

Abstract
The Category B agents, ricin and shiga toxin (Stx), are RNA N-glycosidases that target a highly conserved adenine residue within the sarcin-ricin loop of eukaryotic 28S ribosomal RNA. In an effort to identify small-molecule inhibitors of these toxins that could serve as lead compounds for potential therapeutics, we have developed a simple Vero cell-based high-throughput cytotoxicity assay and have used it to screen ∼81,300 compounds in 17 commercially available chemical libraries. This initial screen identified ∼300 compounds with weak (≥30-<50%), moderate (≥50-<80%), or strong (≥80%) ricin inhibitory activity. Secondary analysis of 244 of these original “hits” was performed, and 20 compounds that were capable of reducing ricin cytotoxicity by >50% were chosen for further study. Four compounds demonstrated significant dose-dependent ricin inhibitory activity in the Vero cell-based assay, with 50% effective inhibitory concentration (EC50) values ranging from 25 to 60 μM. The same 20 compounds were tested in parallel for the ability to inhibit ricin's and Stx1's enzymatic activities in an in vitro translation reaction. Three of the 20 compounds, including the most effective compound in the cell-based assay, had discernible anti-toxin activity. One compound in particular, 4-fluorophenyl methyl 2-(furan-2-yl)quinoline-4-carboxylate (“compound 8”), had 50% inhibitory concentration (IC50) of 30 μM, a value indicating > 10-fold higher potency than is the case for previously described ricin-Stx1 inhibitors. Computer modeling predicted that compound 8 is capable of docking within the ricin active site. In conclusion, we have used a simple high-throughput cell-based method to identify several new small-molecule inhibitors of ricin and Stx.
Keywords: biodefense, toxin, high-throughput screen, therapeutic
1. Introduction
Ricin and shiga toxins (Stx) are members of the A-B family of toxins whose enzymatic A subunits are RNA N-glycosidases that target a highly conserved adenine residue within the sarcin-ricin loop of eukaryotic 28S ribosomal RNA (Endo and Tsurugi 1987, 1988). Both toxins are of public health concern, and they are classified by the Centers for Disease Control and Prevention (CDC) as Category B agents. Ricin is found in the seeds of castor bean plant (Ricinus communis) and is known to have been weaponized for chemical warfare purposes (Centers for Disease Control and Prevention, 2000, 2003; Crompton and Gall, 1980). The toxin is potentially lethal to humans if inhaled, ingested or injected (Audi et al., 2009). By the injection route, the lethal dose in humans is estimated at 0.1-1.0 μg/kg (Miller et al. 2002). The manifestations of ricin poisoning vary depending on the mode of exposure, but may include fever, gastroenteritis, fluid and electrolyte depletion, hypoglycemia, circulatory collapse, and multi-organ failure (Audi et al., 2009). Stx is produced by Shigella dysenteriae type 1 (Strockbine et al., 1988) and by certain strains of Escherichia coli (Calderwood et al., 1987). The A subunit (StxA) shows limited homology with the A subunit of ricin (RTA), although the two proteins catalyzes the same depurination reaction (Calderwood et al., 1987, Endo et al., 1988, Strockbine et al., 1988). Stx-producing E. coli (STEC) strains such as O157:H7, cause gastrointestinal illnesses including bloody diarrhea, hemorrhagic colitis, and life-threatening hemolytic uremic syndrome (HUS). For either ricin or Stx exposure, treatment is strictly supportive; there are currently no specific antidotes against these toxins (Audi et al., 2009; Challoner and McCarron, 1990; Quiňones et al., 2009; Serna and Boedeker, 2008).
RTA is linked via a single disulfide bond to the B subunit (RTB), a galactose-specific lectin that facilitates binding of ricin to host cell surfaces (Baenziger and Fiete, 1979). On binding to cognate cellular glycoprotein and glycolipid receptors, ricin is internalized by endocytosis and then trafficked via the retrograde pathway to the Golgi apparatus and the endoplasmic reticulum (ER) (Sandvig and van Deurs, 2000; Sandvig et al., 2002). The toxin is processed in the ER, and RTA is translocated to the cytoplasm, where a fraction of it escapes degradation by proteosomes and is able to target the host protein biosynthetic machinery (Sandvig and van Deurs, 2000; Sandvig et al., 2002). Stx, following association with its cognate receptor globotriaosylceramide (Gb3), follows a similar intracellular route. Once in the cytoplasm, both StxA and RTA selectively inactivate 28S rRNA (Sandvig and van Deurs, 2000). Ricin's cytoxicity is due to a combination of protein synthesis arrest and triggering of intracellular stress-activated pathways; the result is the induction of apoptosis, with the release of pro-inflammatory mediators (Gonzalez et al., 2006; Hughes et al., 1996; Yoder et al., 2007). Because all of these effects are initiated following ribosome arrest, the most effective inhibitors of ricin and Stx are likely to be those that directly interfere with the toxins' active sites.
The X-ray structure of RTA was solved to resolutions of 2.8Å and 2.5 Å by Montfort et al. (1987) and Rutenber et al. (1991), respectively. Those studies, in combination with site-directed mutagenesis experiments, enabled the identification of the key active site residues, including Glu177, Arg180, Tyr80, Tyr123, and Typ211. Monzingo and Robertus proposed that depurination of adenine involves: Protonation of adenine (N3) by Arg180; delocalization of ring electrons, causing cleavage of C1′-N9 glycosidic bond; and generation of an oxacarbenium ion at C1′. The latter is stabilized by a hydroxide ion that is generated when Glu177 abstracts a proton from a free water molecule in the active site (Monzingo and Robertus, 1992). The authors also reported the crystal structures of RTA bound to two substrate analogues, formycin monophosphate (FMP) and a dinucleotide ApG. The structures of these complexes revealed important ionic and hydrophobic interactions that promote binding of the substrate and its analogues in the active site of RTA (Monzingo and Robertus, 1992). The active site of Stx has key residues that are conserved within the family of ribosome inactivating protein (RIP) and is analogous to the active site of RTA (Fraser et al., 1994; Katzin et al., 1991, Monzingo and Robertus, 1992).
There have been numerous attempts to identify active-site inhibitors of RTA, with the long-term goal of using these molecules as therapeutics against both ricin and Stx. Virtual screening (Shoichet, 2004) has identified substrate analogues and derivatives of pterin, pyrimidine, and guanine as weak to modest RTA inhibitors, with IC50 values ranging from 200 to >2000 μM (Bai et al., 2009; Monzingo et al., 1992; Robertus et al., 1996; Yan et al., 1997). For example, pteroic acid (PTA) and 8-methyl-9-oxaguanine were identified using this method and were confirmed by kinetic measurements to be modest inhibitors of RTA, with respective IC50 values of ∼ 0.6 and 0.4 mM (Miller et al., 2002; Yan et al., 1997). These two bicyclic inhibitors mimic substrate binding in the active site by displacing the side chain of Tyr 80 from a position that makes it partially block the “mouth” of the active site. Occupancy of the active site by adenine or a substrate analogue causes rotation of Tyr 80 by 45°, enabling the phenyl ring of Tyr 80 to π-stack with the ring moiety of the substrate (Miller et al., 2002; Yan et al., 1997). Other inhibitors of RTA have been identified that bind the closed form of RTA. For example, 2,5-diamino-4-6-dihydroxypyridine (DDP), a monocyclic inhibitor with an IC50 of 2.2 mM, was found to stack against the side chain of Tyr80 in its apoenzyme conformation, and failed to enter the RTA specificity pocket (Bai et al., 2009; Miller at al., 2002). Other active-site inhibitors identified thus far include stem-loop oligonucleotides (aptamers) that mimic the oxacarbenium transition state at the RTA depurination site. While these inhibitors form high-affinity complexes with RTA at low pH (∼ 4) (Chen et al., 1998), they do not appear to bind RTA at physiologic pH and likely have limited applicability as drug candidates, due to poor cellular uptake and their inherent susceptibility to degradation in vivo (Bai et al., 2009).
While advances in in silico screening technology will undoubtedly lead to the identification of additional ricin and Stx inhibitors, there is clearly an urgent need for alternative approaches that can accelerate the drug discovery process. Toward this end, Haslam and colleagues recently developed and employed a quantitative luciferase-based assay to identify inhibitors of Stx; positive candidates were then tested against ricin (Saenz et al., 2007; Zhao and Haslam, 2005). The methodology involved transfection of cell lines with cDNA encoding a destabilized derivative luciferase with a short half life. Therefore, cells treated with protein-synthesis inhibitors, including Stx and ricin, demonstrated a rapid decline of luciferase activity. Saenz et al. (2007) exploited the latter finding to screen ∼14,000 compounds, in an HTS format for small-molecule inhibitors of Stx. Those investigators identified two inhibitors of Stx that also showed activity against ricin. Subsequent analysis of the two compounds revealed that they affected intracellular toxin transport. While these particular compounds are unlikely to be developed as therapeutics due to their global effect on intracellular trafficking, they did demonstrate the feasibility of using cell-based HTS to screen for inhibitors of ricin and Stx.
In the present study, we have developed a simplified cell-based luciferase assay that does not require transfection of cells or handling of radioactive material; the assay was easily adapted to an HTS format. Using this method, we screened 17 chemical libraries (>81,000 compounds) and identified a number of compounds that showed weak, moderate, or strong anti-ricin activity. The method was validated through identification of known inhibitors of ricin. Due to the close similarity of the active sites of ricin and Stx, it is likely that ricin inhibitors will have a corresponding inhibitory activity on Stx (Miller et al., 2002). Indeed, an in vitro cell-free translation assay revealed that some of the identified compounds had significant anti-RTA and anti-StxA1 activities. Thus, the HTS method developed in this study can be used to identify inhibitors of ricin and Shiga toxins.
2. Materials and methods
2.1. Cell culture, reagents, and materials
Vero cells (African green monkey adult kidney cells; ATCC CCL-81) were obtained from the American Type Culture Collection (Manassas, VA). The cells were routinely grown and maintained in antibiotic-free Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C in 95% air and 5% CO2. For HTS assays, cells with < 20 passages were used. DMEM, phosphate-buffered saline (PBS, pH 7.4), and calcium- and magnesium-free Dulbecco's PBS (DPBS, pH 7.4) were obtained from the Cell Culture and Media Services Department at the Wadsworth Center. Trypsin (0.05%)-EDTA (0.53 mM) in Hanks' balanced salt solution (HBSS) without sodium carbonate, calcium, and magnesium was obtained from Mediatech, Inc. (Manassas, VA). Cell culture flasks (T75 and T150 cm2 with canted neck and 0.2 μm lid-fitted filter) and microplates (96- and 384- well, white, flat bottom, sterile and polystyrene tissue-culture treated) were from Corning Incorporated (Corning, NY). Ricin (Ricinus communis agglutinin II; 5 mg/ml) and ricin A subunit (RTA) were purchased from Vector Laboratories (Burlingame CA). Ricin was dialyzed in PBS at 4°C for 24 hr to remove sodium azide prior to use. Cycloheximide was purchased from Sigma-Aldrich (St. Louis, MO). Shiga toxin 1 (Stx 1) holotoxin used in cytotoxicity assays was purchased from the Phoenix Laboratory at Tufts-New England Medical Center (Boston, MA). Purified StxA1 was purified from E. coli, as described by Miller et al. (2002). CellTiter-Glo™, control RNA (luciferase mRNA), Rabbit Retic Lysate System (TM232) and Luciferase Assay Reagent were from Promega (Madison, WI). Luminescence was measured using an EnVision® (Perkin Elmer, Waltham, MA) or a SpectraMax® L Molecular Devices (Sunnyvale, CA) microplate luminometer.
2.2. Screening of small molecule libraries
Screening of small molecules was done by the authors in conjunction with the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Diseases (NSRB) at Harvard Medical School (Boston, MA). The screened libraries used for this study are listed in Table 1. The compounds were singly arrayed (apart from natural product extracts that had an undefined number of compounds) in 384-well stock plates as 5 mg/ml dimethyl sulfoxide (DMSO, 100%) solutions. The compounds were screened at a constant dilution (1:300) so as to achieve a final concentration between 10 and 60 μM. The final concentration of DMSO was 0.33%. The purity of the compounds was assured by the manufacturers to be > 90%, as determined by nuclear magnetic resonance (NMR) and liquid chromatography-mass spectrometry (LC-MS). The purity and identity of select compounds was confirmed using one dimensional solution proton NMR (1D 1H NMR) by the Biochemistry Core Facility at the Wadsworth Center. Liquids were dispensed to 384-well microplates using a high-speed Matrix WellMate (Matrix, Hudson, NH) fitted with an 8-channel, small-bore (0.015″ [0.38 mm] inner diameter nozzle orifice) disposable tubing cartridge with a peristaltic pump, and integrated with a stacker for holding plates. Nanoliter volumes of compounds from library stock plates were transferred to assay plates using Seiko or Epson compound transfer robots fitted with stainless steel pin arrays (V&P Scientific, Inc., San Diego, CA).
Table 1.
Chemical libraries screened by HTS in this study.
| Library type | Library name | No. of compounds | 1° hitsa | Cherry picksb | 2° hitsc |
|---|---|---|---|---|---|
| Known bioactive compounds | ActMol TimTec 1 | 480 | 3 | 3 | 3 |
| NINDS Custom Collection 2 | 1040 | 1 | 1 | 0 | |
| Prestwick 1 Collection | 1120 | 4 | 4 | 1 | |
| Selected natural products | ICBG-Fungal extracts | 1760 | 1 | 0 | 0 |
| Commercial compounds | Asinex 1 | 12378 | 65 | 55 | 3 |
| Chembridge 3 | 10560 | 33 | 26 | 2 | |
| ChemDiv 1-4 | 19605 | 35 | 33 | 3 | |
| Enamine 2 | 26576 | 146 | 118 | 10 | |
| Life chemicals 1 | 3893 | 6 | 6 | 1 | |
| Maybridge 1-5 | 3916 | 8 | 6 | 1 | |
| Total compounds | 81328 | 302 | 252 | 24 | |
Number of compounds that gave ≥ 30% cell protection in HTS assay.
Compounds from the primary screen were retested to confirm their anti-ricin activity.
Compounds whose anti-ricin activity was confirmed in the secondary screen.
For the HTS, Vero cells were trypsinized and diluted in DMEM to achieve a density of 4 ×104 cells/ml. The cells (25 μl; ∼1 ×103 cells) were seeded in each well of 384-well plates using a Matrix WellMate, and the plates were then incubated at 37°C for ∼ 20 hr in a 95% air/5% CO2 environment. Compounds (0.1 μl) were transferred by robotics from stock plates to columns 1-22 (or 3-24 depending on the compound stock plate layout) of the assay plates, using stainless steel pin arrays; the plates were then incubated at 37°C for 2 hr. Ricin was dispensed (5 μl per well, resulting in a final concentration of 5 ng/ml) into columns 1-23 of the 384 well plates, while DMEM (no ricin) was added to column 24. Column 23 was defined as the toxin-treated cells, and column 24 was defined as untreated. The assay plates were incubated at 37°C for 48 hr before the addition of 30 μl of CellTiter-Glo™ (diluted 1:5 in PBS). The CellTiter-Glo™ reagent includes luciferin, luciferase, and Mg2+ and when applied to Vero cells the resulting luminescence activity is proportional to the cellular levels of ATP and, therefore cell viability (Neal et al., 2010; Noah et al., 2007; Saenz et al., 2007). The plates were incubated at room temperature for at least 5 min before luminescence was measured with an EnVision® luminometer. Each plate was tested in duplicate. To avoid fluctuations in temperature during the HTS, the plates were transferred and processed in small batches of fewer than 10.
2.3. HTS data analysis
Luminescence data were formatted and analyzed with Microsoft Excel 2003 (Microsoft Corporation, Redmond, WA). The Z′-factor for an individual test plate was calculated according to Zhang et al. (1999): Z′ = 1-((3σp + 3σn)/|μp -μn|), where σp and σn respectively represent the SD values of untreated (“positive”) and toxin-treated (“negative”) control wells, and μp and μn respectively represent the means of untreated and treated (“negative”) control wells. For each test plate there were 32 control wells (16 positive wells and 16 wells for negative wells) positioned in columns 1 and 2 or 23 and 24. Whenever possible, additional controls wells were included within other columns on the plate. As an additional level of quality control, we also preformed full 384 well plate controls (positive and negative) for every 20 duplicate plates that were processed during the HTS screen.
The Z-score for each test well was calculated by subtraction of the median luminescence value obtained from all test wells that were treated with test compounds and ricin (this excluded control wells) on a given plate, followed by division by the SD of those test wells. We used median instead of mean luminescence to reduce the effect of data skewing by outliers as recommended by Malo et al. (2006) and in accordance with the guidelines established by the NSRB. As a correction for residual background luminescence inherent in the assays, the luminescence values obtained from wells that did not contain cells, but did have added CellTiter Glo™ substrate, was subtracted from the entire plate. The inhibitory activity for each test compound was defined as the luminescence value from a single test well × 100, divided by the average luminescence value of the untreated (“positive”) control wells. Compounds from the primary screen were chosen for secondary analysis (“cherry-picked”) if they (i) were present on plates with Z′-factor ≥ 0.5; (ii) demonstrated a Z-score ≥ 2; and (iii) inhibited ricin's cytotoxic activity by ≥ 30%. Compounds that met these criteria were then prioritized, based on ricin inhibitory activity, as being strong (≥ 80% inhibitory activity), medium (≥ 50% < 80%), or weak (< 50% ≥ 30%) inhibitors. The HTS assay was done in duplicate (parallel plates). Compounds were considered “hits” only if they were positive on both test plates, and all other selection criteria were met (as described above). Compounds were not selected in instances where a disparity existed (i.e., positive signal on one plate, and a negative or poor signal on the replicate).
2.4. Secondary and tertiary analysis of small molecule inhibitors
For secondary and tertiary screens, Vero cells (120 μl containing 50,000 cells/ml) were seeded in opaque 96-well plates and maintained overnight in a 37°C incubator with 5% CO2. Cherry-picked compounds (1.2 μl) were supplied by the NSRB at a concentration of 5 mg/ml in DMSO. The compounds were further diluted 10-fold into DMSO, and then a total of 1.2 μl of each compound was added to assay wells in duplicate. As a control, an equal volume of DMSO was added to positive and negative control wells. Cells were then incubated at 37°C for 1 hr, before the addition of ricin to a final concentration of 5 ng/ml. Cell viability was determined 24 hr later using Cell Titer-Glo™ substrate. Compounds that inhibited ricin's cytotoxic effects by ≥ 50% were chosen for tertiary analysis. Tertiary analysis involved dose-response studies using compounds obtained from commercial vendors (see Tables 3, 4). All commercially obtained compounds were solubilized in DMSO.
Table 3.
Selected compounds that showed dose dependent anti-ricin activity.
| Compound no. | Compound structure | EC50 (μM)a | CIDb | Sourcec |
|---|---|---|---|---|
| 1 |  |
29 | 16271106 | Enamine |
| 2 |  |
62 | 644401 | Asinex |
| 3 |  |
59d | 5737931 | Chembridge |
| 4 |  |
23 | 18576762 | Life Chemicals |
Concentration that gave Vero cells 50% protection from ricin toxicity. The IC50 varied with incubation time of ricin with Vero cells
Compound identification number
Supplier of the compound.
Insolubility of the compound was noted at the indicated concentration.
Table 4.
In vitro inhibition of RTA and StxA1 by selected compounds.
| Compound | Structure | CIDa | RTAb IC50 (μM) |
StxA1c IC50 (μM) |
|---|---|---|---|---|
| 1 |  |
16271106 | 1000 | >500 |
| 8 |  |
2399297 | 30 | 40 |
| 13 | 1300461 | 150 | 250 | |
| C17-1 |  |
7531223 | 250 | 500 |
Compound identification number
ricin toxin subunit A
Shiga toxin type 1 subunit A
2.5. In silico analysis of putative RTA inhibitors
Two virtual screening programs, ICM (Abagyan et al., 1994) and GOLD (Jones et al., 1997), were used to dock the structures of the small molecules into RTA's open and closed conformations. The open form corresponds to the conformation of RTA bound to neopterin (PDB: 1BR5) and permits compounds access to the pocket in which adenine substrates and analogs bind (Bai et al., 2009; Miller et al., 2002; Yan et al., 1997). The closed form corresponds to the apoenzyme (PDB: 1RTC) (Mlsna et al., 1993). The docking site in the open form was centered on the known specificity pocket, but also included a second pocket proposed to bind guanine in natural substrates (Monzingo and Robertus, 1992). This second pocket was the major ligand target for the closed form. Compounds identified in the secondary phase of the cell-based screen, as well as known RTA inhibitors, were represented in three-dimensional structure data format (sdf). The compounds were ranked based on approximate ligand binding energy for the ligand-protein complex. PyMol software (DeLano Scientific LLC, Palo Alto, CA) was used to visually examine the best docking pose for each inhibitor (DeLano., 2002). SciFinder Scholar (Chemical Abstracts Service, American Chemical Society, Columbus, OH) was used to identify structurally related molecules.
2.6. In vitro protein translation assay
We used a previously established luciferase-reporter in vitro translation assay to determine whether any of the small-molecule inhibitors of ricin identified in the cell-based assay were capable of interfering directly with the toxin's enzymatic activity (Bai et al., 2009; Neal et al., 2009). Briefly, RTA (12.5 nM) was incubated with the test compound in a solution of BSA (1 mg/ml) and ∼6 % DMSO for 30 min before the addition of rabbit reticulocyte lysate and a translation substrate mixture consisting of amino acids, RNasin ribonuclease inhibitor, and uncapped in vitro transcribed luciferase (luc) mRNA template (20 ng/μl). The final DMSO concentration in the reaction mixture was 1%. The mixture was incubated at 30°C for 90 min before the addition of luciferase assay reagent. The mixture was transferred to an opaque 384-well microplate, and luminescence was measured with a Perkin Elmer Envision luminometer.
3. Results
3.1. Development and validation of a cell-based HTS to identify small-molecule inhibitors of ricin
In an effort to identify novel small-molecule inhibitors of ricin (and Stx), we developed and optimized a cell-based HTS compatible with instrumentation available at the NSRB. An overview of the HTS is depicted in Figure 1. Vero cells (∼1 × 103) were seeded into 384-well plates, and then incubated for ∼16 hr prior to the addition of the compounds (0.1 μl; 10-60 μM final concentration) from available chemical libraries, by robotic pin transfer. Ricin (5 ng/ml) was added to the wells 2 hr later, and cell viability was assessed an additional 48 hr later using a luciferase-based reporter assay (Neal et al., 2010; Noah et al., 2007; Saenz et al., 2007), as described in Section 2.2. The cytotoxicity assay we employed was extremely sensitive and highly reproducible, as evidenced by the fact that signal-to-background and signal-to-noise ratios were generally ≥ 10. In addition, the values obtained from ricin-treated and untreated control wells were separated by a wide margin, as indicated by an average Z′ factor in preliminary studies of >0.6 (Zhang et al., 1999). For plates subjected to the HTS (as described below), the actual Z′ factors were generally >0.7, and frequently >0.8 (data not shown). To ensure uniformity of the assay, we used only low passage Vero cells, and incubation times and assay temperatures were kept constant as described in Section 2.2.
Fig. 1. Schematic of HTS to identify small-molecule inhibitors of ricin.

Vero cells (∼1 × 103) were seeded into 384-well plates, and then incubated for ∼16 hr prior to the addition of the compounds of interest (0.1 μl each; 10-60 μM final concentration) from available chemical libraries, by robotic pin transfer. Ricin (5 ng/ml) was added to the wells 2 hr later, and cell viability was assessed a further 48 hr later, using a luminescent-based readout.
To validate this HTS as a means to identify ricin inhibitors, we performed a pilot screen of 2,640 known bioactive compounds (Table 1). This pilot screen identified four compounds that inhibited the cytotoxic effects of ricin by more than 50% (Table 2). One of the compounds was determined to be brefeldin A, a fungal metabolite known to interfere with the retrograde transport pathway utilized by ricin (and Stx) to traffic to the ER (Klausner et al., 1992; Saenz et al., 2007; Zhao and Haslam, 2005). We confirmed that brefeldin A interfered, in a dose-dependent manner, with ricin cytotoxicity, under our assay conditions (data not shown). The other three compounds identified in this pilot screen were thiostrepton, tosyl phenylalanyl chloromethyl ketone (TPCK), and gliotoxin. While we confirmed that these three compounds did indeed inhibit ricin's cytotoxic activity, they were not pursued further because they are themselves toxic to Vero cells, and/or they were determined not to directly inhibit RTA activity (data not shown). Nonetheless, this pilot study demonstrated the validity of our HTS as a means to identify small molecules that affect the toxicity of ricin.
Table 2.
Known bioactive compounds with significant anti-ricin activity, as identified in primary screen.
| Compound | Source | Bioactivity | Reference |
|---|---|---|---|
| Brefeldin A | Fungi | Inhibits anterograde vesicular transport | Klausner et al. |
| Thiostrepton | Bacteria | Antibiotic and anticancer agent | Kwok et al., Weisblum and Demohn |
| TPCK | Synthetic | Irreversible inhibitor of chymotrypsin | Ong et al. |
| Gliotoxin | Fungi | Antimicrobial agent | Waring et al. |
Compounds were tested at 16.67 μg/ml, TPCK; tosyl phenylalanyl chloromethyl ketone
3.2. Screening of chemical libraries for small-molecule inhibitors of ricin, using the cell-based HTS
We next employed the cell-based HTS to screen 13 chemical libraries (Table 1) available to us through the NSRB, for inhibitors of ricin. These libraries include Asinex 1, Chembridge 3, ChemDiv 1-4, Enamine 2, Life Chemicals 1, and Maybridge 1-5 (Table 1). The majority of compounds in these libraries possess drug-like properties in accordance with Lipinski's rule of five: a median molecular mass < 500 Da, < five hydrogen bond donors, < 10 hydrogen bond acceptors, and log octanol/water partition coefficient (cLogP) < 5 (Lipinski, 2000). An additional library of selected natural products consisting of partially purified extracts from endophytic fungi found in Costa Rica, and free of nuisance compounds (e.g., compounds with high molecular weight and polarity) was also screened (Table 1).
In total, 76,928 pure compounds and 1,760 natural product extracts were screened (Table 1). As detailed in Section 2.3, compounds that conferred ≥ 80% cell viability were considered strong inhibitors of ricin, compounds conferring 50-80% viability were considered to be medium inhibitors of ricin, and compounds conferring 30-50% viability were considered weak inhibitors of ricin. Upon completion of this screen, we had identified 26 (0.03 %) strong inhibitors, 62 (0.08 %) medium inhibitors, and 214 (0.26 %) weak inhibitors of ricin.
NSRB guidelines allowed us to cherry-pick 244 compounds (0.3% of total screened) for secondary screening, as described in Section 2.4. Due to the limited availability of these compounds, the secondary screen consisted of testing each one of them at only a single concentration, ∼10 μM, in a 96-well microtiter plate format. Of the 244 compounds tested in the secondary screen, 20 were able to block the cytotoxic effects of ricin by more than ≥50%, and were therefore chosen for further analysis (Table 3, Suppl. Tables 1 and 2). While the other 224 compounds proved, upon secondary analysis in the cell-based assay, to be relatively weak inhibitors, they were nonetheless examined by computer modeling for the ability to dock within RTA's active site; in some cases they were also tested for the ability to inhibit RTA activity in a cell-free in vitro translation assay (see Section 3.4).
3.3. Characterization of the 20 most active small-molecule inhibitors of ricin
Of the 20 candidate inhibitors identified in the secondary screen, only 7-[(R)-(3-ethoxy-4-hydroxyphenyl)-(pyridin-3-ylamino)methyl]quinolin-8-ol (CID 2977768) was not commercially available. The remaining 19 were purchased from commercial vendors and subjected to dose-response studies at a range of concentrations (∼1-200 μM), as described in Section 2.4. Based on their performance in this assay, the compounds were classified into three groups (I-III): Group I consisted of nine compounds (numbers 11-19) that demonstrated relatively low ricin inhibitory activity and were themselves cytotoxic (Suppl. Table 1). These compounds were not pursued further. Group II consisted of six compounds (numbers 5-10) that showed relatively high ricin inhibitory activity at low concentrations, but were themselves cytotoxic when tested at high concentrations (Suppl. Table 2). Finally, Group III consisted of four compounds (numbers 1-4) that demonstrated a dose-dependent inhibition of ricin activity and were themselves only mildly or moderately cytotoxic (Table 3; Fig. 2). It should be noted that because ricin-cytotoxicity is time- and dose-dependent, all the secondary cell-based assays were done uniformly with 5.0 ng/ml toxin and a 24 hr period of incubation.
Fig. 2. Ricin inhibitory activity of Group III compounds.

Compounds 1-4 (panels A-D) were examined for dose-response inhibitory activity of ricin (5 ng/ml) in a Vero cell cytotoxicity assay, as described in section 2.4. The symbols are as follows: □ with dashed horizontal line represents ricin-treated (“negative”) control cells; ▲, compound- and ricin-treated cells. The data presented represent the average of two experiments each done in triplicate. The error bars (when not hidden by the symbol) reflect the percent deviation from the mean. The best-fit curves were generated using a non-linear regression analysis of data with Graphpad Prism version 5.00 for Windows (San Diego, California USA).
The four compounds in Group III were diverse in terms of overall structure, with the only obvious commonality being the presence of aromatic rings and hydrogen bond donors/acceptors (HBD/HBA). Compound 1, (E)-3-(5-methylfuran-2-yl)-N-(1,2,3,4-tetrahydronaphthalen-1-yl)prop-2-enamide (CID 16271106), demonstrated a dose-dependent capacity to protect Vero cells from ricin intoxication when added 1 hr prior to toxin exposure (Fig. 2A). In these assays, compound 1 had an estimated EC50 of ∼30 μM (Table 3) and conferred maximal protection (∼96%) at 69 μM. Compound 1 did not exhibit significant cytotoxicity, even at ∼100 μM. Compound 2, (4,6-bis(propan-2-ylamino)-1,3,5-triazine-2-carbonitrile (CID 644401) also reduced ricin toxicity in the cell-based assay in a dose-dependent manner (Fig. 2B). Compound 2 had an estimated EC50 of ∼ 60 μM (Table 3), and maximal protection (∼ 69%) was observed at ∼ 89 μM. At this concentration, compound 2 was itself moderately cytotoxic (data not shown). Compound 3, methyl 2-[(5E)-5-[[5-(azepan-1-yl)furan-2-yl]methylidene]-2,4-dioxo-1,3-thiazolidin-3-yl]propanoate (CID 5737931), protected Vero cells from the effects of ricin, although maximal protection never exceeded 60%, due to the fact that the compound was toxic even at concentrations less than 20 μM (Table 3; Fig. 2C). Finally, compound 4, 2-methylsulfonyl-1-(naphthalen-1-ylmethyl)benzimidazole (CID 18576762), had an estimated EC50 of ∼25 μM and conferred maximal cell protection (∼ 97%) at ∼ 58 μM (Table 3; Fig. 2D). However, compound 4 was moderately cytotoxic at concentrations >30 μM. Compounds in Group III had no effect on the ability of cycloheximide to arrest protein synthesis in a Vero cell-based assay, nor did these compounds affect the stability or activity of luciferase (data not shown).
3.4. Identification of compounds that inhibit RTA activity in vitro
The inhibitors selected in the cell-based HTS could potentially interfere with any one of the steps involved in ricin cytotoxicity, including toxin attachment to cell-surface receptors, endocytosis, retrograde transport, dissociation of RTA from RTB, retrotranslocation of RTA across the ER membrane, and/or RTA catalytic activity. In an effort to identify those compounds that specifically interfered with RTA's ribosome inactivating activity, the 20 most effective inhibitors from the HTS were tested in a previously established cell-free in vitro translation assay (Bai et al., 2009; Neal et al., 2010). Compounds were incubated with RTA for 30 min at room temperature and then added to a rabbit reticulolysate mixture in which luciferase (luc) messenger RNA was provided as template. Luminescence was used as an indicator of in vitro translation and protein synthesis.
We identified four compounds that reduced RTA's capacity to inhibit protein synthesis in the cell-free in vitro translation assay (Table 4). The most potent inhibitor was compound 8 (CID 2399297). The dose-response curve for this compound demonstrated that it inhibited RTA with an apparent IC50 of ∼30 μM (Table 4; Fig. 3A). Compound 8 was also capable of interfering with StxA1 activity, with an apparent IC50 of ∼40 μM (Table 4). It should be stressed that these IC50 values are only approximations: The in vitro translation assays revealed that the exponential decay for this compound flattened out at concentrations >40 μM. We suggest that this behavior reflects the poor solubility of the compound, an assumption that is supported by the fact that theoretical calculations of the aqueous solubility of compound 8 estimate a maximum solubility near 40 μM (Tetko et al., 2005). This may also explain why compound 8 was not identified as a particularly effective inhibitor of ricin in the cell-based assays (data not shown). Nonetheless, computer modeling predicted that this compound can associate with RTA (Fig. 3B). Specifically, the quinoline ring of compound 8 was predicted to bind deep in a cleft that is present on RTA when the latter is in the closed conformation (see Sections 1 and 2.5). Efforts to obtain the X-ray structure of RTA complexed with compound 8 have been unsuccessful to date.
Figure 3. Compound 8 inhibits RTA activity in vitro.
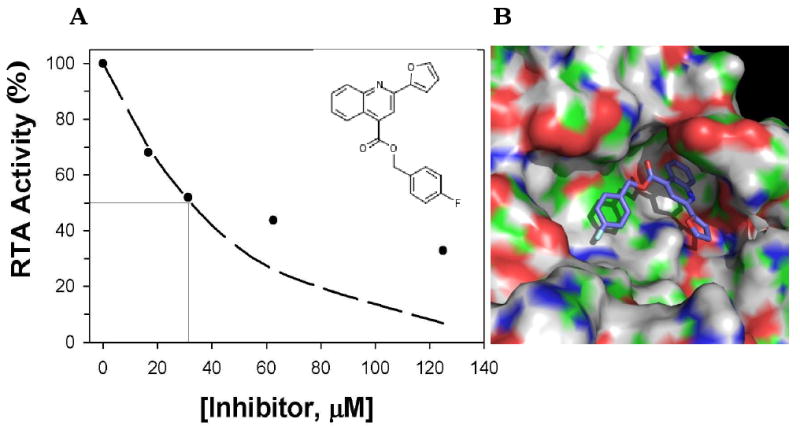
(Panel A) Indicated concentrations (X-axis) of compound 8 (inset) were mixed with RTA and then added to in vitro translation in which luciferase mRNA was present as substrate. Translation of the luciferase mRNA was determined by addition of Bright-Glo™ substrate and measurement of light emission with a luminometer, as described in section 2.6. The values shown from a single representative experiment. (Panel B) Predicted binding of compound 8 with RTA in the closed conformation. The quinoline ring of compound 8 rests deep in the RTA “second pocket” that is present when the molecule in the closed conformation. The two pendant groups also interact with the protein. For example, the aromatic rings on the fluorobenzene group on the extreme left of the compound stack on the side chain of Tyr 80 (green patch beneath the ring). The electronegative fluorine side chain interacts with the cationic side chain of Arg180 (blue band). On the surface of RTA, green corresponds to carbon; red, oxygen; dark blue, nitrogen; white, hydrogen. For the ligand, carbon is dark blue, fluorine is baby blue, and oxygen is red.
The other three compounds capable of inhibiting both RTA and StxA1 in the cell-free in vitro translation assay had apparent IC50 values ranging from 150 to 1000 μM. Compound 1, which was identified as the most effective inhibitor of ricin in the cell-based assay, proved to be only a weak inhibitor of RTA in the cell-free in vitro translation assay, with an apparent IC50 ∼1000 μM. Compound 13 was identified in the initial HTS but was deemed a relatively poor inhibitor of ricin in secondary and tertiary cell-based assays. Nonetheless, it was a modest inhibitor of both RTA and StxA1 in the in vitro assay. Finally, we identified using SciFinder compound C17-1 (N-cyclohexyl-N-[(4-fluorophenyl)methyl]-2-(4H-1,2,4-triazol-3-ylsulfanyl)acetamide; CID 7531223), which blocked RTA and StxA1 activity in vitro with apparent IC50 values of 250 μM and 500 μM, respectively. C17-1 is an analogue of a compound originally identified as a being good ricin inhibitor in the primary screen, but only a modest inhibitor in the secondary screen (data not shown). C17-1 inhibited 50 % of ricin intoxication at a concentration of about 60 μM, but was itself cytotoxic at concentrations > 100 μM (data not shown).
4. Discussion and conclusions
In this study, we have developed and validated a cell-based HTS to identify small-molecule inhibitors of ricin and shiga toxins. The method is sensitive (e.g., signal to background ≥ 10), robust (e.g., average Z′-prime factor of ∼0.6) and reproducible (e.g., the SD between replicates was < 10%). Moreover, it has an advantage over the HTS utilized by Saenz et al. (2007) in that it does not require transfection of the cells prior to seeding in the 384-well plates. Our HTS strategy was validated by the fact that we identified brefeldin A, a known inhibitor of ricin cytotoxicity, in a pilot screen of a collection of known bioactive compounds (Saenz et al., 2007). Using this HTS, we screened 81,328 stock wells in duplicate, from 16 different chemical libraries, as well as from a subset library of fungal extracts. From this screen we identified 302 (∼0.4%) compounds with anti-ricin activities ranging from weak to strong. 244 of these compounds were cherry-picked and subjected to a secondary screen. 20 of the 244 compounds, when tested at a concentration of ∼10 μM, were capable of >50% inhibition of ricin in a cell-based assay. Our hit rate in the secondary screen (0.02% of 81,328 compounds) was similar to that achieved by others who have performed HTS for antiviral compounds (Severson et al., 2008). We conclude that the HTS can be used to screen additional compound libraries, and should therefore accelerate discovery of much-needed inhibitors of both ricin and shiga toxins.
From this HTS, we identified four compounds that had significant dose-dependent anti-ricin activity, coupled with low to moderate cytotoxicity, on Vero cells. The compounds were from four different chemical libraries, and the EC50 values of these compounds ranged from 23 to 62 μM in the cell-based assay (Table 3), potentially classifying them as good lead compounds. The compounds were diverse in terms of overall structure, with the only commonality being the presence of aromatic rings, hydrogen bond donors/acceptors (HBD/HBA), and rotatable/flexible bonds. The solubility of the compounds varied; in the case of compound 3, solubility was likely the factor that limited its efficacy to inhibit ricin in the cell-based assay. One of the four compounds (compound 1) was capable of preventing RTA from inactivating ribosomes in a cell-free assay, suggesting that it targets the toxin's active site (see below). Because the other three compounds (2, 3, and 4) did not appear to act on the active site, they most likely interfere with another step in ricin cytotoxicity, such as intracellular trafficking of the toxin. Indeed, the two compounds identified by Saenz et al. (2007) interfered with intracellular trafficking. One inhibited transport of StxB to perinuclear recycling endosomes, while the second inhibited transport of StxB at a post-recycling endosome stage (Saenz et al., 2007). It is interesting to note that our HTS included the ChemDiv3 library, from which Saenz et al. (2007) identified the above two inhibitors of Stx and ricin. While these two compounds were not among the top 20 compounds identified in our study, they were likely included in the 39 “hits” we obtained in the ChemDiv1-4 series. Unfortunately, this cannot be verified, because the CID numbers of the two compounds were not reported.
Compound 1 is a bicyclic molecule with a flexible side chain linked to a methyl substituted furan ring. The compound has two fused rings like adenine and pteroic acid, but lacks the specific hydrogen bonds that interact in the specificity pocket of the open form of RTA. In particular, it lacks the exocyclic amine group that has been seen to anchor specificity site inhibitors such as adenine, guanine, and pterin based inhibitors (Bai et al., 2009; Monzingo and Robertus, 1992; Yan et al., 1997). Docking studies suggest that compound 1 is more likely to bind RTA in the closed form, where it occupies a second active site pocket (Monzingo and Robertus, 1992). Regardless of the exact mode of binding, in vitro translation assay showed that compound 1 could block RTA catalytic activity, although with only a moderate IC50 (∼ 1 mM). Nonetheless, compound 1 is notable for one more reason: It was capable of inhibiting StxA activity in an in vitro assay with an IC50 of ∼ 0.7 mM. StxA is homologous to RTA and catalyzes the same depurination reaction (Endo et al., 1988). We noted that the EC50 of compound 1 in the cell-based assay was ∼ 33-fold lower than the IC50 of the compound in the cell free translation assay. This discrepancy could be due to the ability of the compound to target processes involved in toxin binding to cognate receptors, endocytosis, trafficking, or processing in the ER. Inhibition of any of these processes could have an additive or synergistic inhibitory effect in a cell-based assay and thus give low EC50.
A number of compounds were identified in the primary screen that subsequently were proved to be toxic to cells and/or poorly soluble, but nonetheless demonstrated the capacity to block RTA activity in vitro. For example, compound 8, which was only a moderate inhibitor of ricin in the cell-based assay, was determined to be the most effective compound to date at inhibiting both RTA and StxA in the in vitro translation assay. Compound 8 had an apparent IC50 of ∼30 μM; it was thus almost 10 times more effective than PBA (Bai et al., 2009). In silico analysis revealed that Compound 8 was the top scorer by ICM, with a score 2.5 σ above the mean for the group (the second best scorer was less than 1σ above the mean). GOLD also ranked compound 8 highly, at position 4 in the list. Modeling studies showed that compound 8 could potentially dock at a pocket near the active site of RTA in its “closed” conformation. The projected binding is reminiscent of how DDP is proposed to interact with RTA. DDP, a monocyclic inhibitor with an IC50 of 2.2 mM, stacked against the side chain of Tyr80 in its apoenzyme conformation, and failed to enter the RTA specificity pocket (Bai et al., 2009; Miller et al., 2002). However, it is not known whether an active site-specific ligand would displace a ligand bound in the second pocket. Overall, these findings demonstrate that the developed HTS method, coupled with in vitro translation assay and in silico screening, will greatly accelerate discovery of ricin inhibitors.
The vast majority of compounds identified as hits in the primary HTS failed to inhibit ricin in the secondary cell-based screen. We believe that this discrepancy is due to two factors. First, the compounds were tested at a 5-fold lower concentration in the secondary assay, as compared to the primary assay (10 μM versus 50 μM). Therefore, the secondary assay was significantly more stringent than the first. Compounds that failed to inhibit ricin at the lower concentration were considered as less effective potential inhibitors and generally were not pursued further. Second, the cherry-picked compounds employed in the secondary screen were not necessarily identical to those tested in the primary screen. The compounds in the primary screen derived from stock plates that are subject to as many as 25 freeze/thaw cycles, and may therefore contain breakdown products or possible contaminants. The cherry-picked compounds, on the other hand, were obtained from a master plate set aside exclusively for secondary screenings.
The primary screen also yielded compounds that likely interfere with ricin in a non specific manner. For example, in the secondary screen, thiostrepton showed a strong dose-dependent protection of Vero cells from ricin intoxication, even though it was highly insoluble in aqueous medium. We observed that thiostrepton alone is capable of stimulating cellular protein synthesis and this may counteract the action of ricin (data not shown). Other compounds may non-specifically block the activity of proteins such as ricin through sequestration or partial unfolding. Those phenomena have been well described in the case of AmpC β-lactamase (Coan et al., 2009; McGovern et al., 2002; Seidler et al., 2003). It should be noted that we found no evidence that any of the Group III compounds promoted the formation of colloidal aggregates (P. Wahome and N. Mantis, unpublished results). Overall, this study underscores the need to perform a tiered screening strategy following HTS, and to couple the cell based assays with both in vitro and in silico analyses.
In conclusion, we have demonstrated the practicality of using a HTS cell-based assay to identify inhibitors of ricin. This method will have application in identification of inhibitors for ricin and other ribotoxins. This method simplifies the screening procedure and likely reduces plate-to-plate variability. The relative simplicity of the assay enabled us to screen >81,000 compounds. In additional to pursuing compounds 1 and 8 as potential lead compounds, we have also completed a screen of an additional ∼120,000 compounds at the NSRB and initiated preliminary characterization of the strongest inhibitors (P. G Wahome and N. J. Mantis, unpublished results).
Supplementary Material
Acknowledgments
We would like to acknowledge Su Chiang, Ren Tao, Ruchir Shah, Andrew Daab, Jen Nale, David Wrobel, and other members of the NSRB staff for assistance with the HTS. We also thank Carolyn McGuinness for assistance in developing the high-throughput screen. This work was supported by grants 5U01AI075509 (JDR) and U54AI057159 from the NIH.
Footnotes
Conflict of interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abagyan RA, Totrov M, Kuznetsov D. ICM—a new method for protein modeling and design: Applications to docking and structure prediction from the disordered native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- Audi J, Belson M, Patel M, Schier J, Osterloh J. Ricin poisoning. JAMA. 2005;294:2342–2351. doi: 10.1001/jama.294.18.2342. [DOI] [PubMed] [Google Scholar]
- Baenziger JU, Fiete D. Structural determinants of Ricinus communis agglutinin and toxin specificity for oligosaccharides. J Biol Chem. 1979;254:9795–9799. [PubMed] [Google Scholar]
- Bai Y, Monzingo AF, Robertus JD. The X-ray structure of ricin A chain with a novel inhibitor. Arch Biochem Biophys. 2009;483:23–28. doi: 10.1016/j.abb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Brugsch HG. Toxic hazards: the castor bean. N Engl J Med. 1960;262:1039–1040. [Google Scholar]
- Calderwood SB, Auclair F, Donohue-Rolfe A, Keusch GT, Mekalanos JJ. Nucleotide sequence of the Shiga-like toxin genes of Escherichia coli. PNAS. 1987;84:4364–4368. doi: 10.1073/pnas.84.13.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Biological and Chemical Terrorism: strategic plan for preparedness and response. Recommendations of the CDC Strategic Planning Workgroup. MMWR Morb Mortal Wkly Rep. 2000;49:1–14. [PubMed] [Google Scholar]
- Centers for Disease Prevention and Control. Investigation of a ricin-containing envelope at a postal facility – South Carolina. MMWR Morb Mortal Wkly Rep. 2003;52:1129–1131. [PubMed] [Google Scholar]
- Challoner KR, McCarron MM. Castor bean intoxication: review of reported cases. Ann Emerg Med. 1990;19:1177–1183. doi: 10.1016/s0196-0644(05)81525-2. [DOI] [PubMed] [Google Scholar]
- Chen XY, Link TM, Schramm VL. Ricin A-chain: Kinetics, mechanism, and RNA stem-loop inhibitors. Biochemistry. 1998;37:11605–11613. doi: 10.1021/bi980990p. [DOI] [PubMed] [Google Scholar]
- Coan KE, Maltby DA, Burlingame AL, Shoichet BK. Promiscuous aggregate-based inhibitors promote enzyme unfolding. J Med Chem. 2009;52:2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J Med Chem. 2009;52:416–424. doi: 10.1021/jm801100v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton R, Gall D. Georgi Markov: death in a pellet. Med Leg J. 1980;48:51–62. doi: 10.1177/002581728004800203. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- Endo Y. Mechanism of action of ricin and related toxins on the inactivation of eukaryotic ribosomes. Cancer Treat Res. 1988;37:75–89. doi: 10.1007/978-1-4613-1083-9_5. [DOI] [PubMed] [Google Scholar]
- Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem. 1988;171:45–50. doi: 10.1111/j.1432-1033.1988.tb13756.x. [DOI] [PubMed] [Google Scholar]
- Endo Y, Tsurugi K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem. 1987;262:8128–8130. [PubMed] [Google Scholar]
- Endo Y, Tsurugi K. The RNA N-glycosidase activity of ricin A-chain: the characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J Biol Chem. 1988;263:8735–8739. [PubMed] [Google Scholar]
- Fan S, Wu F, Martiniuk F, Hale ML, Ellington AD, Tchou-Wong KM. Protective effects of anti-ricin A-chain RNA aptamer against ricin toxicity. World J Gastroenterol. 2007;14:6360–6365. doi: 10.3748/wjg.14.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A, Welsh P, Richardson J, Robertus JD. The role of arginine 180 and glutamic acid 177 of ricin toxin A chain in the enzymatic inactivation of ribosomes. Mol Cell Biol. 1990;10:6257–6263. doi: 10.1128/mcb.10.12.6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ME, Chernaia MM, Kozlov YV, James MNG. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5Å resolution. Nature Struct Biol. 1994;1:59–64. doi: 10.1038/nsb0194-59. [DOI] [PubMed] [Google Scholar]
- Gonzalez TV, Farrant SA, Mantis NJ. Ricin induces IL-8 secretion from human monocyte/macrophages by activating the p38 MAP kinase pathway. Mol Immunol. 2006;43:1920–1923. doi: 10.1016/j.molimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Hughes JN, Lindsay CD, Griffiths GD. Morphology of ricin and abrin exposed endothelial cells is consistent with apoptotic cell death. Hum Exp Toxicol. 1996;15:443–451. doi: 10.1177/096032719601500513. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Katzin BJ, Collins EJ, Robertus JD. The structure of ricin A chain at 2.5Å. Proteins. 1991;10:251–259. doi: 10.1002/prot.340100309. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JMM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EWF. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther. 2008;7:2022–2032. doi: 10.1158/1535-7163.MCT-08-0188. [DOI] [PubMed] [Google Scholar]
- Li Q, Maddox C, Rasmussen L, Hobrath JV, White LE. Assay development and high-throughput antiviral drug screening against Bluetongue virus. Antiviral Res. 2009;83:267–273. doi: 10.1016/j.antiviral.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- Malo N, Hanley JA, Cerquozii S, Pelletier J, Nadon R. Statistical practice in high-throughput screening data analysis. Nat Biotechnol. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- Mantis NJ. Vaccines against the category B toxins: Staphylococcal enterotoxin B, epsilon toxin and ricin. Adv Drug Deliv Rev. 2005;57:1424–1439. doi: 10.1016/j.addr.2005.01.017. [DOI] [PubMed] [Google Scholar]
- McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Ravikumar K, Shen H, Suh JK, Kerwin SM, Robertus JD. Structure-based design and characterization of novel platforms for ricin and Shiga toxin inhibition. J Med Chem. 2002;45:90–98. doi: 10.1021/jm010186s. [DOI] [PubMed] [Google Scholar]
- Mlsna D, Monzingo AF, Katzin BJ, Ernst S, Robertus JD. Structure of recombinant ricin A chain at 2.3 Å. Protein Sci. 1993;2:429–435. doi: 10.1002/pro.5560020315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort W, Villafranca JE, Monzingo AF, Ernst SR, Katzin B, Rutenber E, Nuyhen HX, Hamlin R, Robertus JD. The three-dimensional structure of ricin at 2.8Å. J Biol Chem. 1987;262:5398–5403. [PubMed] [Google Scholar]
- Monzingo AF, Robertus JD. X-ray analysis of substrate analogs in the ricin A-chain active site. J Mol Biol. 1992;227:1136–1145. doi: 10.1016/0022-2836(92)90526-p. [DOI] [PubMed] [Google Scholar]
- Neal LM, O'Hara J, Brey RN, 3rd, Mantis NJ. A monoclonal IgG antibody directed against an immunodominant linear epitope on the ricin A chain confers systemic and mucosal immunity to ricin. 2010;78:552–561. doi: 10.1128/IAI.00796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah JW, Severson W, Noah DL, Rasmussen L, White EL, Jonsson CB. A cell based luminescence assay is effective for high-throughput screening of potential antivirals. Antiviral Res. 2007;73:50–59. doi: 10.1016/j.antiviral.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Ong EB, Shaw E, Schoellmann G. The identification of the histidine residue at the active center of chymotrypsin. J Biol Chem. 1965;240:694–698. [PubMed] [Google Scholar]
- Quiňones B, Massey S, Friedman M, Swimley MS, Teter K. Novel cell-based method to detect Shiga toxin 2 from Escherichia coli O157:H7 and inhibitors of toxin activity. Appl Environ Microbiol. 2009;75:1410–1416. doi: 10.1128/AEM.02230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey GJA, Young JAT. Antitoxins: novel strategies to target agents of bioterrorism. Nat Rev Microbiol. 2004;2:721–726. doi: 10.1038/nrmicro977. [DOI] [PubMed] [Google Scholar]
- Ready MP, Kim YS, Robertus JD. Directed alteration of active site residues in ricin A chain and implications for the mechanism of action. Proteins. 1991;10:270–278. doi: 10.1002/prot.340100311. [DOI] [PubMed] [Google Scholar]
- Robertus JD, Monzingo AF. The structure of ribosome inactivating proteins. Mini Rev Med Chem. 2004;4:477–486. doi: 10.2174/1389557043403837. [DOI] [PubMed] [Google Scholar]
- Robertus JD, Yan X, Ernst S, Monzingo A, Worley S, Day P, Hollis T, Svinth M. Structural analysis of ricin and implications for inhibitor design. Toxicon. 1996;34:1325–1334. doi: 10.1016/s0041-0101(96)00103-1. [DOI] [PubMed] [Google Scholar]
- Rutenber E, Katzin BJ, Collins EJ, Mlsna D, Ernst SE, Ready MP, Robertus JD. The crystallographic refinement of ricin at 2.5 Å resolution. Proteins. 1991;10:240–250. doi: 10.1002/prot.340100308. [DOI] [PubMed] [Google Scholar]
- Saenz JB, Doggett TA, Haslam DB. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect Immun. 2007;75:4552–4561. doi: 10.1128/IAI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B. Entry of ricin and Shiga toxin into cells: molecular mechanisms and medical perspectives. EMBO J. 2000;19:5943–5950. doi: 10.1093/emboj/19.22.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K, Grimmer S, Lauvrak SU, Torgersen ML, Skretting G, van Deurs B, Iversen TG. Pathways followed by ricin and Shiga toxin into cells. Histochem Cell Biol. 2002;117:131–141. doi: 10.1007/s00418-001-0346-2. [DOI] [PubMed] [Google Scholar]
- Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- Schlossman D, Withers D, Welsh P, Alexander A, Robertus JD, Frankel A. Expression and characterization of mutants of ricin toxin A chain in Escherichia coli. Mol Cell Biol. 1989;9:5012–5021. doi: 10.1128/mcb.9.11.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serna A, Boedeker EC. Pathogenesis and treatment of Shiga toxin-producing Escherichia coli infections. Curr Opin Gastroenterol. 2008;24:38–47. doi: 10.1097/MOG.0b013e3282f2dfb8. [DOI] [PubMed] [Google Scholar]
- Severson WE, McDowell M, Ananthan S, Chung DH, Rasmussen L, Sosa MI, White EL, Noah J, Jonsson CB. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2) J Biomol Screen. 2008;13:879–887. doi: 10.1177/1087057108323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strockbine NA, Jackson MP, Sung LM, Holmes RK, O'Brien AD. Cloning and sequencing of the genes for Shiga toxin from Shigella dysenteriae type 1. J Bacteriol. 1988;170:1116–1122. doi: 10.1128/jb.170.3.1116-1122.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, Tanchuk VY, Prokopenko VV. Virtual computational chemistry laboratory-design and description. J Comput-Aided Mol Des. 2005;19:453–463. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- Waring P, Eichner RD, Müllbacher A. The chemistry and biology of the immunomodulating agent gliotoxin and related epipolythiodioxopiperazines. Med Res Rev. 1988;8:499–524. doi: 10.1002/med.2610080404. [DOI] [PubMed] [Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, Lalonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- Weisblum B, Demohn V. Thiostrepton, an inhibitor of 50S ribosome subunit function. J Bacteriol. 1970;101:1073–1075. doi: 10.1128/jb.101.3.1073-1075.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston SA, Tucker AD, Thatcher DR, Derbyshire DJ, Pauptit RA. X-ray structure of recombinant ricin A-chain at 1.8 Å resolution. J Mol Biol. 1994;244:410–422. doi: 10.1006/jmbi.1994.1739. [DOI] [PubMed] [Google Scholar]
- Yan X, Hollis T, Svinth M, Day P, Monzingo AF, Milne GW, Robertus JD. Structure-based identification of a ricin inhibitor. J Mol Biol. 1997;266:1043–1049. doi: 10.1006/jmbi.1996.0865. [DOI] [PubMed] [Google Scholar]
- Yoder JM, Aslam RU, Mantis NJ. Evidence for widespread epithelial damage and coincident production of monocyte chemotactic protein 1 in a murine model of intestinal ricin intoxication. Infect Immun. 2007;75:1745–1750. doi: 10.1128/IAI.01528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhao L, Haslam DB. A quantitative and highly sensitive luciferase-based assay for bacterial toxins that inhibit protein synthesis. J Med Microbiol. 2005;54:1023–1030. doi: 10.1099/jmm.0.46143-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.