a binding domain peptide from the HIV-1 matrix protein p17. Biochemistry. 2008;47:7158–66. doi: 10.1021/bi702416b. [DOI] [PubMed] [Google Scholar]
- 42.Bukrinsky MI, Sharova N, Mcdonald TL, Pushkarskaya T, Tarpley WG, Stevenson M. Association of Integrase, Matrix, and Reverse-Transcriptase Antigens of Human-Immunodeficiency-Virus Type-1 with Viral Nucleic-Acids Following Acute Infection. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:6125–6129. doi: 10.1073/pnas.90.13.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bukrinsky MI, Haggerty S, Dempsey MP, Sharova N, Adzhubei A, Spitz L, Lewis P, Goldfarb D, Emerman M, Stevenson M. A Nuclear-Localization Signal within Hiv-1 Matrix Protein That Governs Infection of Nondividing Cells. Nature. 1993;365:666–669. doi: 10.1038/365666a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haffar OK, Popov S, Dubrovsky L, Agostini I, Tang H, Pushkarsky T, Nadler SG, Bukrinsky M. Two nuclear localization signals in the HIV-1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. Journal of Molecular Biology. 2000;299:359–368. doi: 10.1006/jmbi.2000.3768. [DOI] [PubMed] [Google Scholar]
- 45.Chukkapalli V, Oh SJ, Ono A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hearps AC, Wagstaff KM, Piller SC, Jans DA. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry. 2008;47:2199–2210. doi: 10.1021/bi701360j. [DOI] [PubMed] [Google Scholar]
- 47.Bertani G. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol. 2004;186:595–600. doi: 10.1128/JB.186.3.595-600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 49.Orthaber D, Bergmann A, Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. Journal of Applied Crystallography. 2000;33:218–225. [Google Scholar]
- 50.Jeffries CM, Whitten AE, Harris SP, Trewhella J. Small-angle X-ray scattering reveals the N-terminal domain organization of cardiac myosin binding protein C. J Mol Biol. 2008;377:1186–99. doi: 10.1016/j.jmb.2008.01.080. [DOI] [PubMed] [Google Scholar]
- 51.Claridge JK, Headey SJ, Chow JY, Schwalbe M, Edwards PJ, Jeffries CM, Venugopal H, Trewhella J, Pascal SM. A picornaviral loop-to-loop replication complex. J Struct Biol. 2009;166:251–62. doi: 10.1016/j.jsb.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fournet G, Guinier A. *Letat actuel de la theorie de la diffusion des rayons-x aux petits angles. Journal De Physique Et Le Radium. 1950;11:516–520. [Google Scholar]
- 53.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–1282. [Google Scholar]
- 54.Bergmann A, Fritz G, Glatter O. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA) Journal of Applied Crystallography. 2000;33:1212–1216. [Google Scholar]
- 55.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 1992;25:495–503. [Google Scholar]
- 56.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 57.Whitten AE, Cai SZ, Trewhella J. MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. Journal of Applied Crystallography. 2008;41:222–226. [Google Scholar]
- 58.Voss NR, Gerstein M. Calculation of standard atomic volumes for RNA and comparison with proteins: RNA is packed more tightly. Journal of Molecular Biology. 2005;346:477–492. doi: 10.1016/j.jmb.2004.11.072. [DOI] [PubMed] [Google Scholar]
- 59.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ayed A, Mulder FA, Yi GS, Lu Y, Kay LE, Arrowsmith CH. Latent and active p53 are identical in conformation. Nat Struct Biol. 2001;8:756–60. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- 62.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.