

Abstract
Dendrites, the elaborate processes emerging from neuronal cell bodies, receive most excitatory synaptic inputs. Voltage- and calcium-gated ion channels are abundant in dendrites and modify the shape, propagation and integration of synaptic signals. These ion channels also determine intrinsic dendritic excitability and are therfore important for the induction and manifestation of Hebbian and non-Hebbian plasticity. Revealingly, dendritic channels have distinct expression patterns and biophysical properties from those present in other neuronal compartments. Recent evidence suggests that dendritic ion channels are locally regulated, perhaps contributing to different forms of plasticity. In this review, we will discuss the implications of regulating dendritic ion channel function and trafficking in the context of plasticity and information processing.
1. Introduction
Dendrites are extensive and elaborate processes emerging from the cell body of neurons. They occupy a large surface area and receive the majority of synaptic inputs1. Their predominant function is in processing and transmitting synaptic signals to the cell body and axon initial segment, where, if threshold is reached, action potentials are initiated. This is an active process, as it is now known that dendrites possess an abundance of ion channels that are involved in receiving, transforming and relaying information to other parts of the neuron1. These dendritic ion channels often differ in their biophysical properties and densities from those present in other neuronal compartments. Moreover, ion channel expression and properties may also differ within the dendritic arbor of a particular neuron – e.g. hyperpolarization-activated cation non-selective (HCN) channels are expressed highly in the apical, but not the basal, dendritic tree of layer V cortical pyramidal neurons2-4. This adds an additional layer of complexity to neuronal information processing.
Recent evidence suggests that selective targeting mechanisms determine the distribution and properties of dendritic ion channels5. Specific molecules are involved in the transport of voltage-gated ion channel subunits from the soma to dendrites. Additionally, mRNA can be trafficked into dendrites and locally translated, a process which may be activity-dependent6. Indeed, dendrites contain the necessary machinery that enables local protein synthesis7. Hence, expression of ion channels can be dynamically regulated in dendrites in response to changes in either pre- or post-synaptic activity and may present a sophisticated mechanism by which neurons gate information flow and thereby, neuronal output.
Changes in synaptic strength or connectivity are commonly thought to influence cellular information processing and thus, neural circuitry8. Modifications in synaptic drive can lead to various forms of plasticity, including long term potentiation (LTP) and long term depression (LTD) (see Glossary Box). To keep neuronal activity levels steady, the gain of individual synaptic strengths can be varied to oppose the change in synaptic drive, known as homeostatic plasticity. In addition, the properties and expression of voltage-gated ion channels can be persistently modulated, referred to as intrinsic plasticity (see Glossary Box).
Consequently, neuronal firing patterns may adjust, leading to changes in neural network activity and potentially physiological processes such as learning and memory. Intriguingly, this can occur at the level of individual branches of the dendritic tree9. Moreover, new evidence suggests that these changes can be due to altered trafficking of dendritic ion channels10-19.
In this review, we discuss our current knowledge of dendritic ion channel trafficking and its relationship to cellular and synaptic plasticity and the implications for neuronal function. We argue that to gain insight into neuronal computation, it is essential to understand the regulation of dendritic ion channel expression and function. Since there have been many recent reviews on ligand--gated ion channel trafficking and plasticity (for example see20, 21), here we will focus on dendritic voltage-gated ion channel targeting mechanisms and plasticity.
2 Dendritic K+ channel targeting and plasticity
Potassium (K+) channels are essential for the regulation of neuronal excitability. In neurons, they are involved in a variety of functions including setting of the resting membrane potential, controlling the initiation and shape of single action potentials and determining the amplitude and kinetics of the afterhyperpolarization that follows an action potential22. These K+ channel functions are achieved through their molecular diversity and by dynamic regulation of their membrane distribution and biophysical properties. K+ channels consist of primary pore-forming (α) subunits and auxiliary (β) subunits. Currently, over 70 different genes encoding for the K+ channel α subunit are known. However, based on the structure of the α subunit, K+ channels can be broadly classified into three main families: the six transmembrane (6TM) subunit, the four transmembrane (4TM) subunit and the two transmembrane (2TM subunit families23. The voltage-gated K+ (Kv) and calcium-activated K+ (KCa) channels fall into the 6TM family while the tandem two pore-domain (K2P) and inward rectifiers (Kir) form the 4TM and 2TM families respectively. Dendrites of central neurons contain members of all three families. Here we present recent findings from three of the four major classes of K+ channels, omitting background tandem two-pore K+ channels (K2P), which were the centre of recent reviews24.
2.1 Kv channels
In many central neurons, the densities of most Kv channels appear to be uniform or lower in distal dendrites compared with those present at the soma1. The exception appears to be the Kv4 subunit. Immunohistochemical analysis first showed that the Kv4 subunit was expressed predominantly in dendrites of several central neurons including hippocampal CA1 pyramidal, cerebellar granule and olfactory mitral cells25-27. The Kv4 subunits form a fast activating and inactivating current in heterologous systems, reminiscent of the A-type K+ current (IA) in neurons28. Consistent with the immunohistochemical observations, electrophysiological data together with pharmacology and voltage-sensitive and calcium imaging have shown that the A-type K+ channel is present at a higher density in the apical29-32 radial oblique9, 33-35 and basal4, 9, 36 dendrites than the soma of several types of central neurons. There, they play an important role in determining the amplitude and width of back-propagating action potentials29, 30, 32, 34, 36, 37. They also limit the propagation of local dendritic spikes generated by spatially clustered and temporal synaptic input9, 35. Further, these channels curtail dendritic Ca2+ signals generated by synaptic input or by back-propagating action potentials33, 34, 36, 38. Thus, these channels affect forms of plasticity that depend on back-propagating action potentials or the propagation of local dendritic spikes (i.e. spike timing-dependent plasticity39, 40 9) but not others (i.e. LTP triggered using a 100 Hz stimulation protocol39).
Kv4.2 subunits contain a dileucine motif on the C-terminus that appears to dictate their dendritic localization (Figure 1)41. The kinesin isoform, Kif17 is also involved in dendritic transport of Kv4.242. Kv4.2 cell surface expression is further regulated by a number of auxiliary subunits, Kv channel interacting proteins (KChIPs) and dipeptidyl peptidase-like type II proteins, DPP6 and DPP10 (Figure 1)27. In addition, Kv4.2 subunits are modulated by a variety of kinases such as PKA, PKC and ERK/MAPK (Figure 1)43. Hence, a diverse range of signalling molecules modify Kv4.2 subunit expression and biophysical characteristics.
Figure 1. NMDA receptor-dependent trafficking of K+ channels.
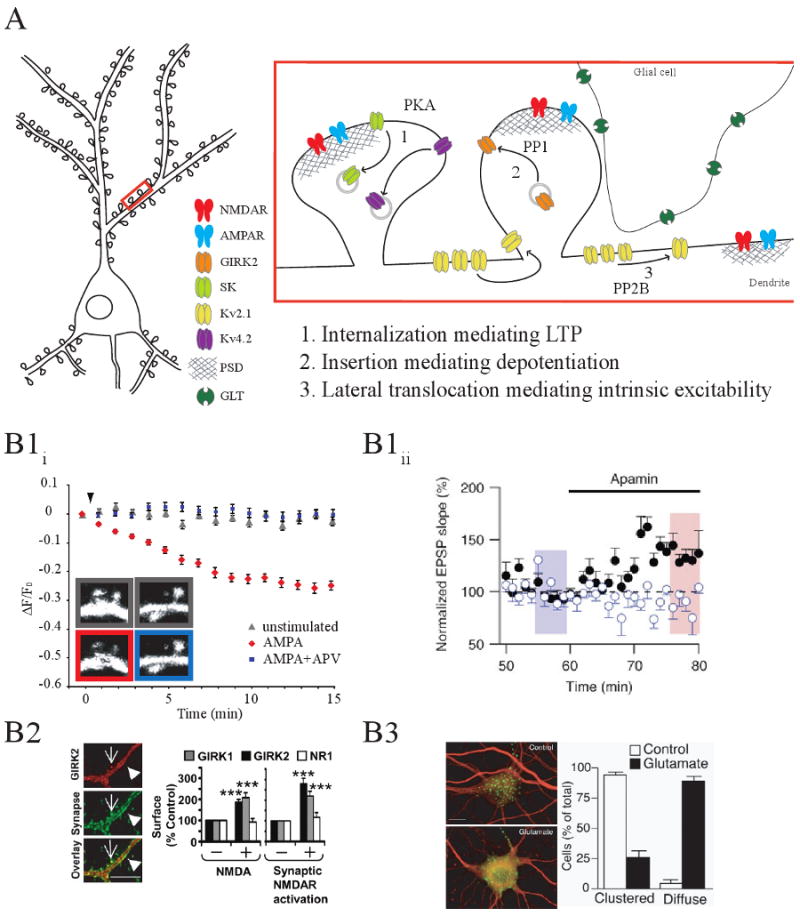
A) Model of NMDAR-dependent K+ channel trafficking that occurs during cellular and synaptic plasticity (1) During LTP, Kv4.2 and SK channels are internalized via PKA activation (2) Depotentiation is mediated by Girk2 synaptic insertion after protein phosphatase-1 activation(3) Intrinsic excitability is altered by the lateral translocation of Kv2.1 channels after protein phosphatase-2B activation. Relevant figures from the original reports are shown below in B1-3. B1) K+ channel internalization during LTP. B1i) Activity-dependent internalization of Kv4.2 requires NMDAR activation. Fluorescence changes are plotted from time-lapse images of spines from hippocampal neurons coexpressing EGFP-tagged Kv4.2 and a soluble red-fluorescent protein (tdTomato). AMPA stimulation progressively decreased Kv4.2 fluorescence intensity in spines, with no significant change in tdTomato fluorescence (inset). Kv4.2 fluorescence intensity was not significantly changed with APV coapplication to block NMDARs. (Adapted from Ref. [46]). B1ii) Activity-dependent internalization of SK channels in hippocampal slices after LTP. EPSPs were evoked from two independent pathways: a control pathway with no LTP induction (closed, black symbols) and a theta-burst pairing (TBP) stimulated pathway with robust LTP induction (open, blue symbols). Forty minutes after induction, stimulus strength was reduced to produce EPSPs of comparable slope in both pathways (blue shaded bar). Internalization of SK channels after LTP is observed by the lack of effect of the SK channel blocker apamin, on EPSP slope in the TBP pathway (blue) compared to the control (black) pathway (Adapted from Ref. [12]). B2) Activity-dependent insertion of GIRK2 in cultured hippocampal neurons. Left, immunostaining of hippocampal neurons with anti-GIRK2 C-terminal (red) and anti-synaptophysin antibodies (green) demonstrating synaptic localization of GIRK2. Arrows point to excitatory synapses on dendritic spines, whereas arrowheads point to inhibitory synapses on dendritic shafts. (Scale bar, 2 μm.). Right, activation of NMDARs t with NMDA and glycine elevated surface expression of GIRK1 and GIRK2 by ∼2-fold but not of the NMDAR subunit NR1. Similar results were found with synaptic NMDAR activation (Adapted from Ref. [15]). B3) Activity-dependent translocation of Kv2.1. Stimulation of cultured hippocampal neurons with glutamate dispersed Kv2.1 (adapted from Ref. [17]).
Since many forms of cellular and synaptic plasticity result in altered activity of kinases and phosphatases, it is perhaps not surprising that activity-dependent changes also affect dendritic Kv4.2 expression and properties. Indeed, in hippocampal CA1 neurons, increased PKA and MAPK activity following spike-timing dependent plasticity shifts the activation curve of IA such that the amplitude of backpropagating action potentials are boosted44, 45. Moreover, dendritic Kv4.2 subunits are internalized after chemical neuronal activation (e.g. AMPA, KCl, or glycine) or LTP induction (Figure 1Bli)46. This activity- dependent internalization of Kv4.2 requires NMDA receptor activation, PKA activity and clatherin-mediated endocytosis10, 46, 47. Since Kv4.2 expression reduces action potential back-propogation, the downregulation of dendritic Kv4.2 during synaptic stimulation is likely to increase back-propagation and the probability of spike-timing dependent plasticity. In addition, activity-dependent alterations in Kv4.2 channel expression influence NR2A/2B subunit expression and consequentially, metaplasticity11. These exciting findings raise the possibility that the input specificity of synaptic plasticity may in part be mediated by alterations in local dendritic Kv channel expression.
Regulation of local protein synthesis and lateral translocation of Kv channels (Figure 1B3) are also mechanisms by which their differential expression occurs. Kv1.1 channels are locally translated in dendrites, and their translation is upregulated upon NMDA receptor inhibition, suggesting that activity-driven suppression of local translation can regulate K+ channel expression19. In pyramidal neurons, clustered somatodendritic Kv2.1 channels disperse laterally along the membrane after neuronal stimulation and dephosphorylation by PP2B (calcineurin)17, 48, 49. This dephosphorylation and translocation is accompanied by a hyperpolarizing shift in the activation and inactivation of Kv2.118, 49. Interestingly, this effect is mediated by the activation of extrasynaptic NMDA receptors, and may be important for the regulation of intrinsic excitability of neurons during excitotoxic events18, 48, 49. Together, these studies highlight the diverse mechanisms that regulate dendritic Kv subunit trafficking and expression following changes in synaptic drive and intrinsic excitability.
2.2 KCa Channels
KCa channels are distinguished from Kv channels by their Ca2+-dependent activation. Three types exist: small-conductance (KCa2.1-3, SK), intermediate-conductance (KCa3.1, IK) and large conductance (KCa1.1, BK)23. KCa1 and KCa2 channels are present in principle dendrites of cerebellar purkinje cells50 and olfactory bulb neurons51, 52 where they are involved in limiting burst firing and dendro-dendritic inhibition respectively. Recent findings suggest that KCa2 subunits may also be present in hippocampal distal dendrites where they are likely to have a role in limiting compartmentalized spikes triggered by strong synaptic input38. Intriguingly, many recent studies have shown that KCa1 and KCa2 are located in close proximity to synaptic and extra-synaptic glutamate receptors in a variety of neuronal subtypes including olfactory bulb neurons51, hippocampal CA1 neurons53, 54, lateral amygdala neurons55 and amacrine cells56. Their location in dendritic spines as well as their calcium dependence makes them ideal modulators of synaptic potential amplitude and kinetics as well as plasticity. Indeed, in the hippocampus and the amygdala, the hyperpolarization mediated by KCa2 channel activation by Ca2+ influx through NMDA receptors has been demonstrated to affect the threshold for synaptic plasticity induction53, 55 and the encoding of memories57, 58.
As yet, very little is known about the mechanisms involved in targeting KCa channels to dendrites and spines. Recent evidence, though, suggests that basal levels of activated PKA may also be involved in synaptic recycling of KCa2.2 channels59. Further, like Kv4.2 subunits (see above), induction of synaptic plasticity in hippocampal neurons results in KCa2.2 internalisation (Figure 1B1ii), potentially enhancing metaplasticity12. This effect is also NMDAR-dependent and mediated by phosphorylation of KCa2.2 by PKA12. Hence, similar mechanisms appear to be engaged in regulation of dendritic Kv4 and KCa channel expression.
2.3 Kir channels
Kir channels are formed by four homo- or heteromeric alpha subunits, each with 2 transmembrane domains23. There are 7 subfamilies, Kir1-7, which differ in the degrees of rectification and their coupling to intracellular molecules60. An internal block conveys the unidirectional inward rectification of Kir channels by polyamines and Mg2+ ions61, with the strength of rectification proportional to the channel's affinity for blocking cations. At membrane potentials more negative than rest, Kir channels pass an inward/depolarizing K+ current, returning the membrane to resting potential. However, at potentials more positive than rest, intracellular cation block prevents outward K+ current from hyperpolarizing the cell membrane. These fundamental rectification properties of Kir channels are essential in maintaining neuronal membrane potential.
There is considerable evidence that Kir3.x channels are situated in the apical dendrites and spines of neocortical and hippocampal CA1 neurons62-65. Kir3.x channels are also unique in that they are activated by G proteins and thus, G-protein coupled receptors such as GABAB receptors66. The synaptic location of Kir channels indicates that their current may be modulated by changes in synaptic strength. Indeed, slow IPSCs mediated by GABAB receptors and GIRK channels are potentiated following low frequency (3 Hz) stimulation67. This effect is NMDA receptor and CaMKII dependent.
Recent evidence shows that GIRK channel expression is also regulated by activity. Enhanced neuronal activity with KCl, glutamate, NMDA or glycine, which reduce Kv4.2 and KCa surface expression, elevate Kir3.1 and Kir3.2 channels in hippocampal dendrites (Figure 1B2)15. This occurs through NMDA receptor-dependent activation of a phosphatase, PP1, which dephosphorylates Kir3.1-2 channels, causing their insertion into the membrane from recycling endosomes. NMDA receptor-dependent insertion of Kir3.1-2 channels mediates the depotentiation (see Glossary Box) of EPSPs after LTP induction68 and may be important in maintaining bidirectional modification of synapses.
3. Dendritic HCN channel function and trafficking: role in plasticity?
Hyperpolarization-activated Cation Non-Selective (HCN) subunits are comprised of four, six-transmembrane spanning α subunits (similar to Kv channels) (Figure 2)69. The channels formed by the subunits are also voltage dependent and usually activate at potentials hyperpolarized to -50 mV. Unlike Kv channels, though, the channels are permeable to both Na+ and K+. They therefore form a depolarizing current, which is involved in maintaining the neuronal resting membrane potential (RMP). Further, they also affect the membrane time constant and membrane resistance69. Hence, alterations in expression and biophysical characteristics of HCN channels can have profound effects on neuronal excitability.
Figure 2. Structure and function of dendritic HCN channels.
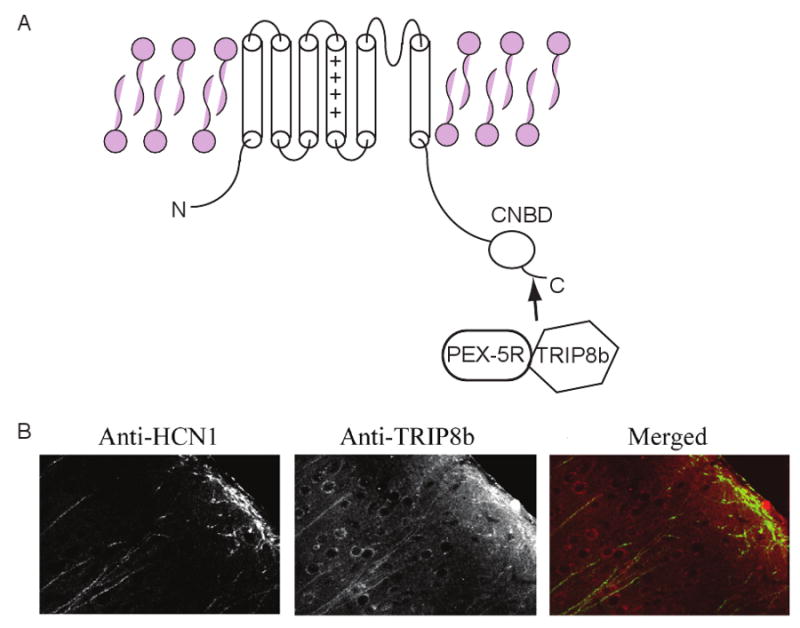
A) Illustration of TRIP8b and PEX5Rp interaction with the C-terminus of HCN subunits. The Cyclic Nucleotide Binding Domain (CNBD) which binds to cAMP is also located on the C-terminus. B) Confocal images showing TRIP8b and HCN1 expression in cortical layer V cell dendrites. The right panel shows the merged image of rhodamine-red labelled anti-TRIP8b and FITC labelled anti-HCN1. Co-localization is depicted as yellow on this image. The double staining was obtained in the same brain section (adapted from Ref. [92]).
Four HCN subunits (HCN1-4) have so far been cloned70. Interestingly, the apical, but not basal, dendrites of adult cortical and hippocampal pyramidal cells are enriched with HCN1 and HCN2 subunits2, 3, 69, 71-73. Their effects on dendritic excitability, though, are complex. Although block or knockdown of HCN channels results in RMP hyperpolarization, significantly greater numbers of dendritic action potentials can be induced by current pulses due to a disproportionate enhancement in membrane resistance74, 75. Additionally, by affecting the membrane resistance, a reduction in Ih increases synaptic potential decay and thereby summation74-79. Consequentially, loss of Ih in distal hippocampal dendrites leads to enhanced LTP80. Further, due to greater EPSP summation, stimulation of distal synapses of cortical pyramidal cell dendrites lacking HCN1 channels elicits somatic action potentials, despite the RMP being more negative than in wildtype neurons74. Thus, as a result of greater pyramidal cell output, neural network excitability is elevated following either pharmacological block or deletion of HCN subunits14, 74. Since alterations in dendritic Ih biophysical characteristics and HCN subunit expression patterns can affect intrinsic cell excitability as well as network stability, it is not surprising that changes in Ih have been associated with physiological processes such as learning and memory14, 80-82 as well as pathophysiological conditions such as epilepsy75, 83-85.
HCN subunits have a cyclic nucleotide binding domain on the C-terminal (Figure 2)69. Indeed, alterations in levels of cAMP shifts the activation curve of Ih 86, 87. cAMP activity is also altered following synaptic and intrinsic plasticity and is one mechanism by which dendritic HCN biophysical properties can change. Indeed, a recent elegant study by Wang et al. (2007)14 demonstrated that stimulation of α2 adrenoreceptors in prefrontal cortical neuron spines reduced HCN function andreinforced working memory. This study highlights how regulation of HCN channel activity can profoundly impact network activity and behavior, an effect dependent on cAMP.
Changes in synaptic strength have also been shown to modify Ih by altering intracellular Ca2+ concentration and associated downstream signalling molecules, including calcium/calmodulin dependent protein kinase II (CaMKII) and protein kinase C (PKC), activity81, 88, 89. Interestingly, pharmacological inhibition of synaptic activity or CaMKII prevents enrichment of HCN1 channels in distal hippocampal CA1 apical dendrites, suggesting that activity-dependent CaMKII activation is involved in dendritic trafficking of HCN channels90, 91.
Several recent studies have identified multiple isoforms of TPR-containing Rab8b interacting protein (TRIP8b) and pex5p-related protein (PEX5Rp) that associate with HCN subunits (Figure 2). Immunohistochemical studies have shown these to be co-localized with HCN1 in hippocampal and cortical apical dendrites (Figure 2) 92-95. These proteins are involved in regulating the expression of HCN channels too92-95. Intriguingly, excessive neuronal activity as occurs during seizures reduces TRIP8b expression in addition to HCN subunit levels in hippocampal neurons13, indicating that selective isoforms of TRIP8b may be subject to activity-dependent regulation. This could therefor be a potential mechanism underlying changes in HCN expression following modified neuronal activity81, 88, 90, 96.
Although multiple pathways have been implicated in HCN dendritic trafficking, are they interconnected? CaMKII and PKC phosphorylation consensus sites are present on HCN subunits as well as on TRIP8b isoforms95, 97. Therefore, it is tempting to speculate that variations in activity of signalling molecules such as CaMKII and PKC locally in dendrites following plasticity, may represent a means by which TRIP8b and HCN channel subunit levels can be dynamically regulated. Since HCN subunits are important for maintaining dendritic excitability and synaptic signal shapes and integration (Figure 2), changes in TRIP8b isoform expression are likely to result in altered neuronal information processing. Further studies, though, are required to determine if the various forms of plasticity (see Glossary Box) lead to differing levels of TRIP8b isoforms and the underlying cellular mechanisms responsible.
3. Voltage-gated Ca2+ channel plasticity in dendrites and spines
To date, ten voltage-gated Ca2+ channel (VGCC) primary subunits have been cloned. These can be further subdivided into 3 families: 1) dihydropyridine-sensitive high voltage-activated, CaV1.x (L-type); 2) dihydropyradine- insensitive high voltage-activated, CaV2.x (N-, P-, Q- and R- types); and 3) low voltage-activated, CaV3.x (T-type)23. Immunohistochemical as well as electrophysiological studies have revealed the presence of all subtypes of VGCC in dendrite shafts98, although the distribution of the various subtypes may differ within these structures across neuronal subtypes. For example in hippocampal CA1 neurons, CaV1.x Ca2+ channels are predominantly present at the base of the apical dendrites99 while CaV2.3 and CaV3.x Ca2+ channels located at a higher density in distal dendrites compared to the soma100, 101. In thalamocortical neurons, on the other hand, CaV3.x channels are found at a high density selectively in proximal dendrites102 where they may play a role in shaping rhythmic bursting103. Further, multiple subtypes of VGCC have been found in dendritic spines in numerous cell types104.
As yet, the factors that permit VGCC to be targeted specifically to dendrites and spines are unknown. However, VGCC activity in dendritic shafts as well as spines is likely to be a mechanism for the induction and maintenance of multiple forms of plasticity, including spike timing dependent plasticity. VGCC opening is enhanced by action potential backpropagation as well as synaptic potential generation98, 104 and Ca2+ entry through VGCC is important for intracellular signalling105. Indeed, Ca2+ entry through dendritic VGCC is necessary for LTD in entorhinal cortical cells106 and hippocampal CA1 neurons107, as well as for LTP at hippocampal CA1- perforant path synapses108 and hippocampal CA1-schaffer collatoral synapses109. Moreover, Ca2+ influx via CaV1.x (L-type) Ca2+ channels in dendritic spines contributes to induction of synapse specific NMDA receptor- dependent LTP in hippocampal neurons110. Like Kv4.2 and KCa channels, these may also form signalling complexes with NMDA receptors in hippocampal spines110. The raised Ca2+ concentration within individual spines can then activate a cascade of intracellular substances including CaMKII and cAMP which can lead to modification of the activity of additional spine VGCC such as CaV2.3 channels111. Further, L-type Ca2+ channel activity in spines is enhanced in a PKA-dependent manner by noradrenaline acting on beta-adrenoreceptors112, perhaps increasing the likelihood of plasticity occurring at selective synapses. Hence, the resting state of VGCC activity in spines and dendrites may be important for neuronal information processing and plasticity.
Concluding remarks
In this article, we have discussed the involvement of dendritic ion channels in determining the threshold and manifestation of various forms of plasticity as well as how plasticity in turn affects the expression and function of these channels. In particular, we have examined how dendritic K+, HCN and Ca2+ channels contribute to an intricate signalling complex within dendriticspines and shafts, which under physiological conditions regulates post-synaptic plasticity and intrinsic excitability. While we have focussed on K+, HCN and Ca2+ channels, dendrites contain all known superfamilies of voltage-gated ion channels1, 98. Much less is known about how these other channel subtypes are selectively targeted to dendrites and whether plasticity affects the dendritic trafficking of these channels. Nonetheless, it is clear that local expression and regulation of dendritic voltage-gated ion channel properties can provide dendrites with a dynamic tuning mechanism to adjust to changes in synaptic strength and thereby modify information processing.
Glossary Box: Neuronal Plasticity.
Since Bliss and Lomo first described the phenomenon of long-term potentiation 1, 2, great strides have been made in understanding synaptic plasticity. The elegance of this discovery was evident when considered in the context of Hebb's postulate for cellular learning3. Here, Hebb described fundamental properties involved in the strengthening of synapses, such as cooperation and coincidence (multiple inputs firing coincidentally cause strengthening) and input specificity (only synapses at active inputs are strengthened). Synaptic plasticity following these principles is often referred to as Hebbian plasticity. Decades later, it is clear that multiple forms of plasticity- both synaptic and cellular (non synapse-specific) coexist in the brain, shaping information processing and storage, and ultimately complex behavior. Here, we provide a glossary of terms for some of these diverse forms of cellular plasticity, with relevant reviews cited for each.
- Synaptic Plasticity4
refers to short-term or long-term changes in the ability of a presynaptic input to alter the postsynaptic response (synaptic efficacy).
- Long-term potentiation (LTP) and Long-term depression (LTD)
are long-term increases or decreases in synaptic efficacy, respectively. Classically, LTP and LTD were examined at Schaeffer collateral-CA1 synapses and characterized as NMDAR-dependent5. This form of synaptic plasticity requires postsynaptic Ca2+ entry through voltage-sensitive NMDA receptors, positioning dendritic ion channels as regulators of NMDAR-dependent synaptic plasticity. However, we now know there are other forms of LTP and LTD with distinct molecular mechanisms. For example, mGluR-dependent LTD is a well described NMDAR-independent LTD 6 found throughout the brain that requires mGluR1/5 activation and protein synthesis for induction.
- Spike-timing dependent plasticity (STDP)7, 8
refers to LTP or LTD elicited through pairing single pre- and post-synaptic action potentials, such as presynaptic firing coincident with a back-propagating action potential (bp-AP). The precise timing of these two events confers both the magnitude and direction of plasticity. Dendritic ion channels are important regulators of STDP, since they maintain membrane properties and control the spread of bpAPs in dendrites9, 10.
- Depotentiation
is a sustained reduction in synaptic efficacy occurring after LTP. This reversal of LTP is thought to function as a synaptic “reset”, preventing LTP saturation and positioning the synapse for further bidirectional modification.
- Metaplasticity11
refers to biological changes that the inducibility of plasticity (or as the prefix suggests, metaplasticity is the “plasticity of plasticity”). With respect to synaptic plasticity, metaplasticity was first described in the BCM model12 as alterations in the threshold for the induction of synaptic plasticity (or modification threshold).
- Intrinsic Plasticity13-15
refers to alterations in local or cell-wide excitability, such that the input / output function of the postsynaptic neuron is changed. Intrinsic plasticity is governed by changes in the expression and activation of dendritic ion channels, which largely control membrane excitability. Interestingly, intrinsic plasticity can occur in parallel with synaptic plasticity and may also serve as a mechanism for metaplasticity (eg: with increased intrinsic excitability reducing the modification threshold, and reduced intrinsic excitability stabilizing the neuron by increasing the modification threshold).
- Homeostatic Plasticity13, 16-18
refers to global, stabilizing changes in the postsynaptic neuron, such as changes in dendritic ion channel expression. Homeostatic plasticity can provide negative feedback control over neuronal strength, and is important for maintaining neuronal activity within a dynamic functional range. This can occur through synaptic scaling, where global changes occur while maintaining relative synaptic weights, through intrinsic plasticity, or through synaptic changes in the modification threshold.
Acknowledgments
MMS is supported by an MRC New Investigator Award (G0700369) and a Wellcome Trust project grant (WT087363MA). This work was supported in part by the Intramural Research Program of the National Institutes of Health and the National Institute of Child Health and Human Development (DH).
References
- 1.Johnston D, Narayanan R. Active dendrites: colorful wings of the mysterious butterflies. Trends in neurosciences. 2008;31:309–316. doi: 10.1016/j.tins.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Larkum ME, et al. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science (New York, N Y. 2009;325:756–760. doi: 10.1126/science.1171958. [DOI] [PubMed] [Google Scholar]
- 3.Nevian T, et al. Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nature neuroscience. 2007;10:206–214. doi: 10.1038/nn1826. [DOI] [PubMed] [Google Scholar]
- 4.Acker CD, Antic SD. Quantitative assessment of the distributions of membrane conductances involved in action potential backpropagation along basal dendrites. Journal of neurophysiology. 2009;101:1524–1541. doi: 10.1152/jn.00651.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nature reviews. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- 6.Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nature reviews. 2007;8:776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- 7.Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annual review of neuroscience. 2001;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- 8.Destexhe A, Marder E. Plasticity in single neuron and circuit computations. Nature. 2004;431:789–795. doi: 10.1038/nature03011. [DOI] [PubMed] [Google Scholar]
- 9.Losonczy A, et al. Compartmentalized dendritic plasticity and input feature storage in neurons. Nature. 2008;452:436–441. doi: 10.1038/nature06725. [DOI] [PubMed] [Google Scholar]
- 10.Hammond RS, et al. Protein kinase a mediates activity-dependent Kv4.2 channel trafficking. J Neurosci. 2008;28:7513–7519. doi: 10.1523/JNEUROSCI.1951-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung SC, et al. Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron. 2008;60:657–671. doi: 10.1016/j.neuron.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin MT, et al. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nature neuroscience. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin M, et al. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol Dis. 2008;32:26–36. doi: 10.1016/j.nbd.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 15.Chung HJ, et al. Neuronal activity regulates phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying potassium channels. Proc Natl Acad Sci U S A. 2009;106:629–634. doi: 10.1073/pnas.0811615106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Misonou H, et al. Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J Neurosci. 2006;26:13505–13514. doi: 10.1523/JNEUROSCI.3970-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Misonou H, et al. Regulation of ion channel localization and phosphorylation by neuronal activity. Nature neuroscience. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- 18.Mohapatra DP, et al. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin, Tex. 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raab-Graham KF, et al. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science (New York, N Y. 2006;314:144–148. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- 20.Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nature reviews. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judge SI, et al. Potassium channel blockers and openers as CNS neurologic therapeutic agents. Recent patents on CNS drug discovery. 2007;2:200–228. doi: 10.2174/157488907782411765. [DOI] [PubMed] [Google Scholar]
- 23.Alexander SP, et al. Guide to Receptors and Channels (GRAC) British journal of pharmacology. (3rd) 2008;153 2:S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotshaw DP. Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell biochemistry and biophysics. 2007;47:209–256. doi: 10.1007/s12013-007-0007-8. [DOI] [PubMed] [Google Scholar]
- 25.Kollo M, et al. Novel subcellular distribution pattern of A-type K+ channels on neuronal surface. J Neurosci. 2006;26:2684–2691. doi: 10.1523/JNEUROSCI.5257-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheng M, et al. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- 27.Vacher H, et al. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiological reviews. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serodio P, et al. Identification of molecular components of A-type channels activating at subthreshold potentials. Journal of neurophysiology. 1994;72:1516–1529. doi: 10.1152/jn.1994.72.4.1516. [DOI] [PubMed] [Google Scholar]
- 29.Christie JM, Westbrook GL. Regulation of backpropagating action potentials in mitral cell lateral dendrites by A-type potassium currents. Journal of neurophysiology. 2003;89:2466–2472. doi: 10.1152/jn.00997.2002. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman DA, et al. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 31.Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nature neuroscience. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- 32.Stuart GJ, Hausser M. Dendritic coincidence detection of EPSPs and action potentials. Nature neuroscience. 2001;4:63–71. doi: 10.1038/82910. [DOI] [PubMed] [Google Scholar]
- 33.Frick A, et al. Normalization of Ca2+ signals by small oblique dendrites of CA1 pyramidal neurons. J Neurosci. 2003;23:3243–3250. doi: 10.1523/JNEUROSCI.23-08-03243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gasparini S, et al. Associative pairing enhances action potential back-propagation in radial oblique branches of CA1 pyramidal neurons. The Journal of physiology. 2007;580:787–800. doi: 10.1113/jphysiol.2006.121343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Kampa BM, Stuart GJ. Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J Neurosci. 2006;26:7424–7432. doi: 10.1523/JNEUROSCI.3062-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, et al. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. The Journal of physiology. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai X, et al. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, et al. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe S, et al. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rivera JF, et al. An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nature neuroscience. 2003;6:243–250. doi: 10.1038/nn1020. [DOI] [PubMed] [Google Scholar]
- 42.Chu PJ, et al. A role for Kif17 in transport of Kv4.2. The Journal of biological chemistry. 2006;281:365–373. doi: 10.1074/jbc.M508897200. [DOI] [PubMed] [Google Scholar]
- 43.Schrader LA, et al. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. American journal of physiology. 2006;290:C852–861. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- 44.Frick A, et al. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nature neuroscience. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- 45.Rosenkranz JA, et al. Kinase-dependent modification of dendritic excitability after long-term potentiation. The Journal of physiology. 2009;587:115–125. doi: 10.1113/jphysiol.2008.158816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, et al. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung SC, Hoffman DA. Biphasic somatic A-type K channel downregulation mediates intrinsic plasticity in hippocampal CA1 pyramidal neurons. PloS one. 2009;4:e6549. doi: 10.1371/journal.pone.0006549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Misonou H, et al. Dynamic regulation of the Kv2.1 voltage-gated potassium channel during brain ischemia through neuroglial interaction. J Neurosci. 2008;28:8529–8538. doi: 10.1523/JNEUROSCI.1417-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulholland PJ, et al. Glutamate transporters regulate extrasynaptic NMDA receptor modulation of Kv2.1 potassium channels. J Neurosci. 2008;28:8801–8809. doi: 10.1523/JNEUROSCI.2405-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Womack MD, Khodakhah K. Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J Neurosci. 2004;24:3511–3521. doi: 10.1523/JNEUROSCI.0290-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron. 2001;31:1027–1034. doi: 10.1016/s0896-6273(01)00428-7. [DOI] [PubMed] [Google Scholar]
- 52.Maher BJ, Westbrook GL. SK channel regulation of dendritic excitability and dendrodendritic inhibition in the olfactory bulb. Journal of neurophysiology. 2005;94:3743–3750. doi: 10.1152/jn.00797.2005. [DOI] [PubMed] [Google Scholar]
- 53.Ngo-Anh TJ, et al. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nature neuroscience. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- 54.Shah MM, Haylett DG. K+ currents generated by NMDA receptor activation in rat hippocampal pyramidal neurons. Journal of neurophysiology. 2002;87:2983–2989. doi: 10.1152/jn.2002.87.6.2983. [DOI] [PubMed] [Google Scholar]
- 55.Faber ES, et al. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nature neuroscience. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- 56.Grimes WN, et al. BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nature neuroscience. 2009;12:585–592. doi: 10.1038/nn.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hammond RS, et al. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stackman RW, et al. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faber ES, et al. Modulation of SK channel trafficking by beta adrenoceptors enhances excitatory synaptic transmission and plasticity in the amygdala. J Neurosci. 2008;28:10803–10813. doi: 10.1523/JNEUROSCI.1796-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bichet D, et al. Merging functional studies with structures of inward-rectifier K(+) channels. Nature reviews. 2003;4:957–967. doi: 10.1038/nrn1244. [DOI] [PubMed] [Google Scholar]
- 61.Ficker E, et al. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science (New York, N Y. 1994;266:1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- 62.Chen X, Johnston D. Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:3787–3792. doi: 10.1523/JNEUROSCI.5312-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takigawa T, Alzheimer C. G protein-activated inwardly rectifying K+ (GIRK) currents in dendrites of rat neocortical pyramidal cells. The Journal of physiology. 1999;517(Pt 2):385–390. doi: 10.1111/j.1469-7793.1999.0385t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ponce A, et al. G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. J Neurosci. 1996;16:1990–2001. doi: 10.1523/JNEUROSCI.16-06-01990.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drake CT, et al. GIRK1 immunoreactivity is present predominantly in dendrites, dendritic spines, and somata in the CA1 region of the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1007–1012. doi: 10.1073/pnas.94.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lujan R, et al. New sites of action for GIRK and SK channels. Nature reviews. 2009;10:475–480. doi: 10.1038/nrn2668. [DOI] [PubMed] [Google Scholar]
- 67.Huang CS, et al. Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell. 2005;123:105–118. doi: 10.1016/j.cell.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 68.Chung HJ, et al. G protein-activated inwardly rectifying potassium channels mediate depotentiation of long-term potentiation. Proc Natl Acad Sci U S A. 2009;106:635–640. doi: 10.1073/pnas.0811685106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- 70.Santoro B, Tibbs GR. The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann N Y Acad Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- 71.Lorincz A, et al. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- 72.Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- 73.Nusser Z. Variability in the subcellular distribution of ion channels increases neuronal diversity. Trends Neurosci. 2009;32:267–274. doi: 10.1016/j.tins.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 74.Huang Z, et al. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009;29:10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shah MM, et al. Seizure-Induced Plasticity of h Channels in Entorhinal Cortical Layer III Pyramidal Neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berger T, et al. High I(h) channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol. 2001;85:855–868. doi: 10.1152/jn.2001.85.2.855. [DOI] [PubMed] [Google Scholar]
- 77.Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poolos NP, et al. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- 79.Williams SR, Stuart GJ. Site independence of EPSP time course is mediated by dendritic I(h) in neocortical pyramidal neurons. J Neurophysiol. 2000;83:3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]
- 80.Nolan MF, et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 81.Fan Y, et al. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- 82.Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen K, et al. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dugladze T, et al. Impaired hippocampal rhythmogenesis in a mouse model of mesial temporal lobe epilepsy. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17530–17535. doi: 10.1073/pnas.0708301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jung S, et al. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen S, et al. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. The Journal of general physiology. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang J, et al. Activity-dependent regulation of HCN pacemaker channels by cyclic AMP: signaling through dynamic allosteric coupling. Neuron. 2002;36:451–461. doi: 10.1016/s0896-6273(02)00968-6. [DOI] [PubMed] [Google Scholar]
- 88.Brager DH, Johnston D. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in I(h) in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:13926–13937. doi: 10.1523/JNEUROSCI.3520-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Welie I, et al. Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5123–5128. doi: 10.1073/pnas.0307711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shin M, Chetkovich DM. Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. The Journal of biological chemistry. 2007;282:33168–33180. doi: 10.1074/jbc.M703736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Richichi C, et al. Mechanisms of seizure-induced ‘transcriptional channelopathy’ of hyperpolarization-activated cyclic nucleotide gated (HCN) channels. Neurobiol Dis. 2008;29:297–305. doi: 10.1016/j.nbd.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santoro B, et al. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zolles G, et al. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 2009;62:814–825. doi: 10.1016/j.neuron.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 94.Lewis AS, et al. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci. 2009;29:6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Santoro B, et al. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62:802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arimitsu T, et al. Activity-dependent regulation of HCN1 protein in cortical neurons. Biochemical and biophysical research communications. 2009;387:87–91. doi: 10.1016/j.bbrc.2009.06.127. [DOI] [PubMed] [Google Scholar]
- 97.Poolos NP, et al. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci. 2006;26:7995–8003. doi: 10.1523/JNEUROSCI.2069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Magee J, et al. Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annual review of physiology. 1998;60:327–346. doi: 10.1146/annurev.physiol.60.1.327. [DOI] [PubMed] [Google Scholar]
- 99.Westenbroek RE, et al. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 100.Magee JC, et al. Subthreshold synaptic activation of voltage-gated Ca2+ channels mediates a localized Ca2+ influx into the dendrites of hippocampal pyramidal neurons. Journal of neurophysiology. 1995;74:1335–1342. doi: 10.1152/jn.1995.74.3.1335. [DOI] [PubMed] [Google Scholar]
- 101.Christie BR, et al. Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. Journal of neurophysiology. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- 102.Williams SR, Stuart GJ. Action potential backpropagation and somato-dendritic distribution of ion channels in thalamocortical neurons. J Neurosci. 2000;20:1307–1317. doi: 10.1523/JNEUROSCI.20-04-01307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cueni L, et al. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nature neuroscience. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- 104.Bloodgood BL, Sabatini BL. Ca(2+) signaling in dendritic spines. Current opinion in neurobiology. 2007;17:345–351. doi: 10.1016/j.conb.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 105.Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004;2004:re15. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- 106.Zhou YD, et al. Increasing Ca2+ transients by broadening postsynaptic action potentials enhances timing-dependent synaptic depression. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19121–19125. doi: 10.1073/pnas.0509856103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Christie BR, et al. The role of dendritic action potentials and Ca2+ influx in the induction of homosynaptic long-term depression in hippocampal CA1 pyramidal neurons. Learning & memory (Cold Spring Harbor, N Y. 1996;3:160–169. doi: 10.1101/lm.3.2-3.160. [DOI] [PubMed] [Google Scholar]
- 108.Takahashi H, Magee JC. Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron. 2009;62:102–111. doi: 10.1016/j.neuron.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 109.Remy S, Spruston N. Dendritic spikes induce single-burst long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17192–17197. doi: 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee SJ, et al. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yasuda R, et al. Plasticity of calcium channels in dendritic spines. Nature neuroscience. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- 112.Hoogland TM, Saggau P. Facilitation of L-type Ca2+ channels in dendritic spines by activation of beta2 adrenergic receptors. J Neurosci. 2004;24:8416–8427. doi: 10.1523/JNEUROSCI.1677-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Bliss TV, Lomo T. Plasticity in a monosynaptic cortical pathway. J Physiol. 1970;207:61P. [PubMed] [Google Scholar]
- 2.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hebb DO. The Organization of Behavior. 1949. [Google Scholar]
- 4.Sjostrom PJ, et al. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- 5.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 6.Anwyl R. Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog Neurobiol. 2006;78:17–37. doi: 10.1016/j.pneurobio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Kampa BM, et al. Dendritic mechanisms controlling spike-timing-dependent synaptic plasticity. Trends Neurosci. 2007;30:456–463. doi: 10.1016/j.tins.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- 9.Johnston D, et al. Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:667–674. doi: 10.1098/rstb.2002.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magee JC, Johnston D. Plasticity of dendritic function. Curr Opin Neurobiol. 2005;15:334–342. doi: 10.1016/j.conb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Mockett BG, Hulme SR. Metaplasticity: new insights through electrophysiological investigations. J Integr Neurosci. 2008;7:315–336. doi: 10.1142/s0219635208001782. [DOI] [PubMed] [Google Scholar]
- 12.Bienenstock EL, et al. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson SB, Turrigiano GG. Strength through diversity. Neuron. 2008;60:477–482. doi: 10.1016/j.neuron.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maffei A, Turrigiano G. The age of plasticity: developmental regulation of synaptic plasticity in neocortical microcircuits. Prog Brain Res. 2008;169:211–223. doi: 10.1016/S0079-6123(07)00012-X. [DOI] [PubMed] [Google Scholar]
- 15.Frick A, Johnston D. Plasticity of dendritic excitability. J Neurobiol. 2005;64:100–115. doi: 10.1002/neu.20148. [DOI] [PubMed] [Google Scholar]
- 16.Desai NS. Homeostatic plasticity in the CNS: synaptic and intrinsic forms. J Physiol Paris. 2003;97:391–402. doi: 10.1016/j.jphysparis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]