

Abstract
Previously it was reported that Alzheimer's disease (AD) patients have reduced amyloid (Aβ1-42) and elevated total tau (t-tau) and phosphorylated tau (p-tau181p) in the cerebrospinal fluid (CSF), suggesting that these same measures could be used to detect early AD pathology in healthy elderly (CN) and mild cognitive impairment (MCI). In this study, we tested the hypothesis that there would be an association among rates of regional brain atrophy, the CSF biomarkers Aβ1-42, t-tau, and p-tau181p and ApoE ε4 status, and that the pattern of this association would be diagnosis specific. Our findings primarily showed that lower CSF Aβ1-42 and higher tau concentrations were associated with increased rates of regional brain tissue loss and the patterns varied across the clinical groups. Taken together, these findings demonstrate that CSF biomarker concentrations are associated with the characteristic patterns of structural brain changes in CN and MCI that resemble to a large extent the pathology seen in AD. Therefore, the finding of faster progression of brain atrophy in the presence of lower Aβ1-42 levels and higher p-tau levels supports the hypothesis that CSF Aβ1-42 and tau are measures of early AD pathology. Moreover, the relationship among CSF biomarkers, ApoE ε4 status, and brain atrophy rates are regionally varying, supporting the view that the genetic predisposition of the brain to amyloid and tau mediated pathology is regional and disease stage specific.
Keywords: MRI, Alzheimer's disease, cerebrospinal fluid, biomarkers, cortical thickness, atrophy, brain tissue volume, ApoE
Introduction
There is an increasing body of evidence from in vivo imaging and post mortem studies indicating that Alzheimer's disease (AD) is associated with a sequence of pathophysiological events that can occur over a long period (approximately 20-years) before clinical symptoms become apparent (Price and Morris, 1999). A slow disease progression provides potentially a window for early interventions to reduce or even stop progression of AD. Histopathological studies showed that the hallmarks of the disease, Aβ-rich amyloid plaques and neurofibrillary tangles formed by abnormal tau, precede neuron loss in presymptomatic AD patients (Price and Morris, 1999). Substantial accumulations of plaques and tangles in the brain can also be found in non-demented subjects with mild cognitive impairment (MCI), a transitional stage between normal aging and dementia (Aizenstein et al., 2008; Jack et al., 2009; Mintun et al., 2006). Consistent with histopathological findings, cerebrospinal fluid (CSF) chemistry studies have pointed to alterations in CSF Aβ (in particular Aβ1-42), total tau (t-tau) and phosphorylated tau (p-tau181p) concentrations preceding clinical symptoms of AD (Fjell et al., 2008). In general, studies found that increased CSF t-tau and p-tau181p were associated with neuronal and axonal damage, whereas reduced CSF Aβ1-42, the form of Aβ that most readily fibrillizes and deposits earliest in plaques, has been implicated to reflect higher amyloid plaque burden in the brain (Clark et al., 2003; Shaw et al., 2009). However, the CSF measures are not easily interpretable because their origins are not exclusively brain derived and they provide no information about the regional spread of brain damage. Despite this, there is considerable agreement that measuring CSF Aβ1-42, t-tau, and p-tau181p improves the diagnostic accuracy for AD (Andreasen et al., 1999).
Independent of biomarker studies, numerous structural MRI studies have shown a characteristic pattern of brain atrophy in AD and a similar pattern in MCI, affecting primarily regions in the parietotemporal lobe, including the hippocampus, which plays a central role in memory formation (Chetelat and Baron, 2003; deToledo-Morrell et al., 2004; Du et al., 2001; Du et al., 2002; Duarte et al., 2006; Hua et al., 2008; Kramer et al., 2004; Morra et al., 2008; Morra et al., 2009a; Morra et al., 2009b; Schroeter et al., 2009; Thompson et al., 2004; Whitwell et al., 2007; Whitwell et al., 2008). In addition, an increasing number of longitudinal MRI studies show that both AD and MCI are also associated with a regional pattern of increased rates of brain tissue loss compared to normal aging (Desikan et al., 2008; Du et al., 2004; Du et al., 2003; Jack et al., 2005; Jack et al., 2008b; Jack et al., 2004; Stoub et al., 2005). With the emerging findings of CSF biomarker and structural imaging alterations in AD, there is considerable interest in utilizing the CSF and MRI measures together to improve detection of early signs of AD, as well as, in unraveling relationships between CSF Aβ1-42, t-tau, and p-tau181p and MRI measures of regional brain alterations. Recently it has been shown that the combination of CSF biomarkers and atrophy rates can provide better prediction of AD than either source of data alone (Brys et al., 2009; Vemuri et al., 2009a, b). However, whether relationships between brain atrophy rates and CSF biomarkers help further to improve predictions has not fully been explored.
Moreover, the role of the apolipoprotein E allele ε4 (ApoE ε4) gene, a major risk factor for AD, ought to be considered for a comprehensive evaluation. Presence of ApoE ε4 is related to abnormal CSF biomarker concentrations (Glodzik-Sobanska et al., 2009; Sunderland et al., 2004), as well as, to higher rates of brain atrophy (Basso et al., 2006; Fleisher et al., 2005; Potkin et al., 2009; Schuff et al., 2009; Sluimer et al., 2008). The relationships among all three factors, CSF biomarkers, ApoE ε4, and rates of regional brain atrophy, might therefore provide important information about the vulnerability of the brain to AD. Our overall goal in this study was therefore to unravel the relationships among all three factors: brain atrophy rates, CSF biomarker concentrations, and presence of ApoE ε4. Toward the goal of identifying an AD biomarker, it will be important to fully understand the relationship between CSF biomarker concentrations and brain degeneration, such as neuron loss, which is thought to underlie the clinical symptoms in AD (Fjell et al., 2008; Jack et al., 2009). While CSF biomarkers relate to cumulative AD pathology in the brain as peripheral measures, MRI as an external tool elucidates the distribution of the AD related neurodegeneration (i.e., brain atrophy in terms of tissue loss and ventricular enlargement). However, relatively few MRI studies so far have reported correlations between CSF biomarkers and the pattern of brain atrophy or the rate of atrophy progression (Fagan et al., 2009; Fjell et al., 2010a; Fjell et al., 2010b; Hampel et al., 2005; Henneman et al., 2009; Herukka et al., 2008; Leow et al., 2009; Schuff et al., 2009). Specifically, in healthy elderly individuals, it has been shown that low CSF levels of Aβ1-42 correlate with ventricular expansion and volumetric reductions in widespread brain areas (Fjell et al., 2010a). In individuals with progressive MCI, low CSF Aβ1-42 concentration and high concentrations of CSF p-tau181p and t-tau are associated with higher subsequent rates of hippocampal atrophy (Hampel et al., 2005; Henneman et al., 2009; Herukka et al., 2008; Schuff et al., 2009). In AD patients, elevated CSF p-tau181p concentrations were associated with higher subsequent rates of hippocampal atrophy and medial temporal atrophy (Hampel et al., 2005; Henneman et al., 2009; Herukka et al., 2008; Leow et al., 2009), while low CSF Aβ1-42 concentrations exhibited larger rates of medial temporal atrophy (Leow et al., 2009). However, the majority of previous MRI studies in this context focused on hippocampal and temporal lobe atrophy and ventricular expansion in MCI and AD patients, while relatively little is known about relations between the CSF biomarker concentrations and atrophy rates of other regions throughout the brain. In addition, variations in these relationships across the spectrum of cognitive impairments have not been comprehensively studied for regions across the brain.
Our main goal in this study was to test the hypothesis that relations between CSF biomarkers (i.e., Aβ1-42, t-tau, and p-tau181p concentrations) and rates of regional brain atrophy not only vary across brain regions but also across the cognitive spectrum, including healthy elderly individuals (CN), individuals with MCI, and AD patients. In particular we tested that (1) low Aβ1-42 and high t-tau and p-tau181p concentrations were associated with smaller mean regional brain tissue volume and cortical thickness in CN, MCI, and AD, (2) low Aβ1-42 and high t-tau and p-tau181p concentrations were associated with increased rates of regional brain atrophy in CN, MCI, and AD, and (3) the patterns of association were group-specific. In addition, we tested whether abnormal CSF biomarker concentrations and ApoE ε4 status separately or together were associated with higher rates of brain atrophy.
Cohort and Methods
We examined the baseline regional tissue volume and cortical thickness, and the rate of change in regional tissue volume and cortical thickness across the brain in CN, individuals with MCI, and AD patients. Structural magnetic resonance imaging (MRI) brain scans at multiple time points (four time point scans - baseline, 6, 12, and 24 months - for CN and AD subjects and five time point scans - baseline, 6, 12, 18, and 24 months - for individuals with MCI) were acquired at multiple Alzheimer's Disease Neuroimaging Initiative (ADNI) sites using 1.5 Tesla MRI scanners. Using FreeSurfer longitudinal processing framework, regional tissue volumes of a total of 108 cortical and sub-cortical regions and local cortical thickness through out the entire cortex were automatically measured at each time point. In each diagnostic group separately, generalized linear mixed effect models followed by pair-wise maximum likelihood test were performed to test: 1) if baseline CSF biomarker concentrations predict absolute volumes and local thickness at baseline; 2) if baseline CSF biomarker concentrations modulate the rates of brain atrophy; and 3) if CSF biomarkers and ApoE ε4 modulate the rates of brain atrophy jointly or independently, after accounting for variations in age, sex, and education. Pursuing a region-of-interest based analysis allows us to study the subcortical structures while surface-based cortical thickness analysis provides better localization of the effects on cortical mantle. Finally, we tested if the observed modulation effects of baseline CSF biomarkers on rates of atrophy differ among groups. The methodological details are explained herein.
Participants
The participants in this study were recruited through the ADNI, a longitudinal, multicenter study launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies, and non-profit organizations, as a $60 million, 5-year public–private partnership to define biomarkers of early Alzheimer's disease for clinical trials (http://www.adni-info.org). The Principal Investigator of this initiative is Michael W. Weiner, MD of the Veteran Affairs Medical Center and University of California in San Francisco.
Briefly, inclusion criteria for the CN group were Mini-Mental State Examination (MMSE) scores between 24 and 30, a Clinical Dementia Rating - Sum of Boxes (CDR-SB) score of 0, and lack of depression, MCI, or dementia. Inclusion criteria for the MCI group followed the Peterson criteria (Petersen et al., 1999) for amnestic MCI, which required a subjective memory complaint, objective memory loss measured by education-adjusted Wechsler Memory Scale-Revised Logical Memory II scores, a CDR-SB of 0.5, absence of significant impairment in other cognitive domains, preserved activities of daily living, and an absence of dementia. AD participants met the National Institute for Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorder Association (NINDS/ADRDA) criteria for probable AD, had an MMSE between 18 and 26, and a CDR-SB of 0.5 to 1.0. Exclusion criteria included history of structural brain lesions or head trauma, significant neurological disease other than incipient AD, and use of psychotropic medications that could affect memory. The full details of the inclusion and exclusion criteria for the ADNI can be found at http://www.adni-info.org. Written consent was obtained from all subjects participating in the study according to the Declaration of Helsinki (Br Med J 1991; 302: 1194), and the study was approved by the institutional review board at each participating site.
The population in this study included ADNI subjects with valid test result for all three CSF biomarkers and successful longitudinal FreeSurfer processing of MR images from at least two time points. Overall, the study population was comprised of 77 CN, 118 MCI, and 53 AD subjects. Details of CSF biomarker concentration measurement and longitudinal structural MR image processing are described in the following sections. The demographic details of each group are given in Table 1.
Table 1.
Demographic features of study groups
| CN | MCI | AD | |
|---|---|---|---|
| N (baseline) | 77 | 119 | 54 |
| N follow-up (6,12,18,24 months) | 77, 76, 0, 63 | 119, 109, 96, 77 | 54, 53, 0, 31 |
| Baseline age (years) | 75 ± 5.0 | 74 ± 7.6 | 74 ± 8.0 |
| Gender (F/M) | 40/37 | 46/73 | 25/29 |
| Baseline CSF Aβ1-42 (pg/ml) | 203 ± 51.8 | 165 ± 57.5 | 142 ± 39.7 |
| Baseline CSF t-tau (pg/ml) | 70 ± 29.5 | 101 ± 50.0 | 128 ± 53.1 |
| Baseline CSF p-tau181p (pg/ml) | 26 ± 15.0 | 35 ± 16.5 | 42 ± 16.8 |
| ApoE ε4 status (carrier %) | 29% | 54% | 67% |
Structural MRI Acquisition
The participants underwent a standardized 1.5 Tesla MRI protocol (http://www.loni.ucla.edu/ADNI/Research/Cores/index.shtml), which included two T1-weighted MRI scans using a sagittal volumetric magnetization prepared rapid gradient echo (MP-RAGE) sequence with the following acquisition parameters: echo time (TE) of 4 ms, repetition time (TR) of 9 ms, flip angle of 8°, acquisition matrix size of 256 × 256 × 166 in the x-, y- and z-dimensions with a nominal voxel size of 0.94 × 0.94 × 1.2 mm3. Only one of the MPRAGE sets was used for analysis. The ADNI MRI quality control center at the Mayo Clinic selected the MP-RAGE image with higher quality and corrected for system-specific image artifacts, as described in (Jack et al., 2008a).
CSF Biomarker Concentrations
CSF samples were obtained from 53% of ADNI participants, while the rest did not undergo lumbar puncture. The demographics of ADNI subjects with CSF samples are comparable with that in the full ADNI patient population (http://www.adni-info.org).
A small sample of CSF from the lower spine of each subject was collected at baseline by lumbar puncture in the morning after an overnight fast. Lumbar puncture was performed with a 20- or 24-gauge spinal needle as described in the ADNI procedures manual (http://www.adni-info.org). In brief, CSF was collected into collection tubes provided to each site, then transferred into polypropylene transfer tubes followed by freezing on dry ice within 1 hour after collection, and shipped overnight to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center on dry ice. 0.5 ml aliquots were prepared from these samples after thawing for 1 hour at room temperature and gentle mixing. The aliquots were stored in bar code–labeled polypropylene vials at -80°C. Aβ1-42, t-tau, and p-tau181p were measured in each aliquots using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium; for research use–only reagents) immunoassay kit–based reagents. Full details of this combination of immunoassay reagents and analytical platform are provided elsewhere (Olsson et al., 2005). The ADNI baseline CSF samples were analyzed over a 14-day period and included test–retest analyses of 29 of the samples that further substantiated the analytical performance (r2 values for comparison of initial test result with retest result of 0.98, 0.90, and 0.85 for t-tau, Aβ1-42, and p-tau181p, respectively for 29 randomly selected samples). Full details of ADNI baseline CSF biomarker measurements are provided elsewhere (Shaw et al., 2009).
FreeSurfer Longitudinal MR Image Processing
Automated cortical thickness measures, cortical parcellation, and subcortical segmentation were performed with FreeSurfer software package, version 4.4 (http://surfer.nmr.mgh.harvard.edu/fswiki). To reduce the confounding effect of intra-subject morphological variability, each subject's longitudinal data series was processed by FreeSurfer longitudinal workflow. The longitudinal workflow was designed to estimate brain morphometry measurements that were unbiased with respect to any time point. Instead of using information from a specific time point as a prior for other time points, a template image volume was created as an unbiased prior for all time points.
FreeSurfer longitudinal workflow consists of four stages: (1) processing of all time points individually with the cross-sectional workflow; (2) creation of a probabilistic template unbiased toward time points from all time points' cross-sectional data; (3) processing of unbiased template with the cross-sectional workflow; and finally (4) re-processing of each time point with the longitudinal workflow, which uses the unbiased template results as initial guess for the segmentation and surface reconstruction. For a full description of the FreeSurfer processing steps, see (Fischl et al., 2002; Fischl et al., 2004), and for a full description of the longitudinal workflow, see http://surfer.nmr.mgh.harvard.edu/fswiki/LongitudinalProcessing.
The Freesurfer measures for each subject at each time point are as follows: (1) Each image voxel in MR image volume was automatically assigned one of 40 neuroanatomical labels and for each anatomical subcortical region-of-interests (ROIs) total tissue volume was estimated (Fischl et al., 2002). (2) Vertex-based cortical thickness measurements were obtained as the distance between the reconstructed surface representations of the gray matter/white matter and white matter/CSF tissue interfaces (Fischl and Dale, 2000). Each cortical surface was spatially normalized to a template cortical surface using a non-rigid high-dimensional spherical averaging method to align cortical folding patterns. Subject cortical thickness maps were mapped onto the template surface based on this spatial normalization and then smoothed by a surface-based Gaussian blurring kernel with a standard deviation of 10mm to remove noise-induced variations in the measurements. (3) Spatial normalization to the template cortical surface was also used to automatically parcellate the subject cortical surfaces at each time point to 34 anatomical regions per cortical hemisphere (Fischl et al., 2004). For each cortical ROI, cortical gray matter volume was estimated based on the cortical parcellation and tissue segmentation.
The surface reconstruction, subcortical segmentation, and cortical parcellation results were visually examined for anatomical accuracy. Although the FreeSurfer software package allows for manual editing to correct registration and segmentation errors, given the large number of subjects in ADNI data set, only the data with accurate results from fully automated processing were used in the subsequent analysis in the interest of a practical total processing time and avoidance of reader bias. 74% of the MR images passed this quality control, 3% of the images failed the quality control completely, and remaining 23% of the images got partial pass on the quality control. Details of the quality control procedure are posted online at http://www.loni.ucla.edu/twiki/pub/ADNI/ADNIPostProc/UCSFFreeSurferMethodsSummary.pdf.
Statistical Analyses
For each subject, variations in brain volumes or cortical thickness were modeled as a function of time starting with the baseline scan (time-point zero) in intervals of subsequent MRI scans in units of years. We employed a general linear mixed effects (GLME) model for analysis of the longitudinal data in which the response variable (i.e., cortical thickness or tissue volumes of cortical and subcortical regions) was regressed against the explanatory variables including time, baseline CSF biomarker concentration (i.e., Aβ1-42, p-tau181p, or t-tau), and the interaction between time and baseline CSF biomarker concentration to estimate the fixed effects in the group, separately from the random effects such as within subject variations in both baseline and longitudinal measures. This concept was used to test the primary hypothesis that variations in CSF biomarker concentrations modulate rates of brain atrophy (i.e., cortical thinning, tissue volume loss, or ventricular expansion). The fixed effect model was formulated as follows:
Here, Vij represents the size of a brain structure (volume or thickness) from subject i at time point j. Accordingly, Tij indicates the time point of the individual MRI scan, Bi0 represents individual CSF biomarker concentrations at baseline and εij is the mixed effects error. Our goal was to test the significance of the coefficient βYears:CSFbio in explaining structural variations (i.e., the moderator) relative to the coefficients β0, βYears, and βCSFbio and independent of random variations in brain structures at baseline and over time. For a significant interaction to occur, CSF biomarker concentration must modulate the relationship between time and the response variable (i.e., local cortical thickness or regional tissue volume). To determine if the addition of an interaction term (βYears:CSFbio) between rates of brain atrophy and CSF biomarkers in the model significantly improves the explanatory power of regional variations in cortical thickness or brain tissue volume as a function of biomarkers, we compared pair-wise GLME models (i.e., with and without the βYears:CSFbio term), fitted by maximum likelihood (ML) via F-tests. These tests were performed separately for each group (i.e., CN, MCI, and AD). Similarly, to determine the significance of CSF biomarker effects on regional tissue volumes and cortical thickness at baseline, the additive term βCSFbio was assessed by pair-wise comparisons of GLME models with and without βCSFbio term, followed by maximum likelihood (ML) via F-tests. Each CSF biomarker was centered on its population mean to reduce colinearity.
To assess if the effect of CSF biomarkers on rates of regional brain atrophy differ across groups (CN, MCI, and AD), we resampled the random effects residual of the fits by 100-fold bootstrap and evaluated differences in distributions by analysis of variance.
Finally, we tested the extent to which ApoE ε4 status contributes to higher brain atrophy rates independently of CSF biomarker concentrations (ApoE ε4 status + CSF biomarker) or via a synergistic interaction with the biomarkers (ApoE ε4 status *CSF biomarker). Again, pair-wise ML tests were performed between models with and without the interaction term (i.e. ApoE ε4 *CSF biomarker) to determine the contribution of the interaction.
Age, gender, and education were included as covariates in each regression model described above. For cortical thickness measures, the GLME models and the corresponding pair-wise ML F-tests were evaluated at each surface vertex independently. All statistical analyses were computed using R (the R Project for Statistical Computing; www.r-project.org). To control for false positive findings given the large number of comparisons per brain map, we used the concept of a false discovery rate (FDR) at the level q=0.05 (Benjamini and Hochberg, 1995). For testing the a-priori hypotheses, which comprised a limited number of planned tests, we used a per comparison error rate of q=0.05 for each test to determine the probability that any one contrast, after passing FDR, is found by chance.
Results
Effects of baseline CSF biomarker concentrations on regional mean tissue volumes
The results of associations between biomarkers and mean regional brain tissue volumes (i.e., βCSFbio coefficient) are summarized in Table 2 for those cortical and subcortical ROIs where the baseline CSF biomarker concentrations had significant effect on mean regional tissue volumes (FDR corrected p<0.05). Listed are both the coefficients from the GLME models (representing change in tissue volume per unit biomarker concentration --- i.e., mm3/(pg/ml)) and the likelihood ratio (LR) of the biomarker effects. Variations in baseline CSF biomarker concentration in the CN group were not significantly associated with mean regional brain volumes. In contrast, higher baseline concentrations of CSF t-tau and p-tau181p in MCI were associated with smaller caudate volumes. In AD, lower baseline concentrations of CSF Aβ1-42 were associated with smaller baseline gray matter volumes in lingual, pericalcarine, and postcentral cortices.
Table 2.
Effects of baseline CSF biomarkers on the mean regional tissue (in units of mm3/(pg/ml)))
| Region | βAβ1-42 | LR | βt-tau | LR | βp-tau181p | LR |
|---|---|---|---|---|---|---|
| Patients with Mild Cognitive Impairment (MCI) | ||||||
| Right caudate | - | - | -4.0 ± 0.9** | 19.6 | -10.1 ± 2.9* | 13.4 |
| Left caudate | - | - | -3.8 ± 0.9** | 19.5 | - | - |
| Patients with Alzheimer's Disease (AD) | ||||||
| Left lingual | 11.0 ± 2.3** | 20.8 | - | - | - | - |
| Right pericalcarine | 3.8 ± 1.0* | 11.7 | - | - | - | - |
| Right cerebral cortex | 144.8 ± 42.8* | 10.9 | - | - | - | - |
| Right lingual | 7.0 ± 2.3* | 10.3 | - | - | - | - |
| Left postcentral | 11.4 ± 3.5* | 9.4 | - | - | - | - |
p-values < 0.05
p-values < 0.005
p-values < 0.0005
LR: Likelihood ratio
-: non-significant effects
Effects of baseline CSF biomarkers concentrations on the rates of regional tissue volume change
Lower concentrations of CSF Aβ1-42 were associated: (i) in CN with increased rates of ventricular enlargement; (ii) in MCI with increased rates of ventricular enlargement and increased rates of atrophy prominently in the left lateral and medial temporal cortices, left hippocampus, left isthmus cingulate, and right amygdala; and (iii) in AD with decreased rates of atrophy in the caudate and accumbens area. The interaction coefficient of the GLME regressions (representing change in the rates of atrophy per unit biomarker concentration --- i.e., mm3/(year*pg/ml)) as well as the likelihood ratio of Aβ1-42 atrophy modulation effects are summarized in Table 3 for cortical and subcortical ROIs with significant effects only (FDR corrected p<0.05).
Table 3.
Effects of baseline CSF biomarkers on rates of regional brain volume loss (in units of mm3/(year*pg/ml)))
| Region | βYears: Aβ1-42 | LR | βYears: t-tau | LR | βYears:p-tau181p | LR | |
|---|---|---|---|---|---|---|---|
| Elderly cognitively normal (CN) | |||||||
| Right lateral ventricle | -4.7 ± 0.8*** | 31.1 | - | - | 12.2 ± 3.0** | 16.1 | |
| Left lateral ventricle | -4.3 ± 0.8*** | 27.0 | - | - | - | - | |
| Right inferior lateral ventricle | - | - | - | - | 1.6 ± 0.4** | 16.8 | |
| Patients with Mild Cognitive Impairment (MCI) | |||||||
| Right lateral ventricle | -5.6 ± 1.2*** | 21.9 | - | - | - | - | |
| Left temporal pole | 0.4 ± 0.1** | 17.4 | - | - | -1.1 ± 0.3** | 13.1 | |
| Left lateral ventricle | -5.6 ± 1.4** | 16.3 | - | - | - | - | |
| Left fusiform | 0.9 ± 0.2** | 16.3 | - | - | - | - | |
| Left inferior temporal | 1.1 ± 0.3** | 15.9 | - | - | -3.7 ± 1.0** | 14.1 | |
| Left entorhinal | 0.3 ± 0.1** | 15.9 | - | - | -1.0 ± 0.2** | 16.7 | |
| Left isthmus cingulate | 0.2 ± 0.1* | 12.5 | - | - | - | - | |
| Left hippocampus | 0.3 ± 0.1* | 11.7 | - | - | -0.9 ± 0.3* | 10.8 | |
| Right middle temporal | 1.0 ± 0.3* | 10.1 | - | - | -4.3 ± 1.1** | 15.6 | |
| Left parahippocampal | 0.2 ± 0.1* | 10.0 | - | - | -0.9 ± 0.2** | 13.2 | |
| Left inferior lateral ventricle | -0.5 ± 0.2* | 9.4 | - | - | - | - | |
| Right inferior lateral ventricle | -0.5 ± 0.2* | 9.2 | - | - | - | - | |
| Left cerebral cortex | 9.3 ± 3.1* | 9.1 | - | - | - | - | |
| Left middle temporal | 0.8 ± 0.3* | 9.1 | - | - | -2.7 ± 0.9* | 8.2 | |
| Right cerebral cortex | 8.8 ± 2.9* | 9.0 | - | - | -31.5 ± 10.3* | 0.4 | |
| Right fusiform | 0.8 ± 0.2* | 8.7 | - | - | - | - | |
| Third ventricle | -0.2 ± 0.1* | 7.8 | - | - | - | - | |
| Right amygdala | 0.2 ± 0.1* | 7.0 | - | - | - | - | |
| Left amygdala | - | - | - | - | -0.7 ± 0.2* | 12.7 | |
| Right inferior temporal | - | - | - | - | -3.7 ± 1.0** | 13.6 | |
| Patients with Alzheimer's Disease (AD) | |||||||
| Left caudate | -2.6 ± 0.5*** | 24.2 | -1.3 ± 0.4* | 9.6 | - | - | |
| Right caudate | -2.3 ± 0.5** | 18.7 | -1.3 ± 0.4* | 10.5 | - | - | |
| Right accumbens area | -0.6 ± 0.2** | 14.7 | - | - | - | - | |
| Right isthmus cingulate | - | - | 0.6 ± 0.1** | 17.6 | - | - | |
| Right posterior cingulate | - | - | 0.7 ± 0.2* | 12.4 | - | - | |
| Right precuneus | - | - | 1.5 ± 0.5* | 11.1 | - | - | |
| Left lateral ventricle | - | - | -8.0 ± 2.4* | 11.0 | - | - | |
p-values < 0.05
p-values < 0.005
p-values < 0.0005
LR: Likelihood ratio
-: non-significant effects
In contrast to baseline CSF Aβ1-42 concentrations, baseline concentrations of CSF t-tau were not significantly associated with any rate of brain volume changes in CN and MCI groups. In AD, however, higher concentrations of baseline CSF t-tau were associated with decreased rates of atrophy in the right posterior cingulate, isthmus cingulate, and precuneus cortices and ventricular enlargement, and increased rates of caudate atrophy as summarized in Table 3.
Finally, atrophy modulation effects of baseline CSF p-tau181p concentration largely mirrored the pattern seen for Aβ1-42 in CN and MCI, though associations with baseline CSF p-tau181p involved fewer regions in MCI. Specifically, higher concentrations of CSF p-tau181p were associated: (i) with increased rates of ventricular enlargement in CN; (ii) with increased rates of regional brain atrophy prominently in the left temporal lobe, hippocampus, and amygdala in MCI. In AD, however, baseline CSF p-tau181p concentrations were not associated with rate of any brain volume changes. Significant interaction coefficient of the GLME models as well as the likelihood ratio of p-tau modulation effects are summarized in Table 3.
Effects of baseline CSF biomarker concentrations on mean regional cortical thickness
Next, we report effects of CSF biomarker concentrations on mean cortical thickness after accounting for variations in age, gender, and education across subjects. Figure 1 depicts the regional distribution of CSF Aβ1-42 effects on mean cortical thickness for CN. Also shown in Figure 1 is the regional distribution of the corresponding p-values of the CSF Aβ1-42 effects based on the ML F-tests. In CN, lower baseline CSF Aβ1-42 was associated with a thinner cortex in the left frontal pole, left rostral-middle frontal left superior frontal, left pars opercularis, left pars triangularis, left supramarginal, left inferior parietal, left superior temporal, left middle temporal, left inferior temporal, left posterior cingulate, left precuenus, left fusiform, right frontal pole, right rostal middle frontal, right supramarginal, right inferior parietal, right superior parietal, right middle temporal, right inferior temporal, right medial orbito-frontal, right posterior cingulate, right paracentral lobule, right precuneus, and right fisuform cortices. In MCI and AD, no statistically significant association between the baseline CSF biomarker concentrations and the mean cortical thickness measures were observed.
Figure 1.

Association between baseline CSF Aβ1-42 concentrations and absolute cortical thickness in CN group. [top row: cortical maps of regression coefficients βAβ1-42 from GLME models. bottom row: resulting FDR corrected p-value map from pair-wise ML F-tests.]
Neither t-tau nor p-tau181p showed significant association with mean cortical thickness in CN, MCI, and AD groups.
Effects of baseline CSF biomarkers concentrations on the rates of regional cortical atrophy
Similarly, we report next the modulation effects of CSF biomarkers on the rates of cortical thinning after accounting for variations in age, gender, and education across subjects. Figure 2 depicts the regional distribution of a CSF Aβ1-42 modulation effects on the rates of cortical thinning for MCI. Also shown in Figure 2 is the regional distribution of the corresponding p-values of the CSF Aβ1-42 effects based on the ML F-tests. In MCI, lower concentrations of CSF Aβ1-42 were associated with increased rates of cortical thinning throughout the cortex. The effects were statistically significant (FDR corrected; p<0.05) in the left temporal pole, left inferior temporal, left middle temporal, left inferior parietal, left paracentral lobule, left cingulate, left isthmus cingulate, left precuneus, left entorhinal, left fusiform, right inferior temporal, and right middle temporal cortices.
Figure 2.
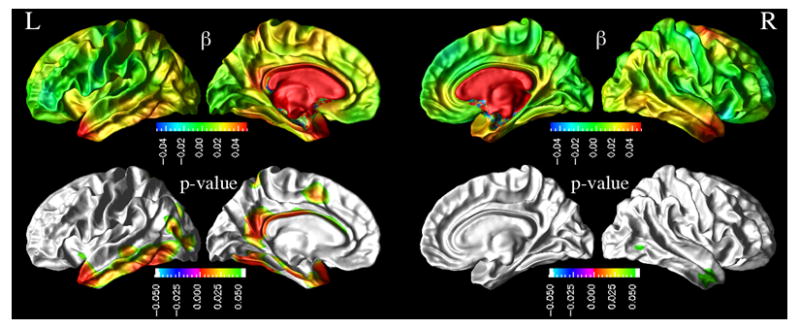
Top row: Distribution of effects of lower baseline CSF Aβ1-42 concentrations on higher rate of cortical thinning in MCI group, expressed in terms of a regression coefficient beta (in mm3/(year*pg/ul)). Bottom row: Also displayed are the corresponding significance maps (p-values, FDR corrected) derived from pair-wise maximum likelihood F-tests between GLME models with and without an interaction between biomarker concentration and atrophy rate.
In Figure 3 are shown the regional distribution of CSF p-tau181p effects on the rates of cortical thinning for MCI as well as the corresponding significance maps. The results indicate that higher baseline CSF p-tau181p concentrations in MCI were associated with higher rates of cortical thinning, significantly in the left temporal pole, left superior temporal sulcus, left entorhinal gyrus, right inferior and middle temporal cortices. Similarly, higher baseline CSF t-tau concentrations were associated with higher cortical atrophy rates in the left entorhinal gyrus in MCI patients, as shown in Figure 4.
Figure 3.

Effects of baseline CSF p-tau181p concentrations on the rates of cortical atrophy in MCI group. [top row: cortical maps of regression coefficients βYears:p-tau181p from GLME models. bottom row: resulting FDR corrected p-value map from pair-wise ML F-tests.]
Figure 4.
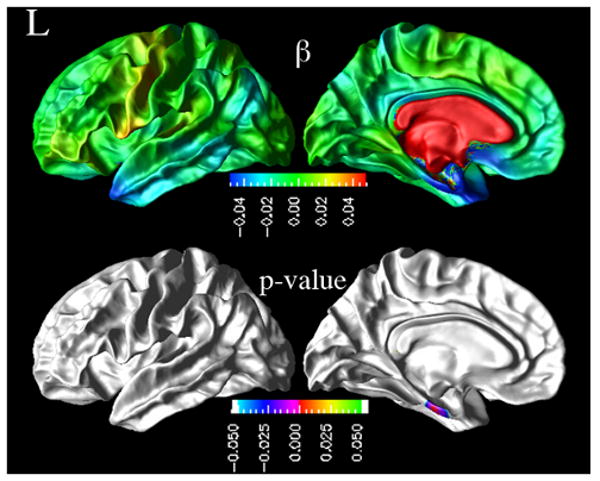
Effects of baseline CSF t-tau concentrations on rate of cortical atrophy in MCI group. [top row: cortical maps of regression coefficients βYears: t-tau from GLME models. bottom row: resulting FDR corrected p-value map from pair-wise ML F-tests.]
Neither CN nor AD patients showed significant modulation effects of baseline CSF biomarkers on the cortical atrophy rates after correcting for multiple comparison. Although they are not statistically significant, cortical maps of regression coefficients of βYears: CSFbiomarker for CN and AD groups are provided in the Supplementary Figure.
Group differences in CSF biomarker effects on rates of cortical thinning
By bootstrapping the random effects residuals of the GLME fits with time and baseline CSF biomarker concentration interaction term, we tested if the estimates of the association between biomarkers and atrophy rates significantly differ across populations. Based on pair-wise group comparison, we found that differences in the estimations between the groups were all significant (p= 0.05 level).
ApoE ε4 Analyses
In our data set, presence of ApoE ε4 alleles correlated significantly with the CSF Aβ1-42 concentrations (r=-0.50, p<10-5 in CN, r=-0.49, p<10-8 in MCI, and r=-0.53, p<10-5 in AD), after controlling for age. A partial correlation between presence of ApoE ε4 alleles and p-tau181p and t-tau after controlling for age was significant only in MCI group (r=0.34 with p<10-3 and r=0.39 with p<10-6, respectively).
In CN group, ML F-test showed that a higher rate of ventricular enlargement was associated independently with higher t-tau as well as presence of ApoE ε4, while lower CSF Aβ1-42 and higher p-tau181p alone explained the increased rate of ventricular enlargement without a significant contribution from ApoE ε4.
In individuals with MCI, CSF biomarker concentrations (i.e., lower Aβ1-42, higher p-tau181p and t-tau) and ApoE ε4 were independently associated with higher rates of ventricular enlargement and tissue volume loss in bilateral hippocampus, right amygdala, bilateral temporal lobe (i.e, middle temporal, inferior temporal, temporal pole, fusiform), right lingual, left entorhinal, left parahippocampal, left isthmus cingulate, and left precuneus cortices. Similarly, lower CSF Aβ1-42 and ApoE ε4 together were associated with higher rates of cortical thinning in temporoparietal cortex, including precuneus and posterior cingulate (Supplementary Figure II). CSF tau and ApoE ε4 together were associated with higher rates of cortical thinning in the entorhinal cortex, precuneus and temporal pole (Supplementary Figure II). In contrast, lower Aβ1-42 alone explained higher rates of tissue volume loss as well as cortical thinning in left entorhinal, fusiform, inferior temporal, temporal pole, and parahippocampal cortices without a significant contribution from ApoE ε4. Similarly, higher CSF tau explained higher rates of cortical thinning in entorhinal cortex without a significant contribution from ApoE e4.
In patients with AD, lower Aβ1-42 and higher t-tau concentrations as well as presence of ApoE ε4 were associated with higher rates of tissue volume loss in the left and right caudate. However, a higher rate of volume loss in the caudate was explained by lower Aβ1-42 alone without a significant contribution of ApoE ε4 status. On the other, ApoE ε4 alone was associated with a higher rate of volume loss in caudate, independent of the CSF t-tau concentration.
In no case did an interaction between ApoE ε4 status and CSF biomarker concentration (i.e. ApoE ε4 status * CSF biomarker) approach significance in predicting rate of volume loss or cortical thinning.
Discussion
We have four major findings: (1) In controls, an association between CSF Aβ1-42 and baseline cortical thickness was observed prominently in regions that generally appear affected in AD. Furthermore, lower CSF Aβ1-42 and higher p-tau181p concentrations were associated with an increase in the rate of ventricular expansion in controls. (2) In MCI subjects, an association was observed between increased CSF tau and decreased baseline caudate volume as well as between lower CSF Aβ1-42 and increased ventricular enlargement. In addition, lower CSF Aβ1-42 and higher p-tau181p and t-tau concentrations were associated with higher rates of brain volume loss in regions of the temporal and parietal cortices and in subcortical regions implicated in AD pathology. (3) In AD patients, lower CSF Aβ1-42 concentration was associated with a decrease in volumes of brain regions not typically implicated in the amyloid pathology of AD. Specifically, lower CSF Aβ1-42 was associated with reduced atrophy rates in caudate and accumbens areas. On the other hand, higher t-tau concentration was associated with increased atrophy rates in regions of the posterior cingulate and precuneus, decreased atrophy rate in caudate, and decreased rate of ventricular enlargement. (4) The relationship among CSF biomarkers, ApoE ε4 status and brain atrophy rates was regionally specific and varied across the clinical groups.
Our finding in controls, demonstrating that low baseline CSF Aβ1-42 biomarker concentration is associated with thinner cortex predominantly in the inferior temporal, parietal, frontal, precuneus, and posterior cingulate cortices, provides evidence for a link between variations in peripheral CSF chemistry and regional brain size. Moreover, the result suggests that the link between CSF markers and regional brain size is already established in absence of any apparent clinical symptoms of cognitive deficits. A previous study in cognitively normal elderly also found an association between low levels of CSF Aβ1-42 and smaller whole-brain volume (Fagan et al., 2009). It has been shown that in healthy elderly individuals, reduction in CSF Aβ1-42 is a predictor of cognitive decline and development of AD (Skoog et al., 2003; Stomrud et al., 2007); therefore, the association between low baseline CSF Aβ1-42 concentration and thin cortex in controls could reflect preclinical AD pathology. However, pathological conditions other than AD might also contribute to the relationship between low CSF Aβ1-42 concentration and cortical thinning.
Another interesting observation in controls is the association between variations in CSF Aβ1-42 and p-tau181p concentrations and the rate of ventricular dilation. There are several explanations for this finding. First, the associations may reflect early brain changes associated with AD pathology before clinical symptoms of dementia become apparent and ventricular dilation may reflect the combinations of all these brain changes, which are likely distributed diffusely throughout the brain at this early stage and therefore difficult to detect with conventional techniques of imaging statistics. Another explanation is that the association between CSF and structural changes in controls are not specifically related to AD pathology but merely reflect a general trend of a relationship between CSF biomarker concentrations and brain structures, including other pathologies that can lead to neurodegeneration such as cerebrovascular diseases. It will be important in future studies to determine the predictive value of the relationship between low CSF Aβ1-42 and high CSF p-tau181p and ventricular dilatation in controls for the development of AD pathology.
Individuals with MCI displayed associations between CSF p-tau181p and t-tau concentrations and baseline volume of subcortical structures, specifically the caudate, while other baseline regional brain volumes or cortical thickness measures had no significant association with CSF biomarker concentrations. This finding implies a dissociation between CSF biomarkers and their effects on the brain in individuals with MCI. The finding in MCI, showing that variations in CSF biomarker concentrations are associated with a characteristic pattern of altered rates of regional brain atrophy, similar to the pattern seen in AD, further supports the view that these relations reflect brain alterations presymptomatic to AD and could be useful for staging disease severity and assessing disease progression. Specifically, lower CSF Aβ1-42 and increased CSF p-tau181p and t-tau concentrations in MCI were associated with higher atrophy rates both in terms of tissue volume loss and cortical thinning involving primarily inferior and medial temporal, parietal, precuneus, and posterior cingulate cortices and tissue volume loss in subcortical structures, including hippocampus, amygdala, and enlargement of ventricles. Structural MRI studies in AD consistently revealed a pattern of neuroanatomic abnormalities that predominantly involves structures in the medial temporal cortex (i.e., hippocampus and the entorhinal cortex (deToledo-Morrell et al., 2004; Du et al., 2001; Du et al., 2004; Du et al., 2003; Hampel et al., 2005; Morra et al., 2008; Morra et al., 2009a; Morra et al., 2009b; Schroeter et al., 2009; Stoub et al., 2005; Thompson et al., 2004)) where the early pathological changes are seen, then gradually extends to temporoparietal cortical areas (Chetelat and Baron, 2003; Desikan et al., 2008; Hua et al., 2008; Whitwell et al., 2007; Whitwell et al., 2008) as severity of AD progresses (DeCarli et al., 2007; Jack et al., 2005; Jack et al., 2008b; Jack et al., 2004; Whitwell et al., 2007; Whitwell et al., 2008). Our finding that lower CSF Aβ1-42 and higher CSF p-tau181p and t-tau concentrations were associated with higher atrophy rates of the temporal horn and inferior temporal lobe regions points to a selective vulnerability of these regions to AD pathology, consistent with histopathological findings. The finding that lower CSF Aβ1-42 is associated with a characteristic pattern of brain atrophy in MCI that resembles the atrophy pattern seen in AD is encouraging for the use of CSF Aβ1-42 as an early indicator of AD. Most importantly, elucidating the detrimental relationship between CSF biomarkers and rates of brain atrophy is of great interest to detect AD pathology in early stage, which is fundamental for an accurate early diagnosis of the disease, development of new treatment interventions, and evaluation of clinical trials in AD. The synergistic relationship between CSF biomarker and neurodegeneration patterns are of clinical interest as they may not only improve monitoring AD progression and evaluation of new AD therapies but also aid enrichment of clinical trial cohorts by identifying specific subsets of patients with MCI especially at high risk of developing AD (Blennow and Hampel, 2003; Hampel et al., 2003; John, 2001). Such a custom tailored cohort selection is desirable since drugs with disease-arresting effects have better efficacy in the preclinical and early phase of the disease before the synaptic and neuronal loss become widespread (Shaw et al., 2007).
The spatial extent of baseline CSF Aβ1-42 modulation effects on brain atrophy and ventricular expansion observed in individuals with MCI is consistent with previous volumetric studies on patterns of increased atrophy rate in AD patients compared to elderly healthy controls (Scahill et al., 2002). In addition, a prior autopsy study on AD patients (Arnold et al., 1991) reported neuritic plaques distributed throughout the cortex with the highest densities in the temporal and occipital lobes, while relatively lower plaque densities were found in the parietal lobe. This is consistent with our CSF Aβ1-42 modulation effect findings in MCI. Compared to the prior autopsy study in AD patients (Arnold et al., 1991), the spatial extents of CSF p-tau181p and t-tau modulation effects are consistent with the neurofibrillary tangle distributions in AD pathology. In particular, hippocampus and cortical regions surrounding entorhinal cortex and amygdala were reported as the most severely affected areas by neurofibrillary tangles. However, since we do not know how many of the MCI subjects in this study will ultimately develop AD, we cannot determine the predictive value of CSF biomarker concentrations for AD. Another observation in the MCI group was the left hemisphere dominance of the CSF biomarkers' atrophy modulation effects. This observation is consistent with the asymmetric loss of GM (i.e., the left hemisphere atrophies faster than the right hemisphere) in AD (Thompson et al., 2003) and yet again supporting the hypothesis that CSF Aβ1-42, p-tau181p, and t-tau are measures of early AD pathology.
In AD patients, lower baseline CSF Aβ1-42 concentration is associated with smaller baseline GM tissue volumes in lingual, pericalcarine, and post central cortices, which are not traditionally associated with AD pathology. Another surprising finding in AD patients was that abnormal CSF biomarker levels were not significantly associated with rates of cortical thinning, despite positive findings of an association between CSF Aβ1-42, as well as, t-tau levels and rates of volume loss in the caudate, posterior cingulate and precuneus. Similarly, abnormal CSF biomarker levels in controls were also not significantly associated with rates of cortical thinning but showed an association with ventricular enlargement. The difference between the CSF biomarker relations to cortical thinning and volume loss could be related to differences in MRI sensitivity for detection of volume loss versus cortical thinning. It is also plausible, however, that the difference in AD is the result of a complex relationship between advanced AD pathology in most cortical regions and peripheral biomarker concentration, while the difference in controls might reflect a threshold effect of minimum biomarker concentration on cortical thinning and volume loss. More studies are warranted to further investigate these issues.
Compared to CN and MCI groups, CSF biomarker concentrations have opposite modulation effects on rates of brain tissue volume change in AD. Specifically, we observed decreased rates of atrophy in the caudate and accumbens area in the presence of lower concentrations of CSF Aβ1-42 (relative to the CSF Aβ1-42 concentration distribution in AD group), as well as, decreased rates of atrophy in the right posterior cingulate and precuneus cortices and ventricular enlargement in the presence of higher concentrations of baseline CSF t-tau (relative to the CSF t-tau concentration distribution in AD group). Although not statistically significant, similar trends were observed in cortical atrophy analysis as well, as shown in the supplementary figure. One explanation of this finding could be the disease-stage specific effects of CSF biomarkers on brain atrophy. AD subjects with lower CSF Aβ1-42 and higher CSF t-tau concentrations (relative to the biomarker concentration distributions in AD group) probably have advanced AD pathology where they reach a plateau in their rate of volume loss, which appears as a slower progression of the brain atrophy.
An interesting finding was that relations between the rate of brain atrophy and CSF biomarker concentrations varied across the CSF Aβ1-42, p-tau181p, and t-tau. This is not unexpected because it is known that amyloid plaques and tau containing tangles are distributed discordantly in brain at early stages of the disease. Specifically, the accumulation of amyloid plaques occurs in cortical regions whereas tangles appear in subcortical structures, predominantly involving the hippocampus (Arnold et al., 1991; Braak and Braak, 1991; Price and Morris, 1999). Our findings reflect this pattern to some extent. However, the association between elevated CSF tau concentration and caudate atrophy in MCI and AD is an unexpected finding. Recent amyloid imaging studies using PiB-PET reported substantial amyloid deposition in the striatum, including the caudate, in symptomatic and asymptomatic subjects carrying the presenilin-1 (PS1) mutation gene for familial AD (Klunk et al., 2007). It is therefore possible that our finding with respect to caudate atrophy can be explained by the heterogeneity of MCI and AD study groups, which might include subjects with early onset AD.
Finally, the finding of regional variations among CSF biomarkers, ApoE ε4 status, and brain atrophy rate relationships support the view that the genetic predisposition of the brain to amyloid and tau mediated pathology is region and disease stage specific. Interestingly, the most prominent region associated with CSF biomarker regardless of ApoE ε4 status in MCI included the entorhinal cortex, which is thought to be affected early by AD. Moreover, the effect of ApoE ε4 status on the relationships could be dose dependent (Andersson et al., 2007; Glodzik-Sobanska et al., 2009; Sunderland et al., 2004).
Our findings in this study are largely consistent with several similar studies on relationships between CSF biomarkers and brain alterations in MCI and AD (Hampel et al., 2005; Henneman et al., 2009; Herukka et al., 2008). Specifically, increased concentrations of CSF p-tau181p are associated with higher subsequent rates of hippocampal atrophy in the progressive MCI and AD patients (Hampel et al., 2005; Henneman et al., 2009) (Herukka et al., 2008), of medial temporal atrophy in AD patients (Leow et al., 2009), of the temporal and parietal atrophy in MCI (Fjell et al., 2010b), and of right posterior ventricular horn expansion (Chou et al., 2009). In contrast, low CSF Aβ1-42 concentration exhibited an association with increased rate of left hippocampal atrophy in the progressive MCI patients (Fjell et al., 2010b; Herukka et al., 2008), of the medial temporal atrophy in AD (Leow et al., 2009), of the temporal and parietal atrophy in MCI (Fjell et al., 2010b), and of ventricular expansion (Chou et al., 2009). Elevated CSF t-tau concentrations are associated with higher rates of hippocampal atrophy in stable MCI patients (Herukka et al., 2008). In controls, it has been shown that low CSF Aβ1-42 concentration correlates with ventricular expansion and volumetric reductions in widespread brain areas, including inferior temporal, inferior parietal, frontal, posterior cingulate, precuneus, caudate, and amygdala regions (Fjell et al., 2010a). Fjell et al. reported generally larger effects of CSF biomarkers on brain tissue change than what we found on the same cohort (Fjell et al., 2010a; Fjell et al., 2010b). However, several methodological differences between our study and that by Fjell et al. complicate direct comparisons. For example, whereas Fjell et al. aimed to evaluate potentially accelerated rates between the first and second scan intervals while accepting mixed effects on rates, we aimed to separate random from fixed effects on rates in order to boost sensitivity while ignoring the possibility of accelerated rates. Since each approach has its estimation bias, the different findings are difficult to interpret.
The majority of previous MRI studies, except (Fjell et al., 2010a; Fjell et al., 2010b), in this context focused on the hippocampal and temporal lobe atrophy and ventricular enlargement, while our approach was generalized by assessing various other brain regions. Based on this, we discovered that the association between CSF biomarkers and structural changes are regionally differential. Although this observation is not entirely surprising, given that the pathological processes of plaque and tangle formation, which CSF Aβ1-42 and p-tau181p and t-tau indirectly represent, respectively. The finding of faster progression of brain atrophy in presence of lower baseline concentrations of Aβ1-42 and higher concentrations of p-tau181p/t-tau in MCI together with the similarities between the MCI pattern of CSF biomarker atrophy modulation effects and distribution of tangles and plaques in AD support the hypothesis that CSF biomarkers are measures of early AD pathology. MCI pattern of relations between rate of brain atrophy and CSF biomarker concentrations should be further explored to identify possible pre-symptomatic AD pathology. This finding also suggests a strategy for the potential use of biomarkers in clinical trials. For example, CSF Aβ1-42 concentration could be used to assess the effect of disease modifying interventions on cortical regions while CSF p-tau181p and t-tau concentrations could be used to assess effects on subcortical structures, while both biomarkers could be used together to determine if interventions affect cortical and subcortical brain structures differentially. CSF biomarker cut-offs to select fastest progressing cohorts could greatly improve the power of AD prevention trials on healthy elderly and MCI.
Several limitations of our study ought to be mentioned. First, MCI and AD subjects were diagnosed clinically; therefore other pathologies may have contributed to their symptoms and the relationships between CSF biomarkers and brain alterations may be unrelated to AD pathology. Another limitation is that CSF biomarkers, especially CSF Aβ1-42, have been shown to be saturated and may not accurately reflect severity of brain amyloid deposition or plaque density in the later stages of the disease (Andreasen et al., 1999; Stefani et al., 2006). Therefore, structural brain changes may still occur secondarily to ongoing amyloid deposition or plaque accumulation. Restriction to linear, time-invariant brain atrophy rates is a technical limitation of our study. This is likely a gross simplification because the loss of brain tissue may be compounding and furthermore neurodegeneration in AD may be a dynamic process, which varies during disease progression. Therefore, models with nonlinear atrophy rate characteristics might lead to different results; however, such models are not always robust, given the limited number of serial MRI measurements and they also require careful validation. Finally, another technical limitation is that our study included fewer CN than MCI subjects despite expectations that power to detect atrophy will be higher for MCI than in CN because of higher atrophy rates in MCI. Therefore, comparisons between CN and MCI (and AD) could be biased toward lower sensitivity to detect a change in CN.
In summary, our findings demonstrate that alterations in CSF Aβ1-42, p-tau181p, and t-tau are each associated with characteristic patterns of structural brain changes (cross-sectionally or longitudinally) in CN and MCI that resembles to a large extent the pattern seen in AD pathology. Specifically, the finding of faster progression of brain atrophy in individuals with MCI in the presence of lower baseline CSF Aβ1-42 and higher CSF tau levels supports the view that these CSF biomarkers reflect AD brain pathology. Since the CSF Aβ1-42 and tau levels were also associated with a systematic pattern of regional brain atrophy rates that resembled the pattern known in AD, our findings further support the view that CSF Aβ1-42 and tau reflect brain damage due to AD pathology. Overall, the findings imply that CSF Aβ1-42 and tau taken together with MRI measures of rates of brain atrophy progression are promising candidates as biomarkers for early detection of AD.
Supplementary Material
Acknowledgments
This work is funded by the National Institutes of Health (NIH), National Institute of Biomedical Imaging and Bioengineering (NIBIB) [T32 EB001631-05].
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., and Wyeth, as well as non-profit partners the Alzheimer's Association and Alzheimer's Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org/). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation.
Footnotes
Disclosure Statement: Dr. Tosun, Ms. Truran-Sacrey, Dr. Shaw, and Dr. Trojanowski report no disclosures.
Dr. Aisen has served as a consultant to Pfizer, Merck, and Novartis.
Dr. Schuff received honorary from the Michael J Fox foundation, the British Research Council and Elsevier Publishing company; receives research support from M.J. Fox foundation, Department of Defense (WX), P41 RR023953 (Coinvestigator); P50AG23501 (Coninvestigator).
Dr. Petersen serves as a consultant to Elan Pharmaceuticals, Wyeth Pharmaceuticals, and GE Healthcare; receives royalties from publishing Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIA [AG 06786 (PI) and AG 16574 (PI)].
Dr. Weiner serves on scientific advisory boards for Bayer Schering Pharma, Eli Lilly, Nestle, CoMentis, Neurochem, Eisai, Avid, Aegis, Genentech, Allergan, Lippincott, Bristol Meyers Squibb, Forest, Pfizer, McKinsey, Mitsubishi, and Novartis. He has received non–industry-supported funding for travel; serves on the editorial board of Alzheimer's & Dementia; received honoraria from the Rotman Research Institute and BOLT International; receives research support from Merck & Co, Avid, NIH [U01AG024904 (PI), P41 RR023953 (PI), R01 AG10897 (PI), P01AG19724 (Coinvestigator), P50AG23501(Coinvestigator), R24 RR021992 (Coinvestigator), R01 NS031966 (Coinvestigator), and P01AG012435 (Coinvestigator)], the Department of Defense [DAMD17-01-1-0764 (PI)], and the Veterans Administration [MIRECC VISN 21 (Core PI)]; and holds stock in Synarc and Elan Pharmaceuticals.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE. Frequent Amyloid Deposition Without Significant Cognitive Impairment Among the Elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson C, Blennow K, Johansson SE, Almkvist O, Engfeldt P, Lindau M, Eriksdotter-Jonhagen M. Differential CSF Biomarker Levels in APOE- e4-Positive and -Negative Patients with Memory Impairment. Dementia and Geriatric Cognitive Disorders. 2007;23:87–95. doi: 10.1159/000097354. [DOI] [PubMed] [Google Scholar]
- Andreasen N, Minthon L, Vanmechelen E, Vanderstichele H, Davidsson P, Winblad B, Blennow K. Cerebrospinal fluid tau and A[beta]42 as predictors of development of Alzheimer's disease in patients with mild cognitive impairment. Neuroscience Letters. 1999;273:5–8. doi: 10.1016/s0304-3940(99)00617-5. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The Topographical and Neuroanatomical Distribution of Neurofibrillary Tangles and Neuritic Plaques in the Cerebral Cortex of Patients with Alzheimer's Disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- Basso M, Gelernter J, Yang J, MacAvoy MG, Varma P, Bronen RA, van Dyck CH. Apolipoprotein E epsilon4 is associated with atrophy of the amygdala in Alzheimer's disease. Neurobiology of Aging. 2006;27:1416–1424. doi: 10.1016/j.neurobiolaging.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. The Lancet Neurology. 2003;2:605–613. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brys M, Glodzik L, Mosconi L, Switalski R, De Santi S, Pirraglia E, Rich K, Kim BC, Mehta P, Zinkowski R, Pratico D, Wallin A, Zetterberg H, Tsui WH, Rusinek H, Blennow K, de Leon MJ. Magnetic Resonance Imaging Improves Cerebrospinal Fluid Biomarkers in the Early Detection of Alzheimer's Disease. Journal of Alzheimer's Disease. 2009;16:351–362. doi: 10.3233/JAD-2009-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat Ga, Baron JC. Early diagnosis of alzheimer's disease: contribution of structural neuroimaging. NeuroImage. 2003;18:525–541. doi: 10.1016/s1053-8119(02)00026-5. [DOI] [PubMed] [Google Scholar]
- Chou YY, LeporÈ N, Avedissian C, Madsen SK, Parikshak N, Hua X, Shaw LM, Trojanowski JQ, Weiner MW, Toga AW, Thompson PM. Mapping correlations between ventricular expansion and CSF amyloid and tau biomarkers in 240 subjects with Alzheimer's disease, mild cognitive impairment and elderly controls. NeuroImage. 2009;46:394–410. doi: 10.1016/j.neuroimage.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, Morris JC, McKeel DW, Jr, Farlow M, Weitlauf SL, Quinn J, Kaye J, Knopman D, Arai H, Doody RS, DeCarli C, Leight S, Lee VMY, Trojanowski JQ. Cerebrospinal Fluid Tau and {beta}-Amyloid: How Well Do These Biomarkers Reflect Autopsy-Confirmed Dementia Diagnoses? Arch Neurol. 2003;60:1696–1702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- DeCarli C, Frisoni GB, Clark CM, Harvey D, Grundman M, Petersen RC, Thal LJ, Jin S, Jack CR, Jr, Scheltens P, Alzheimer's Disease Cooperative Study Group Qualitative Estimates of Medial Temporal Atrophy as a Predictor of Progression From Mild Cognitive Impairment to Dementia. Arch Neurol. 2007;64:108–115. doi: 10.1001/archneur.64.1.108. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Fischl B, Cabral HJ, Kemper TL, Guttmann CRG, Blacker D, Hyman BT, Albert MS, Killiany RJ. MRI measures of temporoparietal regions show differential rates of atrophy during prodromal AD. Neurology. 2008;71:819–825. doi: 10.1212/01.wnl.0000320055.57329.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deToledo-Morrell L, Stoub TR, Bulgakova M, Wilson RS, Bennett DA, Leurgans S, Wuu J, Turner DA. MRI-derived entorhinal volume is a good predictor of conversion from MCI to AD. Neurobiology of Aging. 2004;25:1197–1203. doi: 10.1016/j.neurobiolaging.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Du AT, Schuff N, Amend D, Laakso MP, Hsu YY, Jagust WJ, Yaffe K, Kramer JH, Reed B, Norman D, Chui HC, Weiner MW. Magnetic resonance imaging of the entorhinal cortex and hippocampus in mild cognitive impairment and Alzheimer's disease. Journal of Neurology, Neurosurgery & Psychiatry. 2001;71:441–447. doi: 10.1136/jnnp.71.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Kramer JH, Ganzer S, Zhu XP, Jagust WJ, Miller BL, Reed BR, Mungas D, Yaffe K, Chui HC, Weiner MW. Higher atrophy rate of entorhinal cortex than hippocampus in AD. Neurology. 2004;62:422–427. doi: 10.1212/01.wnl.0000106462.72282.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Laakso MP, Zhu XP, Jagust WJ, Yaffe K, Kramer JH, Miller BL, Reed BR, Norman D, Chui HC, Weiner MW. Effects of subcortical ischemic vascular dementia and AD on entorhinal cortex and hippocampus. Neurology. 2002;58:1635–1641. doi: 10.1212/wnl.58.11.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Zhu XP, Jagust WJ, Miller BL, Reed BR, Kramer JH, Mungas D, Yaffe K, Chui HC, Weiner MW. Atrophy rates of entorhinal cortex in AD and normal aging. Neurology. 2003;60:481–486. doi: 10.1212/01.wnl.0000044400.11317.ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte A, Hayasaka S, Du A, Schuff N, Jahng GH, Kramer J, Miller B, Weiner M. Volumetric correlates of memory and executive function in normal elderly, mild cognitive impairment and Alzheimer's disease. Neuroscience Letters. 2006;406:60–65. doi: 10.1016/j.neulet.2006.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased Cerebrospinal Fluid A beta(42) Correlates with Brain Atrophy in Cognitively Normal Elderly. Annals of Neurology. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11050–11055. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole Brain Segmentation: Automated Labeling of Neuroanatomical Structures in the Human Brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM. Automatically Parcellating the Human Cerebral Cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Amlien I, Bjornerud A, Reinvang I, Gjerstad L, Cappelen T, Willoch F, Due-Tonnessen P, Grambaite R, Skinningsrud A, Stenset V, Fladby T. Morphometric Changes in the Episodic Memory Network and Tau Pathologic Features Correlate with Memory Performance in Patients with Mild Cognitive Impairment. AJNR Am J Neuroradiol. 2008;29:1183–1189. doi: 10.3174/ajnr.A1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Blennow K, Brewer JB, Dale AM, Alzheimer's Disease Neuroimaging Initiative Brain Atrophy in Healthy Aging Is Related to CSF Levels of A{beta}1-42. Cereb Cortex. 2010a:279. doi: 10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM, Alzheimer's Disease Neuroimaging Initiative CSF Biomarkers in Prediction of Cerebral and Clinical Change in Mild Cognitive Impairment and Alzheimer's Disease. J Neurosci. 2010b;30:2088–2101. doi: 10.1523/JNEUROSCI.3785-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher A, Grundman M, Jack CR, Jr, Petersen RC, Taylor C, Kim HT, Schiller DHB, Bagwell V, Sencakova D, Weiner MF, DeCarli C, DeKosky ST, van Dyck CH, Thal LJ, Alzheimer's Disease Cooperative Study Sex, Apolipoprotein E {varepsilon}4 Status, and Hippocampal Volume in Mild Cognitive Impairment. Arch Neurol. 2005;62:953–957. doi: 10.1001/archneur.62.6.953. [DOI] [PubMed] [Google Scholar]
- Glodzik-Sobanska L, Pirraglia E, Brys M, de Santi S, Mosconi L, Rich KE, Switalski R, Louis LS, Sadowski MJ, Martiniuk F, Mehta P, Pratico D, Zinkowski RP, Blennow K, de Leon MJ. The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer's disease. Neurobiology of Aging. 2009;30:672–681. doi: 10.1016/j.neurobiolaging.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Burger K, Pruessner JC, Zinkowski R, DeBernardis J, Kerkman D, Leinsinger G, Evans AC, Davies P, Moller HJ, Teipel SJ. Correlation of Cerebrospinal Fluid Levels of Tau Protein Phosphorylated at Threonine 231 With Rates of Hippocampal Atrophy in Alzheimer Disease. Arch Neurol. 2005;62:770–773. doi: 10.1001/archneur.62.5.770. [DOI] [PubMed] [Google Scholar]
- Hampel H, Teipel SJ, Fuchsberger T, Andreasen N, Wiltfang J, Otto M, Shen Y, Dodel R, Du Y, Farlow M, Moller HJ, Blennow K, Buerger K. Value of CSF [beta]-amyloid1-42 and tau as predictors of Alzheimer's disease in patients with mild cognitive impairment. Mol Psychiatry. 2003;9:705–710. doi: 10.1038/sj.mp.4001473. [DOI] [PubMed] [Google Scholar]
- Henneman WJP, Vrenken H, Barnes J, Sluimer IC, Verwey NA, Blankenstein MA, Klein M, Fox NC, Scheltens P, Barkhof F, van der Flier WM. Baseline CSF p-tau levels independently predict progression of hippocampal atrophy in Alzheimer disease. Neurology. 2009;73:935–940. doi: 10.1212/WNL.0b013e3181b879ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herukka SK, Pennanen C, Soininen H, Pirttilä T. CSF Aβ42, Tau and Phosphorylated Tau Correlate with Medial Temporal Lobe Atrophy. Journal of Alzheimer's Disease. 2008;14:51–57. doi: 10.3233/jad-2008-14105. [DOI] [PubMed] [Google Scholar]
- Hua X, Leow AD, Lee S, Klunder AD, Toga AW, Lepore N, Chou YY, Brun C, Chiang MC, Barysheva M, Jack CR, Jr, Bernstein MA, Britson PJ, Ward CP, Whitwell JL, Borowski B, Fleisher AS, Fox NC, Boyes RG, Barnes J, Harvey D, Kornak J, Schuff N, Boreta L, Alexander GE, Weiner MW, Thompson PM, Alzheimer's Disease Neuroimaging, I 3D characterization of brain atrophy in Alzheimer's disease and mild cognitive impairment using tensor-based morphometry. NeuroImage. 2008;41:19–34. doi: 10.1016/j.neuroimage.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, Whitwell JL, Ward C, Dale AM, Felmlee JP, Gunter JL, Hill DLG, Killiany R, Schuff N, Fox-Bosetti S, Lin C, Studholme C, DeCarli CS, Krueger G, Ward HA, Metzger GJ, Scott KT, Mallozzi R, Blezek D, Levy J, Debbins JP, Fleisher AS, Albert M, Green R, Bartzokis G, Glover G, Mugler J, Weiner MW, Study A. The Alzheimer's disease neuroimaging initiative (ADNI): MRI methods. Journal of Magnetic Resonance Imaging. 2008a;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC, Alzheimer's Disease Neuroimaging Initiative Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Shiung MM, Weigand SD, O'Brien PC, Gunter JL, Boeve BF, Knopman DS, Smith GE, Ivnik RJ, Tangalos EG, Petersen RC. Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology. 2005;65:1227–1231. doi: 10.1212/01.wnl.0000180958.22678.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Weigand SD, Shiung MM, Przybelski SA, O'Brien PC, Gunter JL, Knopman DS, Boeve BF, Smith GE, Petersen RC. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology. 2008b;70:1740–1752. doi: 10.1212/01.wnl.0000281688.77598.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Shiung MM, Gunter JL, O'Brien PC, Weigand SD, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Cha RH, Tangalos EG, Petersen RC. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology. 2004;62:591–600. doi: 10.1212/01.wnl.0000110315.26026.ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John HG. Incorporating biomarkers into clinical drug trials in Alzheimer's disease. Journal of Alzheimer's Disease. 2001;3:287–292. doi: 10.3233/jad-2001-3303. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST. Amyloid Deposition Begins in the Striatum of Presenilin-1 Mutation Carriers from Two Unrelated Pedigrees. J Neurosci. 2007;27:6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Schuff N, Reed BR, Mungas D, Du AT, Rosen HJ, Jagust WJ, Miller BL, Weiner MW, Chui HC. Hippocampal volume and retention in Alzheimer's disease. Journal of the International Neuropsychological Society. 2004;10:639–643. doi: 10.1017/S1355617704104050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leow AD, Yanovsky I, Parikshak N, Hua X, Lee S, Toga AW, Jack CR, Jr, Bernstein MA, Britson PJ, Gunter JL, Ward CP, Borowski B, Shaw LM, Trojanowski JQ, Fleisher AS, Harvey D, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Alzheimer's Disease Neuroimaging Initiative: A one-year follow up study using tensor-based morphometry correlating degenerative rates, biomarkers and cognition. NeuroImage. 2009;45:645–655. doi: 10.1016/j.neuroimage.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun MA, LaRossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Validation of a fully automated 3D hippocampal segmentation method using subjects with Alzheimer's disease mild cognitive impairment, and elderly controls. NeuroImage. 2008;43:59–68. doi: 10.1016/j.neuroimage.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jr, J CR, Schuff N, Weiner MW, Thompson PM, Initiative, a.t.A.s.D.N Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer's disease, mild cognitive impairment, and elderly controls. Human Brain Mapping. 2009a;30:2766–2788. doi: 10.1002/hbm.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Toga AW, Jack CR, Jr, Schuff N, Weiner MW, Thompson PM. Automated mapping of hippocampal atrophy in 1-year repeat MRI data from 490 subjects with Alzheimer's disease, mild cognitive impairment, and elderly controls. NeuroImage. 2009b;45:S3–S15. doi: 10.1016/j.neuroimage.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K. Simultaneous Measurement of {beta}-Amyloid(1-42), Total Tau, and Phosphorylated Tau (Thr181) in Cerebrospinal Fluid by the xMAP Technology. Clin Chem. 2005;51:336–345. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild Cognitive Impairment: Clinical Characterization and Outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH, Saykin AJ, Orro A, Lupoli S, Salvi E, Weiner M, Macciardi F, Alzheimer's Disease Neuroimaging, I Hippocampal Atrophy as a Quantitative Trait in a Genome-Wide Association Study Identifying Novel Susceptibility Genes for Alzheimer's Disease. PLoS ONE. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and ldquopreclinicalrdquo Alzheimer's disease. Annals of Neurology. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC. Mapping the evolution of regional atrophy in Alzheimer's disease: Unbiased analysis of fluid-registered serial MRI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter ML, Stein T, Maslowski N, Neumann J. Neural correlates of Alzheimer's disease and mild cognitive impairment: a systematic and quantitative meta-analysis involving 1351 patients. NeuroImage. 2009;47:1196–1206. doi: 10.1016/j.neuroimage.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR, Jr, Weiner MW, Alzheimer's Disease Neuroimaging Initiative MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009;132:1067–1077. doi: 10.1093/brain/awp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Korecka M, Clark CM, Lee VMY, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007;6:295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VMY, Trojanowski JQ, Initiative, A.s.D.N. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Annals of Neurology. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog I, Davidsson P, Aevarsson ì, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal Fluid Beta-Amyloid 42 Is Reduced before the Onset of Sporadic Dementia: A Population-Based Study in 85-Year-Olds. Dementia and Geriatric Cognitive Disorders. 2003;15:169–176. doi: 10.1159/000068478. [DOI] [PubMed] [Google Scholar]
- Sluimer JD, Vrenken H, Blankenstein MA, Fox NC, Scheltens P, Barkhof F, van der Flier WM. Whole-brain atrophy rate in Alzheimer disease: Identifying fast progressors. Neurology. 2008;70:1836–1841. doi: 10.1212/01.wnl.0000311446.61861.e3. [DOI] [PubMed] [Google Scholar]
- Stefani A, Martorana A, Bernardini S, Panella M, Mercati F, Orlacchio A, Pierantozzi M. CSF markers in Alzheimer disease patients are not related to the different degree of cognitive impairment. Journal of the Neurological Sciences. 2006;251:124–128. doi: 10.1016/j.jns.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Stomrud E, Hansson O, Blennow K, Minthon L, Londos E. Cerebrospinal Fluid Biomarkers Predict Decline in Subjective Cognitive Function over 3 Years in Healthy Elderly. Dementia and Geriatric Cognitive Disorders. 2007;24:118–124. doi: 10.1159/000105017. [DOI] [PubMed] [Google Scholar]
- Stoub TR, Bulgakova M, Leurgans S, Bennett DA, Fleischman D, Turner DA, deToledo-Morrell L. MRI predictors of risk of incident Alzheimer disease: A longitudinal study. Neurology. 2005;64:1520–1524. doi: 10.1212/01.WNL.0000160089.43264.1A. [DOI] [PubMed] [Google Scholar]
- Sunderland T, Mirza N, Putnam KT, Linker G, Bhupali D, Durham R, Soares H, Kimmel L, Friedman D, Bergeson J, Csako G, Levy JA, Bartko JJ, Cohen RM. Cerebrospinal fluid [beta]-amyloid1-42 and tau in control subjects at risk for Alzheimer's disease: The effect of APOE [epsilon]4 allele. Biological Psychiatry. 2004;56:670–676. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, de Zubicaray G, Janke AL, Rose SE, Semple J, Herman D, Hong MS, Dittmer SS, Doddrell DM, Toga AW. Dynamics of Gray Matter Loss in Alzheimer's Disease. J Neurosci. 2003;23:994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, de Zubicaray GI, Janke AL, Rose SE, Semple J, Hong MS, Herman DH, Gravano D, Doddrell DM, Toga AW. Mapping hippocampal and ventricular change in Alzheimer disease. NeuroImage. 2004;22:1754–1766. doi: 10.1016/j.neuroimage.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Jr, Alzheimer's Disease Neuroimaging Initiative MRI and CSF biomarkers in normal, MCI, and AD subjects: Diagnostic discrimination and cognitive correlations. Neurology. 2009a;73:287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Jr, Alzheimer's Disease Neuroimaging Initiative MRI and CSF biomarkers in normal, MCI, and AD subjects: Predicting future clinical change. Neurology. 2009b;73:294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer's disease. Brain. 2007;130:1777–1786. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Shiung MM, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology. 2008;70:512–520. doi: 10.1212/01.wnl.0000280575.77437.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.