

Abstract
Reticulons are a group of membrane-bound proteins involved in diverse cellular functions, and are suggested to act as inhibitors of β-secretase enzyme 1 (BACE1) activity that cleaves amyloid precursor protein. Reticulons are known to accumulate in the dystrophic neurites of Alzheimer disease, and studies have suggested that alterations in reticulons, such as increased aggregation, impair BACE1 binding, increasing amyloid-β production, and facilitating reticulon deposition in dystrophic neurites. To further characterize the cellular distribution of reticulon, we examined reticulon-3 expression in cases of Alzheimer disease, Parkinson disease, and diffuse Lewy body disease. A more widespread cellular distribution of reticulon-3 was noted than in previous reports, including deposits in dystrophic neurites, neuropil threads, granulovacuolar degeneration, glial cells, morphologically normal neurons in both hippocampal pyramidal cell layer and cerebral neocortex, and specifically neurofibrillary tangles and Lewy bodies. These results are compatible with reticulon alterations as nonspecific downstream stress responses, consistent with its expression during periods of endoplasmic reticulum stress. This emphasizes the increasing recognition that much of the AD pathological spectrum represents a response to the disease rather than cause, and emphasizes the importance of examining upstream processes, such as oxidative stress, that have functional effects prior to the onset of structural alterations.
Keywords: Alzheimer’s disease, amyloid, diffuse Lewy body disease, dystrophic neurite, oxidative stress, Parkinson disease, reticulon
INTRODUCTION
Reticulons (RTN) are a group of membrane-bound proteins located in the endoplasmic reticulum (ER) and on the cell membrane which have been shown to influence a diverse range of cellular functions, including retrograde transport of proteins from the golgi complex to the ER1 and nuclear envelope assembly2. They also appear to play a role in the regulation of apoptosis3, 4. RTN4, also known as Nogo, consists of several isoforms and has been shown to be an inhibitor of neurite regeneration5, 6 and a regulator of vascular proliferation7. Further, RTNs are decreased in areas of atherosclerotic plaque, thus reflecting the various mechanisms by which RTNs maintain healthy vasculature. RTN3 and RTN4 have both been shown to be involved with regulation of β-secretase AβPP cleaving enzyme 1 (BACE1) activity8, a protease responsible for cleavage of amyloid-β protein precursor (AβPP) resulting in formation of amyloid-β (Aβ). In particular, it has been shown that reduced levels of RTN3 by RNA interference resulted in an increase in Aβ levels in HEK-293 cells9. Another study showed that over-expression of RTN3 and RTN4-B/C in the same cell lines resulted in a 30–50% decrease in the release of Aβ10.
Previous studies demonstrated accumulation of RTN3 aggregates in dystrophic neurites in senile plaques of Alzheimer disease (AD)11. In the same study, transgenic mice with enrichment of human RTN3 demonstrated formation of dystrophic neurites within the brain which correlated with decreased spatial memory acquisition. Alterations in the structure of RTN3, specifically the formation of RTN3 aggregates, results in decreased binding to BACE1 and may lead to the increased accumulation of Aβ12. While it has been suggested that altered RTN3 specifically localizes to neuritic pathology13, another contradictory study reported only neuronal localization, without accumulation of RTN3 in disease-specific inclusions14.
To expand upon prior reports and also clear any controversy, in this study we sought to further examine RTN3 in a variety of human neurodegenerative diseases in vivo and further characterize the detailed histological and cellular distribution of RTN3.
MATERIALS AND METHODS
Tissue
For this study, brain tissue samples from a variety of neurodegenerative diseases and non-diseased individuals were examined. Tissues were collected using IRB approved protocols, fixed in either routine formalin or methacarn (methanol:chloroform:acetic acid; 6:3:1), embedded in paraffin, and sectioned at 6 μm. Cases used included hippocampus sections from cases of neuropathologically defined AD (n=13; aged 59–96 years, post-mortem interval 4–19 hours), and from 2 young control cases (aged 20 years) and 5 age-matched controls (aged 53–78 years). Also, brainstem sections from cases of Parkinson disease (n=5) and progressive supranuclear palsy (n=3), as well as hippocampus and adjacent cortex from cases of AD with Lewy body disease (n=2), one case of pure form Lewy body disease (age 70 years), Pick disease (n=4), and one case of subacute sclerosing panencephalitis were also examined.
Immunohistochemistry
For all cases, routine immunohistochemistry was performed. No antigen retrieval processes were utilized. Sections were deparaffinized through 2 changes of xylene and rehydrated through a graded ethanol series to Tris buffered saline (TBS; 50 mM Tris, 150 mM NaCl, pH=7.6). Endogenous peroxidases were abolished with a 30 minute incubation in 3% H2O2. After blocking sections with 10% normal goat serum (NGS) in TBS for 30 minutes, primary antibodies were applied for 16 hours at 4°C. Sections were then rinsed in 1% NGS in TBS, followed by 10 min in 10% NGS, and a species specific secondary antibody made in goat was applied for 30 min. After rinsing again as previous, the species-specific peroxidase-anti-peroxidase complex was applied for 1 hour at RT. Sections were rinsed 2 times in Tris buffer and developed with 3′-3′-diaminobenzidine (Dako). Single stained sections were dehydrated and mounted with permount. For some experiments, sections were double stained for both reticulon and markers for disease pathology. For these experiments, the mouse antibodies were detected using the alkaline-phosphatase-anti-alkaline-phosphatase method with Fast blue as chromagen. These sections were mounted using crystalmount media.
Antibodies used included the rabbit antisera directed against the N-terminus (R454) and C-terminus of RTN3 (R458) at 1/1000 dilution1. These antisera have been characterized previously in both human and mouse brain tissue using both immunohistochemistry and Western blot analysis (1,8,11). Antibodies against phosphorylated tau (AT8, Pierce Endogen; PHF1, gift of S. Greenberg) were also used to detect neurofibrillary pathology and a rabbit polyclonal antisera against α-synuclein (Chemicon) was used to localize Lewy bodies as previously described15, 16.
RESULTS
In control individuals, significant neuronal staining in the hippocampus was seen only in the 3 control cases older than 65 years (Fig 1C), while 4 control cases under the age of 60 years old showed very little neuronal labeling (Fig 1A). This observation appears consistent with the observed age-dependent increased expression of RTN3 in mouse brains17. On adjacent serial sections stained for phosphorylated tau using the monoclonal antibody AT8, the younger controls had no AD-type pathology (Fig 1B), while the older controls showed some neuritic pathology (Fig 1D). Dystrophic neurites which accumulated around senile plaques were positive for both RTN3 and AT8 (Fig 1C, D). However, while a small number of neurons contained AT8-positive pretangles, those same neurons displayed only cytoplasmic labeling for RTN3, indistinguishable from the RTN3 levels in all other neurons (Fig 1C, D).
Figure 1.
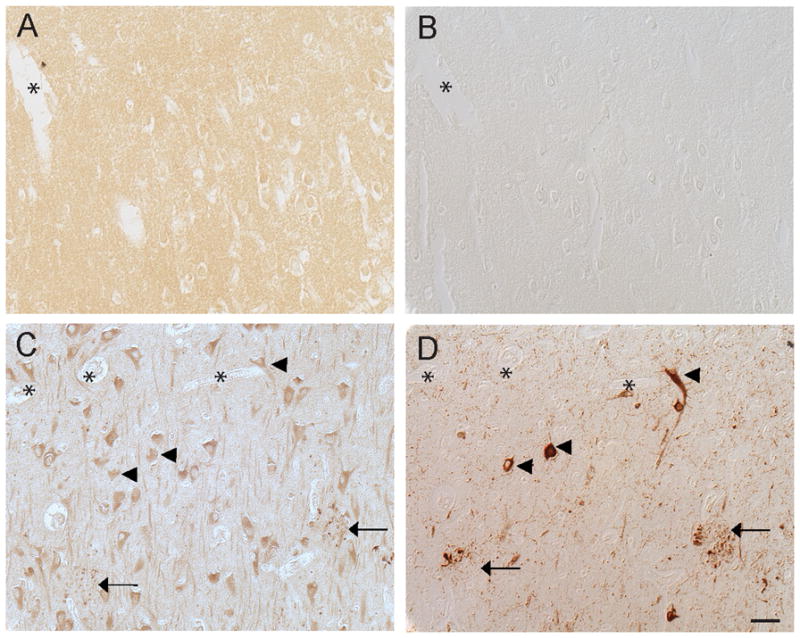
RTN3 in non-diseased brain. In control individuals under the age of 60, we did not detect any RTN3 in the pyramidal neurons of the hippocampus (A). On adjacent serial sections, no phosphorylated tau, using antibody AT8, was detected in these cases either (B). In the older control cases over the age of 65, however, significant neuronal accumulation of RTN3 was seen (C), as well as small dystrophic neurites around senile plaques (arrows). On an adjacent section (D), AT8 labeled the senile plaques and a small number of pretangles (arrowheads). The NFT-containing neurons were indistinguishable from other neurons in the RTN3 stained section (arrowheads,C). *= landmark vessels (A,B) and (C,D). Scale bar= 50 μm.
The majority of AD cases (8/11) also showed significant neuronal labeling (Fig 2A,D). The other 3 AD cases with minimal neuronal labeling also had the fewest pathological structures immunolabeled, and also the least amount of phosphorylated tau-containing NFT. However, in the majority of AD cases, RTN3 immunohistochemistry on both methacarn and formalin fixed tissue showed labeling for dystrophic neurites, as well as neurofibrillary tangles (NFT), neuropil threads, and granulovacuolar degeneration, in both the hippocampal pyramidal cell layer and in the cerebral cortex (Fig 2A,B,C). Generally, within specific cases, the CA4 and CA3 regions presented normal neuronal labeling, while the CA2 and CA1 showed significant neuritic and NFT stains. Labeling of astrocytic cytoplasm in both gray and white matter was also present. No endothelial or vascular smooth muscle cell labeling was detected.
Figure 2.
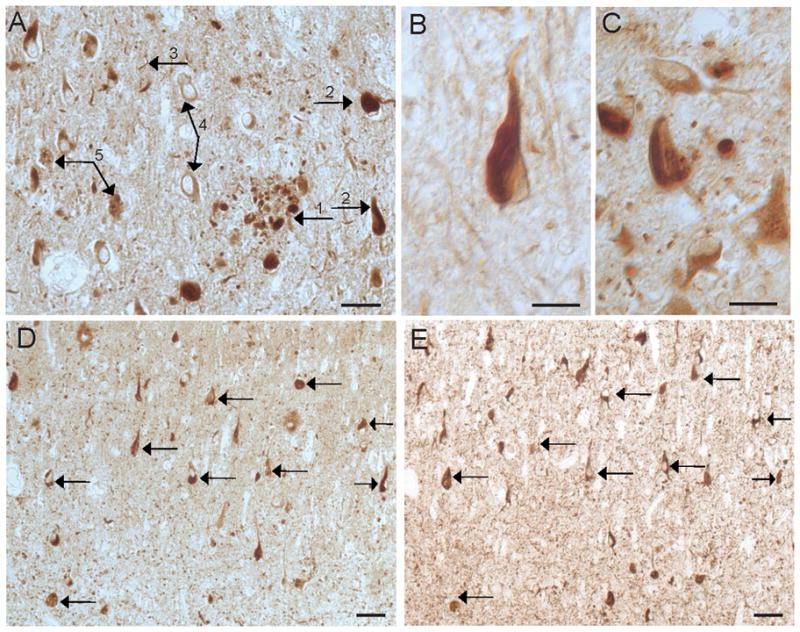
RTN3 in AD. In the hippocampus of a 67 year old AD case (A), RTN3 localized to dystrophic neurites around senile plaques (1), NFTs (2), neuropil threads (3), neuronal cytoplasm (4), and GVD (5). NFT are prominent in the majority of AD cases, including a 72 year old patient (B) and a 59 year old patient (C). On adjacent serial sections from an 80 year old case of AD stained for RTN3 (C) and AT8 (D), many of the same NFT contain both proteins (arrows). Scale bars= 50 μm (A,D,E), 20 μm (B,C,).
Using adjacent serial sections, many of the NFT that contain RTN3 colocalize with phosphorylated tau (Fig 2D,E). Double labeling in AD with phosphorylated tau (PHF1) and anti-reticulon emphasized the presence of reticulon in morphologically normal neurons and also highlighted a subset of neurites that did not contain phosphorylated tau (Fig 3).
Figure 3.

Double staining of RTN3 and phosphorylated tau. A,B. RTN3 (brown) colocalizes with phosphorylated tau (PHF1, blue) in many NFT-containing neurons. Many dystrophic neurites, neuropil threads, and neurons not containing NFT display only RTN3 immunoreactivity. Some cells distinctly contain only phosphorylated tau (blue only). Scale bar = 50 μm.
RTN3 immunohistochemistry on sections of midbrain from patients affected by Parkinson’s disease showed distinct immunolabeling of Lewy bodies, variously involving the outer rim of the Lewy body and within the core (Fig 4A,B). The antibodies specifically recognizing either the N or C termini of RTN3 labeled the same Lewy body on serial sections (Fig 4C,D). No labeling of Lewy neurites was detected in any of the cases analyzed. In cases of cortical Lewy body disease, LBs were specifically labeled with the antisera against RTN3 (Fig 5A). The same LBs contained α-synuclein as shown using adjacent serial sections (Fig 5B). In another case of LBD, many of the same α-synuclein positive structures (Fig 5D) also contained RTN3 (Fig 5C).
Figure 4.
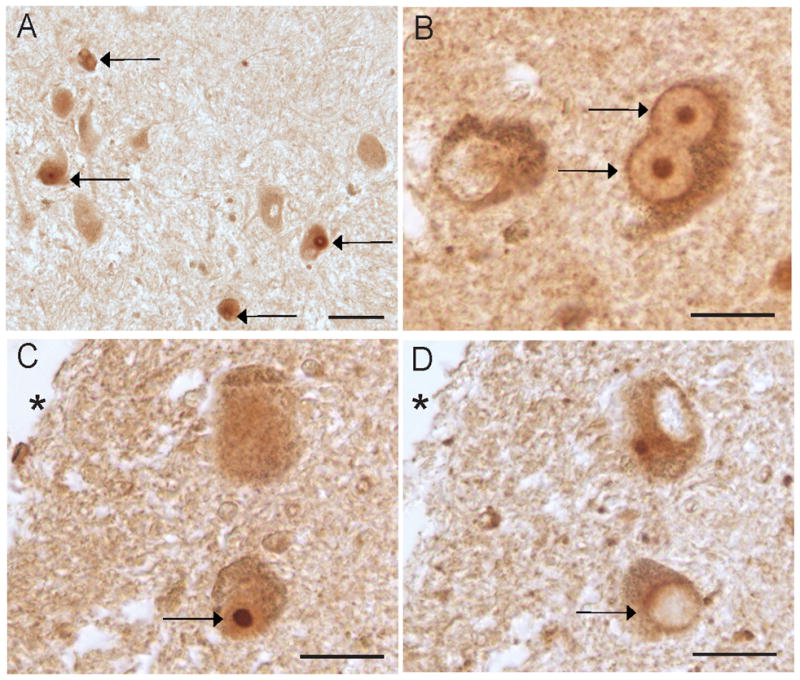
RTN3 in Parkinson disease. In Parkinson disease midbrain, RTN3 distinctly labeled Lewy bodies (A, arrows). At higher magnification, R458 distinctly labels both the core and rim of Lewy bodies (B, arrows). On adjacent serial sections, the same Lewy body (arrows) is recognized by both the C terminal specific antibody R458 (C), and the N terminal specific antibody R454 (D). * denotes landmark. Scale bars= 50 μm (A), 20 μm (B,C,D).
Figure 5.

RTN3 in Lewy body disease. In temporal cortex serial sections from a case of LBD, the same LB contains both RTN3 (A) and α-synuclein (B). In another case, many of the same structures that contain RTN3 (arrows, C), also contain α-synuclein (arrows, D). *= landmark vessels (A,B) and (C,D). Scale bars = 50 μm.
No labeling of Pick bodies, PSP tangles, or SSPE tangles was observed using the RTN3 antibody R458. Further, in PSP cases, no RTN3 immunoreactivity of any tufted astrocytes or coiled bodies was noted.
DISCUSSION
In this study, we found diffuse labeling of RTN in a variety of pathological inclusions as well as within normal neurons. Among the inclusions containing RTN3 were neurofibrillary tangles, senile plaques, Lewy bodies, Hirano bodies, and granulovacuolar degeneration. Diffuse cytoplasmic staining of neurons was also noted in both disease and aged controls. In the brains of young individuals between the ages of 20 and 60 years, minimal RTN3 was present in the neurons. One function of RTN3 is to modulate BACE1 activity, and it has been shown that BACE activity significantly increases with age in both human cortical and cerebellar brain regions18. Thus, the lack of neuronal RTN3 in the young cases analyzed could be due to the fact that BACE1 activity has not reached sufficient levels to incur RTN3 modulatory activity.
Our findings expand previous experimental studies by Hu et al., suggesting specificity for neuritic pathology11. Our use of a non-crosslinking fixative (methacarn) for many of the AD cases used in the present study may have facilitated the exposure of RTN epitopes that are obscured using traditional fixatives, and the examination of a large series of AD cases revealed the additional localization in most of the more advanced cases. These findings are also in disagreement with Kume et al., which again may be attributed to differences in primary fixative, although use of an antibody to a slightly different epitope likely contributed to this discrepancy14. The finding of RTN3 strongly associated with Lewy body pathology, but not the tau pathology associated with Pick disease or PSP, suggests RTN3 may have additional functions.
The results of our study, in particular the RTN accumulation in diseased brains, suggest that RTN is among the host of indicators of neuronal injury or neuronal stress, and is most likely a reactive or adaptive event rather than a causative agent. In this respect, it is interesting that RTN expression is linked to Bcl-2 regulation during endoplasmic reticulum stress4, which is a generalized process precipitated by such things as ischemia and oxidative stress19, 20.
The fact that pathological lesions such as neuritic plaques occur late in disease in general, and a have a capricious relationship with clinical disease, argues against neuritic damage as a rate limiting factor for disease21. The issue of the fundamental misconception of the relationship between lesion (e.g., dystrophic neurites) and pathogenesis has been raised21–24. Our data, demonstrating that reticulon 3 is most likely a downstream process furthers our contention that the focus in etiopathogenesis in AD should be on processes that precede lesions.
In conclusion, we demonstrate a wider cellular distribution of reticulon protein than previously recognized, suggesting that reticulon expression may be a nonspecific, downstream stress response in the AD brain.
Acknowledgments
Work in the authors’ laboratories is supported by the National Institutes of Health (AG031852) and the Alzheimer’s Association (IRG-07-60196).
References
- 1.Wakana Y, Koyama S, Nakajima K, Hatsuzawa K, Nagahama M, Tani K, et al. Reticulon 3 is involved in membrane trafficking between the endoplasmic reticulum and Golgi. Biochem Biophys Res Commun. 2005;334:1198–205. doi: 10.1016/j.bbrc.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Kiseleva E, Morozova KN, Voeltz GK, Allen TD, Goldberg MW. Reticulon 4a/NogoA locates to regions of high membrane curvature and may have a role in nuclear envelope growth. J Struct Biol. 2007;160:224–35. doi: 10.1016/j.jsb.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. A novel protein, RTN-XS, interacts with both Bcl-XL and Bcl-2 on endoplasmic reticulum and reduces their anti-apoptotic activity. Oncogene. 2000;19:5736–46. doi: 10.1038/sj.onc.1203948. [DOI] [PubMed] [Google Scholar]
- 4.Wan Q, Kuang E, Dong W, Zhou S, Xu H, Qi Y, et al. Reticulon 3 mediates Bcl-2 accumulation in mitochondria in response to endoplasmic reticulum stress. Apoptosis. 2007;12:319–28. doi: 10.1007/s10495-006-0574-y. [DOI] [PubMed] [Google Scholar]
- 5.GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–44. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- 6.Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, et al. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383–4. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- 7.Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, et al. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–8. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 8.He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat Med. 2004;10:959–65. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Liu BP, Budel S, Li M, Ji B, Walus L, et al. Blockade of Nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble Nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci. 2004;24:10511–20. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murayama KS, Kametani F, Saito S, Kume H, Akiyama H, Araki W. Reticulons RTN3 and RTN4-B/C interact with BACE1 and inhibit its ability to produce amyloid beta-protein. Eur J Neurosci. 2006;24:1237–44. doi: 10.1111/j.1460-9568.2006.05005.x. [DOI] [PubMed] [Google Scholar]
- 11.Hu X, Shi Q, Zhou X, He W, Yi H, Yin X, et al. Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J. 2007;26:2755–67. doi: 10.1038/sj.emboj.7601707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Q, Prior M, He W, Tang X, Hu X, Yan R. Reduced amyloid deposition in mice overexpressing RTN3 is adversely affected by preformed dystrophic neurites. J Neurosci. 2009;29:9163–73. doi: 10.1523/JNEUROSCI.5741-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He W, Shi Q, Hu X, Yan R. The membrane topology of RTN3 and its effect on binding of RTN3 to BACE1. J Biol Chem. 2007;282:29144–51. doi: 10.1074/jbc.M704181200. [DOI] [PubMed] [Google Scholar]
- 14.Kume H, Konishi Y, Murayama KS, Kametani F, Araki W. Expression of reticulon 3 in Alzheimer’s disease brain. Neuropathol Appl Neurobiol. 2009;35:178–88. doi: 10.1111/j.1365-2990.2008.00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, et al. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol. 1994;145:42–7. [PMC free article] [PubMed] [Google Scholar]
- 16.Castellani R, Smith MA, Richey PL, Perry G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996;737:195–200. doi: 10.1016/0006-8993(96)00729-9. [DOI] [PubMed] [Google Scholar]
- 17.Shi Q, Hu X, Prior M, Yan R. The occurrence of aging-dependent reticulon 3 immunoreactive dystrophic neurites decreases cognitive function. J Neurosci. 2009;29:5108–15. doi: 10.1523/JNEUROSCI.5887-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol. 2004;164:719–25. doi: 10.1016/s0002-9440(10)63159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banhegyi G, Mandl J, Csala M. Redox-based endoplasmic reticulum dysfunction in neurological diseases. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05571.x. [DOI] [PubMed] [Google Scholar]
- 20.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA. Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol. 2008;67:523–31. doi: 10.1097/NEN.0b013e318177eaf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321:823–9. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 23.Nunomura A, Castellani RJ, Lee HG, Moreira PI, Zhu X, Perry G, et al. Neuropathology in Alzheimer’s disease: awaking from a hundred-year-old dream. Sci Aging Knowledge Environ. 2006;2006:pe10. doi: 10.1126/sageke.2006.8.pe10. [DOI] [PubMed] [Google Scholar]
- 24.Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol. 2006;111:503–9. doi: 10.1007/s00401-006-0071-y. [DOI] [PubMed] [Google Scholar]