


Abstract
Bulged-G motifs are ubiquitous internal RNA loops that provide specific recognition sites for proteins and RNAs. To establish the common and distinctive features of the motif we determined the structures of three variants and compared them with related structures. The variants are 27-nt mimics of the sarcin/ricin loop (SRL) from Escherichia coli 23S ribosomal RNA that is an essential part of the binding site for elongation factors (EFs). The wild-type SRL has now been determined at 1.04 Å resolution, supplementing data obtained before at 1.11 Å and allowing the first calculation of coordinate error for an RNA motif. The other two structures, having a viable (C2658U•G2663A) or a lethal mutation (C2658G• G2663C), were determined at 1.75 and 2.25 Å resolution, respectively. Comparisons reveal that bulged-G motifs have a common hydration and geometry, with flexible junctions at flanking structural elements. Six conserved nucleotides preserve the fold of the motif; the remaining seven to nine vary in sequence and alter contacts in both grooves. Differences between accessible functional groups of the lethal mutation and those of the viable mutation and wild-type SRL may account for the impaired elongation factor binding to ribosomes with the C2658G•G2663C mutation and may underlie the lethal phenotype.
INTRODUCTION
Deciphering the principles that govern ribonucleic acid structure is central to understanding the molecular underpinnings of the diverse roles that RNAs play in cellular function. These principles are beginning to become known, due in part to the recent determinations of RNA structures; notable examples are those from the two subunits of the ribosome (1–4). Folded RNAs are composed of A-form helices that are frequently interrupted by other RNA structural elements. These include terminal loops (relevant examples are T-loops, hook turns and tetraloops), internal loops (relevant examples are bulged-G motifs and K-turns) and the A-minor motif, which is the most common RNA tertiary contact (5,6). To fold RNA and create RNA and protein binding sites, the motifs rely on the twists and turns in the phosphodiester backbone and the distinctive surfaces that arise from non-canonical interactions. To investigate the structural basis of sequence-specific recognition and how RNA elements stack on one another, extensive structural comparisons have been made for T-loops (7), hook turns (8), GNRA tetraloops (9), K-turns (10) and A-minor interactions (5,6), but are lacking for bulged-G motifs. This paper provides a comprehensive comparison of high-resolution bulged-G motif structures.
The bulged-G motif is also referred to as the Loop E motif (11), the sarcin/ricin motif (12), the bulged-G cross-strand A stack (13) and the S-motif (14). The bulged-G motif designation was first used in a review of RNA motifs by Moore (15) and is used throughout because it describes a feature of the motif that is essential for protein recognition (16) and has been used by others (6,17). NMR studies provided the first view of the motif in loop E from Xenopus laevis 5S ribosomal RNA (rRNA) (11), and shortly thereafter in the sarcin/ricin loop (SRL) from Escherichia coli 23S rRNA (18). Crystallographic studies subsequently determined the structures of a related cross-strand A stack that lacks a bulged G (13) and two variants of the bulged-G motif derived from the rat and E.coli SRL RNA (19,20). The bulged G of the motif forms a base triple that is flanked on either side by non-Watson–Crick base pairs (non-WC bps) that are followed by WC bps. Six of these nucleotides are conserved. The remaining seven to nine nucleotides vary in identity, and include two or three non-WC bps that stack on the 5′-side of the bulged G (Fig. 1). It is unknown how variation in sequence changes the structure and surface features of this motif. Based on structural comparison between the rat and the E.coli bulged-G motifs (20) and visual inspection of the seven occurrences in the structure of the 50S ribosomal subunit from Haloarcula marismortui (21,22), bulged-G motifs share a common geometry. The phosphodiester backbone surrounding the bulged G forms a distinctive S-shape that is referred to as the S-turn (19,20). As an internal loop, the motif stacks on other RNA structural elements, with bend angles at each junction. The bend angles and their variance were unknown.
Figure 1.
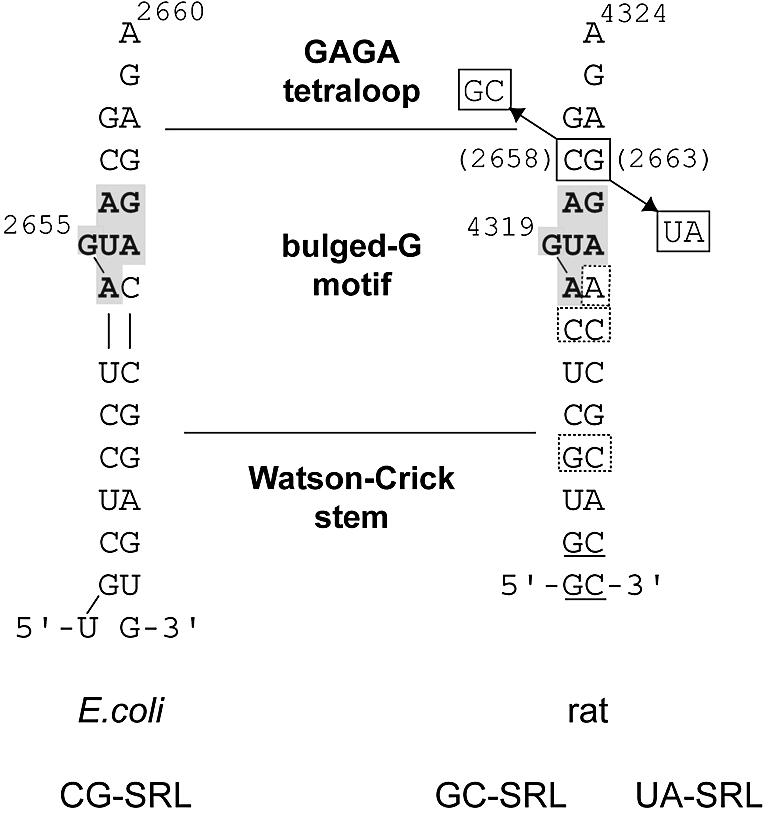
Sequences used for crystallization of the E.coli CG-SRL (left), and rat GC-SRL and UA-SRL sequences (right), showing the six conserved nucleotides of the bulged-G motif (gray box). The terminal nucleotides in the rat sequence are designed to form a paired stem (underlined). Mutations of the C•G bp in the rat 28S rRNA sequence (E.coli 23S rRNA numbering in parentheses) are boxed. Differences between the bulged-G motif of the E.coli and rat sequences are shown (dashed boxes on the rat sequence).
Our studies center on the archetypal bulged-G motif found in a stem–loop structure conserved in all ribosomes—the SRL RNA. The name derives from the toxins sarcin and ricin that target the SRL. Cleavage of a single covalent bond by either toxin inhibits binding of elongation factors (EF-G and EF-Tu) to the ribosome, thereby inactivating translation (23,24); cell death then results from a caspase III-dependent apoptotic pathway that is triggered by an unknown mechanism (25). The SRL RNA is an essential part of the binding site for EFs (26,27), and possibly for other GTPase translation factors such as initiation factor IF2 (28,29). Synthetic oligonucleotides with the SRL sequence (∼30mers) mimic the form and function of the SRL in the ribosome (1,30). SRL mimics have served as a minimal substrate for EF-G binding (31), for sarcin and ricin activity (32) and for other structural studies (18–20,33–35). The SRL RNA folds into two motifs (18–20,33): a GAGA tetraloop and a bulged-G motif, separated by an invariant C2658•G2663 WC bp (E.coli 23S rRNA numbering is used throughout). Biochemical and structural studies suggest that the EF binding site includes the major groove face of both motifs (20,27,36,37) and may therefore include the intervening C2658•G2663 WC bp. Juxtaposition of these motifs, separated by one invariant WC bp, is thought to present a unique site on the ribosome surface that is recognized by IF2, EFs and toxins alike (20,21,27,36,37).
Mutational studies evaluated the role of the invariant C2658•G2663 bp in EF-G binding (38). The C2658G•G2663C mutation has a dominant lethal phenotype. Consistent with a lethal phenotype, ribosomes and oligonucleotide mimics of the SRL with the C2658G•G2663C mutation have decreased binding to EFs compared with their wild-type counterparts; similar results were obtained for the C2658A•G2663U mutation. Mutations of invariant nucleotides are expected to be detrimental to growth, otherwise sequence variation would be observed in nature. Surprisingly, the C2658U•G2663A mutation affects neither cell growth nor ribosome function (38), yet has not been observed in nature (39,40). In an A-form helix, substitution of one WC bp with another is isosteric; however, the C2658•G2663 bp is flanked on both sides by G•A mismatches. NMR studies have shown that the structure of tandem G•A mismatches can be altered by flanking WC bps (41). Mutation of the C2658•G2663 bp to another WC bp may affect EF binding in a direct or indirect manner. The effect would be direct if the mutation disrupts one or more energetically significant contacts to the EFs by an isosteric replacement of one base with another. The effect would be indirect if the mutation alters the SRL RNA fold, thereby simultaneously altering several potential contacts to the EFs.
To provide insight into the structural basis of a lethal mutation, we compared the structures of a viable (C2658U• G2663A) and a lethal mutation (C2658G•G2663C) of the SRL RNA. The only significant differences between the structures are the functional groups that are presented to the major and the minor grooves. Two Hoogsteen edge groups are identical in the wild-type and the viable mutation but differ in the lethal mutation. Importantly, the two groups are part of the putative EF binding surface. The difference may therefore account for the impaired binding of EF-G to ribosomes with the C2658G•<G2663C mutation. We have determined the wild-type SRL RNA at sufficiently high resolution to establish the atomic coordinate error, hydration and disorder of a typical example of the bulged-G motif. Extensive structural comparisons reveal a common hydration and geometry for the bulged-G motif. Unexpectedly, the bend angles between the motif and its flanking structural elements vary, providing the rigid internal loop with flexible junctions.
MATERIALS AND METHODS
RNA synthesis and purification
The CG-SRL 27mer 5′-UGCUCCUAGUACGAGAGGACCGGAGUG, the UA-SRL 27mer 5′-GGUGCUCAGUAUGAGAAGAACCGCACC and the GC-SRL 27mer 5′-GGUGCUCAGUAGGAGACGAACCGCACC were synthesized at the Yale Keck Oligonucleotide Synthesis Facility (mutant nucleotides are underlined). The CG-SRL RNA reproduces the nucleotides at positions 2647–2673 in E.coli 23S rRNA. In GC-SRL and UA-SRL, nucleotides 1–2 and 26–27 were designed to pair and create blunt ends, whereas nucleotides 3–25 correspond to Rattus norvegicus 28S rRNA nucleotides 4315–4337 (DDBJ/EMBL/GenBank accession no. V01270). To remain consistent with previous publications, nucleotides 4315–4337 are referred to as 4313–4335 (Fig. 1). Each RNA was deprotected with tetra-butyl ammonium fluoride and purified by electrophoresis in urea-denaturing gels.
RNA crystallization
Prior to crystallization, 2.5 mg/ml RNA in 50 mM K•<MOPS (pH 7.0) and 5 mM MgCl2 was heated for 10 min at 60°C then cooled at room temperature for 10 min. Crystals of each variant grew in 1–4 days by vapor diffusion at room temperature in drops having 2 µl of the annealed RNA and 1 µl of well solution. For CG-SRL, the well solution contained 3.0–3.2 M (NH4)2SO4 and buffer X [50 mM K•<MOPS (pH 7.0), 10 mM MgCl2, 10 mM MnCl2]. For GC-SRL and UA-SRL, the well solution contained 3.0–3.2 M (NH4)2SO4 and buffer Y [50 mM K•<MOPS (pH 7.0), 20 mM MgCl2, 1 mM spermine and 2 mM CoCl2]. Before freezing the crystals in propane, they were transferred first to a stabilizer containing 3.5 M (NH4)2SO4 and buffer X or buffer Y, and then to a cryostabilizing solution that also contained 15% (w/v) xylitol.
To obtain crystals of GC-SRL and UA-SRL, we varied the length and the helical twist of the mutant SRL RNAs. The aim of the variation was to enable end-to-end stacking and the formation of pseudo-continuous helices, which are features of many RNA and protein–RNA crystal structures, including those of the rat and E.coli SRL RNA (19,20). To vary the overall helical twist of the SRL RNA hairpin we screened RNAs with both the eukaryotic and the bacterial SRL RNA sequences. These sequences differ primarily by the presence or the absence of one C•<C bp (Fig. 1). For GC-SRL, we screened stems with 6, 7, 8 and 9 bp of the E.coli 23S SRL rRNA sequence; we also screened stems with 5, 6 and 7 bp of the rat 28S SRL rRNA sequence. For UA-SRL, we screened stems with 5 and 6 bp of the rat 28S SRL rRNA sequence. Crystals for each mutant were obtained only with a 5 bp stem of the rat SRL RNA sequence, totaling 27 nt.
Data collection and reduction
Data from the UA-SRL and CG-SRL crystals were collected at the Structural Biology Center (SBC) 19-ID beamline at the Advanced Photon Source (APS) at Argonne National Laboratories using a charge-coupled device (CCD) designed and built by SBC staff. Diffraction data from the GC-SRL crystal were collected at the BioCARS 14-BM-C beamline at the APS using an ADSC 4K CCD. Frozen crystals were maintained at 100K during data collection. Integration and merging of intensities were done with DENZO and SCALEPACK, respectively (42).
Structure determination and refinement
The 1.04 Å resolution data set for the CG-SRL structure was refined starting with the structure (20) of the SRL RNA from E.coli 23S rRNA (PDB no. 483D). The SHELX-97 program (43) was used to refine the structure in which individual anisotropic thermal parameters were restrained to avoid over-fitting the data. Riding hydrogen atoms were used during the final cycles of refinement. Coordinate errors were determined by a blocked full-matrix inversion during the final refinement cycle. Coordinate errors can be used by SHELX-97 to provide standard errors for the resulting torsion angles (Tables 2 and 3) and for contact bond lengths. In the bulged-G motif of the CG-SRL structure, errors for hydrogen bond lengths within the RNA vary from 0.015 to 0.05 Å, and those between RNA and solvent vary from 0.016 to 0.1 Å; errors in torsion angles vary from 1.1° to 18°. In contrast to structures determined at lower resolution where only an average coordinate error is attainable, this analysis clearly distinguishes regions that are well defined from those that are not. The CG-SRL structure contained four regions that were modeled as multiple conformations: 2647–2650, 2654, 2655 and 2671–2673. The latter is not found in the 483D structure. The RMSD between the structure from PDB entry 483D and that of CG-SRL is 0.103 Å. For the CG-SRL structure we used more stringent criteria to determine solvent molecules than those used for the 483D structure. As a result, the 483D structure contains 49 additional solvent molecules that were predominantly located in the second shell of hydration.
Table 2. Bulged-G motif torsion angles.
| Positiona | αCGb,c | αavgd | βCG | βavg | γCG | γavg | δCG | δavg | εCG | εavg | ζCG | ζavg | χCGe | χavg | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C2652 | 1 | 293.3 (1.3) | 295 (7) | 179.4 (1.1) | 173 (5) | 52.4 (1.9) | 58 (6) | 81.4 (1.8) | 80 (2) | 211.2 (1.1) | 212 (4) | 300.8 (1.2) | 295 (4) | 202.9 (1.2) | 199 (4) |
| U2653 | 3af | 291.7 (1.3) | 295 (5) | 172.3 (1) | 168 (3) | 52.4 (1.9) | 56 (4) | 85.9 (1.7) | 86 (3) | 202.4 (1.1) | 210 (6) | 56.3 (1.6) | 52 (5) | 212.5 (1.2) | 214 (4) |
| 3b | 258.3 (1.4)g | 259 (2) | 301 (3) | 326 (18) | |||||||||||
| A2654 | 4a | 170 (1.9) | 164 (8) | 141 (2) | 140 (5) | 49 (2) | 59 (6) | 148 (5) | 151 (2) | 256 (10) | 256 (10) | 165 (10) | 156 (20) | 224.6 (1.6) | 220 (6) |
| 4b | 188 (4) | 187 (4) | 221 (4) | 188 (24) | 63 (2) | 149 (6) | 272 (7) | 126 (7) | |||||||
| G2655 | 5a | 44 (7) | 46 (2) | 253 (6) | 264 (11) | 275 (7) | 267 (11) | 147 (8) | 154 (10) | 194 (6) | 194 (4) | 142 (8) | 139 (5) | 288 (18) | 268 (10) |
| 5b | 268 (7) | 275 (9) | 96 (6) | 85 (9) | 179 (7) | 177 (2) | 170 (7) | 192 (7) | 128 (7) | 263 (16) | |||||
| U2656 | 6 | 291 (3) | 294 (5) | 151.9 (1.9) | 151 (4) | 39 (3) | 36 (4) | 89 (3) | 87 (2) | 215.6 (1.9) | 217 (1) | 296 (2) | 292 (4) | 182.8 (1.6) | 186 (4) |
| 286 (2) | |||||||||||||||
| A2657 | 7 | 284 (2) | 292 (6) | 178.7 (1.6) | 176 (3) | 53 (3) | 52 (2) | 81 (3) | 82 (2) | 212.2 (1.8) | 222 (8) | 306 (2) | 304 (4) | 193.1 (1.4) | 194 (3) |
| C2658 | 8 | 289.1 (1.9) | 296 (8) | 173.3 (1.2) | 169 (6) | 53 (3) | 51 (7) | 79 (2) | 81 (2) | 211.9 (1.5) | 210 (7) | 296.7 (1.5) | 295 (3) | 196.9 (1.4) | 200 (5) |
| G2663 | 9b | 144.4 (1.4) | 139 (9) | 223.2 (1.8) | 231 (8) | 179 (2) | 187 (9) | 85 (3) | 86 (3) | 221.9 (1.5) | 223 (6) | 300.4 (1.5) | 301 (3) | 186.7 (1.4) | 187 (2) |
| 9ch | 209 | 318 | |||||||||||||
| G2664 | 10 | 295.3 (1.6) | 297 (5) | 172.8 (1.1) | 174 (2) | 57 (2) | 47 (9) | 79 (2) | 78 (2) | 201.1 (1.3) | 202 (5) | 281 (1.3) | 284 (6) | 195.3 (1.3) | 202 (6) |
| A2665 | 11a | 253.9 (1.3) | 253 (5) | 77 (2) | 74 (12) | 166.3 (1.5) | 174 (12) | 81.8 (1.9) | 86 (4) | 209.7 (1.2) | 208 (2)i | 285.3 (1.3) | 284 (6) | 178.8 (1.4) | 181 (4) |
| 11b | 234 (4)i | ||||||||||||||
| C2666 | 12a | 303.8 (1.4) | 303 (6) | 170.7 (1.3) | 163 (8) | 62 (2) | 58 (6) | 84 (2) | 83 (1) | 224.8 (1.9) | 205 (5) | 293.3 (1.9) | 290 (5) | 193.7 (1.7) | 197 (3) |
| 12b | 224 (3) | ||||||||||||||
| C2667 | 13 | 296 (2) | 294 (4) | 163.6 (1.9) | 170 (6) | 54 (3) | 53 (5) | 85 (3) | 81 (3) | 217 (2) | 218 (5) | 302 (2) | 298 (4) | 195 (2) | 199 (6) |
| G2668 | 15a | 291 (2) | 296 (5) | 174.6 (1.7) | 173 (2) | 53 (3) | 55 (5) | 76 (3) | 79 (4) | 211 (2) | 214 (2) | 290 (2) | 286 (3) | 192.4 (1.5) | 189 (4) |
| 15bh | 58 | 75 | 19 | 37 | |||||||||||
| Aj | 285 | 184 | 53 | 81 | 214 | 289 | 193 |
aPositions are defined in Figure 3.
bBackbone torsion angles: 5′→O3′n–1–P–αO5′–βC5′–γC4′–δC3′–εO3′–ζPn+1–O5′n+1→3′
cValues with subscript CG are taken from CG-SRL structure with torsion angle errors derived from coordinate errors in parentheses.
dValues with subscript ‘‘avg’’ are the average values for all structures at better than 2 Å resolution with standard deviation in parentheses.
eGlycosidic torsion angle: O4′–C1′–N1–C2 (C or U); O4′–C1′–N9–C4 (A or G)
fMultiple conformers are notated using different letters.
gValues in bold text differ from standard A-form by >36°.
hThese values are from PDB entry 1MSY (9).
iThe two conformations in the kinked strand arise from these alternate torsion angles.
jStandard A-form values (63).
Table 3. Average η and θ values for both types of S-turns.
| Positiona | S1 (bulged-G motif) | S2e | |||||
|---|---|---|---|---|---|---|---|
| ηCGb,c | ηavgd | θCG | θavg | η | θ | ||
| C2652 | 1 | 173.3 (0.4) | 173 (6) | 228 (0.3) | 224 (5) | 171 (11) | 226 (13) |
| NPf | 2ag | 169 (4) | 227 (3) | NPh | NPh | ||
| 2b | |||||||
| U2653 | 3a | 176.9 (0.4) | 174 (6) | 338.8 (0.4) | 341 (2) | 165 (9) | 336 (9) |
| 3b | 162.8 (0.4) | 311.1 (0.6) | 321 (7) | ||||
| A2654 | 4a | 57.6 (1.1) | 57 (14) | 175.4 (1.6) | 177 (6) | 57 (15) | 159 (12) |
| 4b | 79.5 (0.9) | 169 (1.6) | |||||
| G2655 | 5a | 319.3 (2.3) | 311 (8) | 23 (2.5) | 28 (7) | S2I: 26 (6) | S2I and S2II: 329 (15) |
| 5b | 298.1 (1.5) | 41.9 (1.6) | S2II and SIII :335 (5) | SIII: 65 (16) | |||
| U2656 | 6a | 164.8 (1) | 169 (3) | 239.1 (0.5) | 233 (6) | NPh | NPh |
| 6b | 171 (1.1) | ||||||
| A2657 | 7 | 162.4 (0.5) | 168 (5) | 234.8 (0.5) | 241 (6) | S2I: 264 (6) | 221 (16) |
| S2II and SIII: 143 (28) | |||||||
| C2658 | 8 | 160.9 (0.6) | 159 (2) | 225.1 (0.5) | 226 (6) | 162 (9) | 213 (37) |
| G2663 | 9a | 165.4 (0.7) | 175 (9) | 220.8 (0.7) | 219 (5) | i | |
| 9bj | 193 | 356 | i | ||||
| G2664 | 10 | 165.8 (0.5) | 158 (7) | 172.5 (0.5) | 170 (3) | i | |
| A2665 | 11 | 197.5 (0.8) | 202 (4) | 149.3 (0.8) | 159 (11) | i | |
| C2666 | 12a | 187.4 (0.5) | 163 (3) | 236.4 (0.5) | 215 (2) | i | |
| 12b | 185 (3) | 236 (1) | |||||
| NPf | 13 | 165 (4) | 232 (8) | i | |||
| C2667 | 14 | 160.1 (0.5) | 167 (4) | 240.7 (0.6) | 229 (5) | i | |
| G2668 | 15a | 158.5 (0.6) | 166 (7) | 218 (0.6) | 215 (4) | i | |
| 15bj | 356 | 141 | |||||
aPosition numbers are defined in Figure 3.
bVirtual torsion angles: C4′n-1–P–ηC4′–θPn+1–C4′n+1
cValues with subscript CG are taken from CG-SRL structure with torsion angle errors derived from coordinate errors in parentheses.
dValues with subscript ‘‘avg’’ are the average value for structures at better than 2 Å resolution with standard deviation in parentheses.
eVariants of S2 S-turns that have distinctive η and θ values are indicated by superscript roman numerals. A representative backbone trace for each variant is shown in Figure 3d.
fNot present in the CG-SRL structure.
gPositions with multiple values for specific torsion angles reflect alternate conformations at that position.
hNot present in S2 S-turns (Fig. 3f).
iη and θ values for the opposing strand of S2 S-turns are highly variable and average values cannot be calculated.
jThese values are from PDB entry 1MSY (9).
The GC-SRL and UA-SRL structures were determined by molecular replacement with the program EPMR (44) using the structure from PDB entry 483D as the search model. While these structures contain three molecules per asymmetric unit, non-crystallographic symmetry restraints were not used during refinement. The program CNS was used for refinement (45). See Table 1 for final refinement statistics. The UA-SRL structure contained three regions that were modeled as multiple conformations: the phosphate of nucleotide 9 in chain A; the backbone of nucleotide 20 and nucleotides 21–23 of chain B; and nucleotides 20–22 of chain C. Models of the structure were built with the program O (46) using maps calculated with SIGMAA-weighted coefficients (47). Figures were generated with RIBBONS (48), RASTER3D (49) or GRASP (50).
Table 1. Crystallographic data.
| Data set | GC-SRL | UA-SRL | CG-SRL |
|---|---|---|---|
| Resolution range (Å) (outer shell) | 40–2.25 (2.29–2.25) | 40–1.75 (1.78–1.75) | 40–1.04 (1.06–1.04) |
| Space group | P4322 | P4322 | P43 |
| a = b (Å) | 42.14 | 42.25 | 29.529 |
| c (Å) | 336.17 | 336.92 | 76.574 |
| Copies in asymmetric unit | 3 | 3 | 1 |
| Unique reflections (fold redundancy) | 15 569 (9.5) | 29 876 (6.6) | 29 848 (5.1) |
| Completeness (%) | |||
| overall (outer shell) | 98.8 (98.0) | 91.3 (80.0) | 94.9 (82.5) |
| I/σI (outer shell) | 41.1 (6.8) | 31.5 (2.0) | 55.3 (2.0) |
| Rmergea (outer shell) | 0.077 (0.473) | 0.052 (0.468) | 0.037 (0.446) |
| R-factorb (outer shell) | 0.225 (0.295) | 0.201 (0.269) | 0.1361 (0.319) |
| Rfreeb (outer shell) | 0.266 (0.316) | 0.241 (0.294) | 0.1774 |
| % of data used to calculate Rfree | 9.7 | 8.8 | 10.0 |
| RNA atoms | 1734c | 1872c | 708 |
| Water atoms | 99 | 306 | 107 |
| Sulfate ions | 8 | 4 | 0 |
| Sodium ions | 1 | 0 | 0 |
| Coordinate error (Å) | 0.31d | 0.17d | 0.065e |
| RMSD from ideality | |||
| bonds (Å) | 0.004 | 0.003 | 0.007 |
| angles | 0.8° | 0.8° | 0.023 Å |
| Average B-factors (Å2) RNA overall | 33.8 | 19.3 | 17.32 |
aRmerge = ΣhklΣ|Ii – <I>| / ΣhklΣIi for all data with I/σI > –3.
bR factor = Σhkl | |Fobs| – k|Fcalc| | / Σhkl |Fobs|.
cThe mutant structures differ in number of RNA atoms because of the mutation and alternate conformations observed in the UA-SRL mutant structure.
dEstimated coordinate error from SIGMAA (47), using low resolution data truncated to 5 Å.
eAverage radial atomic positional error from SHELXL (43).
DDM (difference distance matrix) analysis of RNA
For each structure, the distance, r, between each atom, i, and its neighboring atom, j, is calculated, giving a distance matrix, rij. The difference between distance matrices from two different structures is the DDM. For proteins, which have two degrees of freedom in the backbone (φ and ψ), distances are calculated using only the Cα atoms. To simplify representation of RNA geometry, which has six degrees of freedom in the backbone (α, β, γ, δ, ε and ζ), distances were calculated using two RNA backbone atoms (C4′ and P), reducing the torsion angles from six to two. The two-atom simplification of the RNA backbone geometry is expected to retain distinguishing structural details (51–54).
Identification of motifs
We wrote the program CHI2 to identify motifs from η and θ values that were calculated with the AMIGOS script (54). A similar analysis can be performed using the program PRIMOS (14). Each motif was identified by η and θ values from the minimum number of nucleotides that define its distinctive geometric features. For the bulged-G motif, we used η and θ values of four nucleotides including and flanking the bulged G. These include the two nucleotides on the 5′-side of the bulged G and one on its 3′-side. For each sequential stretch of four nucleotides in the target structure, a χ2 value was calculated as the sum (for η and θ values of each target and reference nucleotide) of the squared difference between target and reference values that were each normalized by the reference value. The reference included η and θ values from four nucleotides: the bulged G, one nucleotide on its 5′-side and two nucleotides on its 3′-side. The χ2 values were typically <100 for matches with the target. Motifs were identified as the nucleotide stretches with the lowest χ2 values.
Structures used for comparison
The 19 bulged-G motif structures used for comparison are: the CG-SRL structure; the three copies of the bulged-G motifs in the GC-SRL and the UA-SRL structures; the 1.4 Å SRL RNA structure from PDB entry 1MSY (9); the seven examples in the 2.4 Å structure of the 50S subunit of the ribosome from PDB entry 1JJ2 (10); the two copies of the bulged-G motifs in the 2.2 Å resolution restrictocin co-crystal structure from PDB entry 1JBR (21); and the two copies of the bulged-G motifs in the 1.97 Å resolution restrictocin co-crystal structure from PDB entry 1JBS (21). One of the bulged-G motifs in the 50S subunit structure is in the 5S rRNA (75–81 and 158–164); the remaining six bulged-G motifs are in the 23S rRNA (171–178 and 158–164; 210–216 and 224–229; 355–362 and 290–296; 585–591 and 567–572; 1367–1373 and 2052–2057; 2689–2695 and 2700–2705). The numbering for the S-turn strand is listed first; numbers begin and end with nucleotides that pair to form the punctuating WC bps. For analysis of hydration, we used seven structures that were determined to 2.0 Å or higher resolution. After superimposing the equivalent atoms of the bulged-G motif, solvent sites were considered structural if a cluster of at least four of the structures had equivalent solvent sites.
Previous studies (21) indicated that there are two types of S-turns; the S-turn in the bulged-G motif is referred to as S1 and the alternate variety as S2 (14). Seven S2 S-turns from PDB entry 1JJ2 were used for comparison. Each falls into one of three classes of S2 variants, as defined by the average η and θ values for the S-turn strand (Table 3): residues 891–896 and 766–770 (S2I); 1162–1167 and 1179–1184 and 1192 (S2I); 1622–1627 and 1570–1574 (S2III); 1774–1780 and 1764–1769 (S2I); 1869–1874 and 1856–1860 (S2II); 1981–1987 and 1977 and 2001–2003 (S2I); 2887–2892 and 2864–2870 (S2III). The numbering for the S-turn strand is listed first. S2II and S2III variants had not been previously identified (14).
The S-turn is made of two turns: the first reverses the chain direction and involves the two nucleotides on the 5′-side of the bulged G (designated herein as the 5′-turn); the second restores the chain direction and involves the bulged G and the 3′-adjacent nucleotide. The 18 examples of 5′-turns in structures determined at 2.4 Å or better were used for comparison: nine from PDB entry 1JJ2 (residues 84–86, 1069–1071, 1228–1230, 1531–1533, 1662–1664, 2035–2037, 2131–2133, 2389–2391, 2783–2785), two from the 2.3 Å structure of a mutant P4–P6 domain of Tetrahymena thermophila group I intron from PDB entry 1HR2 (55) (two molecules per asymmetric unit; residues 165–167), and seven from a vitamin B12-binding RNA aptamer structure determined at 2.3 Å resolution from PDB entry 1ET4 (56) (five molecules per asymmetric unit; residues 28–30 in chains A–E and residues 12–14 from chains B and C). A least-squares superposition of each 5′-turn includes six residues flanking either side of the core turn residues listed above.
Calculation of bend angles
Bend and torsion angles were calculated with a Python script based on the Crystallography Concept Library (CCL) and Crystallography Protocol Library (CPL) (http://renzresearch.com/cpl.html). Angles were calculated only for flanking structural elements that contained at least 4 bp.
RESULTS
Structure determination and overview
The CG-SRL RNA structure was determined at 1.04 Å resolution (Table 1 and Materials and Methods). The data were refined to a final Rfree of 0.177 and a conventional R factor of 0.136. The data reported here are of better quality than those reported before (20): the Rmerge is lower (0.037 compared with 0.047), the signal-to-noise ratio is higher (I/σI is 55.3 compared with 35.4) and the resulting Rfree is lower (0.177 compared with 0.202). Collection of better data was made possible by using a more brilliant beamline equipped with an undulator and microfocusing capability; previously, a bending magnet beamline was used (20).
Both mutant structures (GC-SRL and UA-SRL) were determined by molecular replacement (Table 1 and Materials and Methods). The GC-SRL crystal data, which extend to 2.25 Å resolution, were refined to a final Rfree of 0.265, with a conventional R factor of 0.225. The UA-SRL crystal data, which extend to 1.75 Å resolution, were refined to a final Rfree of 0.242, with a conventional R factor of 0.202. The GC-SRL and UA-SRL structures, with mutant SRL RNA sequences from the rat 28S rRNA, are appropriate for determining how mutations at the equivalent positions in the E.coli SRL affect its structure, because the site of mutation is flanked by the same base pairs and base triple (Fig. 1). The structures are determined at sufficient resolution to allow detection of small changes in the geometry and hydration that are the result of the mutations. Moreover, each mutant structure has three independent copies of the SRL RNA in the asymmetric unit. Analysis of the variation between each mutant structure and among the three independent copies in each asymmetric unit is helpful in determining whether subtle changes are a result of lattice packing forces or of the mutations. Analysis of the CG-SRL, GC-SRL and the UA-SRL structures shows that they share a common fold; atoms superimpose with a pair-wise RMSD ranging from 0.4 to 0.8 Å (Fig. 2). Each structure consists of a GAGA tetraloop and a bulged-G motif that is connected to the stem region by two water-mediated non-WC bps (19,20).
Figure 2.
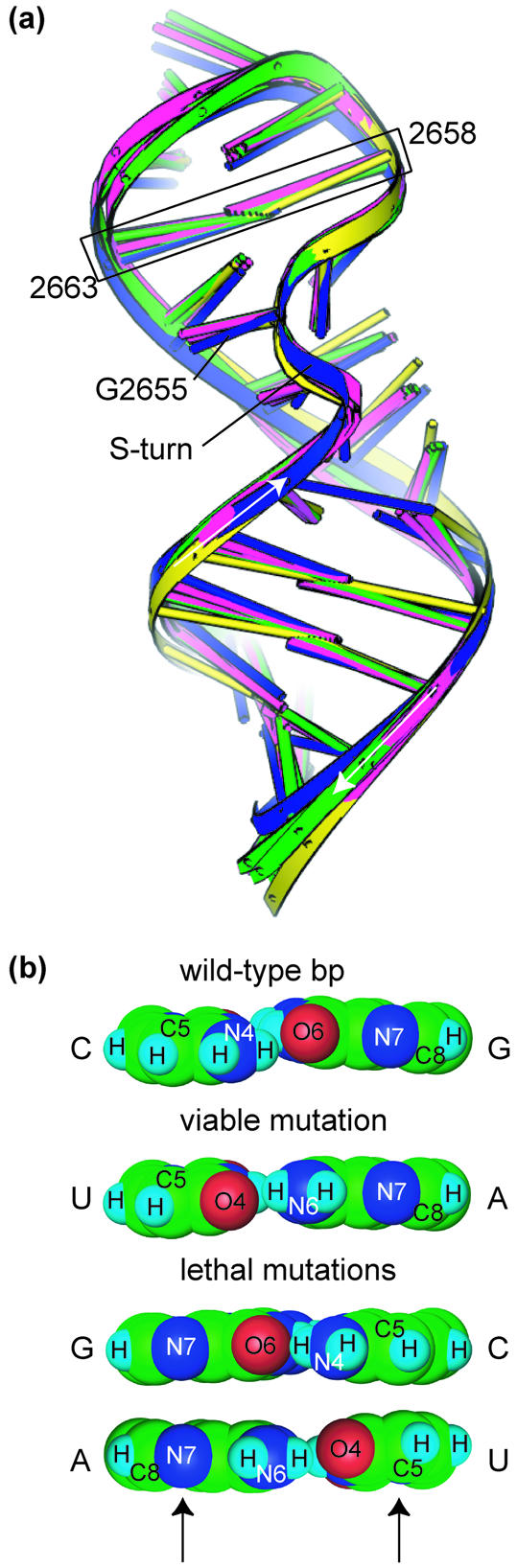
Structural differences between C2658G•G2663C and C2658U•G2663A mutations. (a) Superposition of CG-SRL (blue), GC-SRL (green), UA-SRL (magenta) and wild-type rat SRL RNA [yellow; PDB entry 480D (19)]. The mutated bp is boxed, and the bulged G and the S-turn are shown. Arrows indicate the 5′→3′ direction of the phosphodiester backbone. (b) Space-filling models of the Hoogsteen edges of the viable and lethal mutations of the C2658•G2663 bp; arrows indicate key functional groups that are different in the lethal and the viable mutations.
Structural basis of the lethal mutation
Within a given bulged-G motif such as the SRL, sequence conservation may arise to preserve the fold or surface features essential for sequence-specific recognition. The invariant C2658•<G2663 bp in the SRL RNA structure is an example of the latter. Our structural studies show that the C2658G•<G2663C and the C2658U•<G2663A mutants have the same fold as the wild-type SRL RNA, thereby maintaining most features of the EF binding surface (Fig. 2a). To detect subtle changes in the structure, we carried out DDM analyses (57) on RNA (Materials and Methods). Although DDM analyses have been widely used for comparisons of protein structures, such analyses had not been done for comparisons of RNA structures. Subtle differences can be accentuated or minimized by the choice of which atoms to superimpose. DDM is independent of which atoms are chosen for superposition and is therefore an objective way to assess differences between two structures. Based on DDM, pair-wise variation of the three structures in the asymmetric unit (0.3–0.4 Å) was greater than pair-wise variation between any of the three GC-SRL mutant structures and any of the three UA-SRL mutant structures (0.1–0.2 Å). Apparently, the small deviations in the structure resulting from different lattice environments are larger than those resulting from mutation. The structures with the viable and lethal mutation are therefore the same, except for the functional groups that are presented to the major and the minor grooves. Two functional groups—C5 of C2658 and N7 of G2663—of the WC bp are identical in the wild-type and the viable C2658U•<G2663A mutation but are different in the lethal C2658G•G2663C mutation (Fig. 2b). The difference is significant because the groups are part of the surface that is expected to contact the EFs. Oligonucleotides that mimic the E.coli SRL RNA and ribosomes with the C2658G•G2663C mutation bind to EF-G less well than those with the wild-type sequence (38). The differences of the two functional groups may therefore account for the impaired binding of the elongation factor to ribosomes with the C2658G•G2663C mutation in 23S rRNA and may underlie the lethal phenotype.
Common features
Comparison of seven bulged-G motif structures determined at better than 2.0 Å resolution reveals that the motif has a common hydration and geometry (Fig. 3); atoms superimpose with a pair-wise RMSD that varies between 0.3 and 0.6 Å. The geometry of the S-turn strands is strikingly similar, whereas that of the opposing strand has two similar but distinct trajectories (see below). The resolution cutoff was necessary to adequately define the hydration and the torsion angles of the bulged-G motifs. The structures were determined from crystals grown under low and high salt conditions and were exposed to a variety of lattice environments, thereby minimizing the effect of crystallization artifacts on our analyses.
Figure 3.

Comparisons of bulged-G motifs and variants. (a) The common geometry and hydration of bulged-G motifs among the seven bulged-G motif structures determined at better than 2.0 Å (Materials and Methods) as shown in a stereodiagram of the superposition with AC/YY (blue) and the AA/YY/(YY) variants (purple). (b) Superposition of A•C (blue) and A•A (purple) bps. (c) Superposition of U•C (blue) and C•C (purple) bps. Common solvent molecules in (a), (b) and (c) are colored light blue. (d) Stereo diagram of superimposed S2 motifs with the backbone highlighted for four representative variants (Table 3 and Materials and Methods): two for S2I (black and red); S2II (blue) and S2III (green). The two examples of S2I S-turns have different geometries in the strand that opposes the S-turn. The S2II and S2III variants have not been previously identified (14). (e) Stacking diagram for bulged-G motifs with the consensus sequence. The termini form WC bps; N is A, C, G or U. Sometimes positions 3 and 13 are purines, not pyrimidines. Positions are numbered as a reference for torsion angle values in Tables 2 and 3. Standard symbols are used to classify the bps (62); a circled W represents water-mediated interactions. Dashed boxes indicate nucleotides present only in certain variants. (f) Stacking diagram for S2 motifs. Positions are numbered as a reference for torsion angle values given in Table 3. Standard symbols are used to classify the bps (62). Dashed boxes indicate nucleotides present only in certain variants. The base at position 7 stacks in one of two ways (asterisk and double asterisk).
The CG-SRL structure is a typical bulged-G motif because its torsion angles are typical of those observed for the other related structures (Table 2). The details described for the CG-SRL structure are therefore applicable to other bulged-G motifs. Standard errors for bond distances and torsion angles result directly from the atomic coordinate error. This is the first time that these error values have been determined for an RNA motif. Based on analysis of equivalent hydrogen bonds (58), the non-WC base interactions in CG-SRL are ideal: the angles between the N-H bond and acceptor atoms lie near 180° and none have values <141°; moreover, the distances between hydrogen bonding donor and acceptor atoms range from 2.67 to 3.19 Å (Fig. 4). The non-WC interactions in the motif are stabilized by up to six direct and solvent-mediated base-to-base and base-to-backbone interactions, three more than possible in WC bps. As observed before (20), the S-turn on one strand and the kink on the opposite strand arise from deviations in a few specific torsion angles. Of the 91 possible backbone and glycosidic torsion angles in each of these bulged-G motifs, only 18 deviate by >36° from A-form geometry. The poorer quality of electron density in G2655 relative to the other bases is a result of static and/or dynamic disorder (Fig. 4d). The highest anisotropic thermal parameters are associated with the bulged G and the surrounding S-shaped backbone, which are sites known to be important for protein recognition (16,21,31). As a result, the standard errors of the torsion angles associated with G2655 are higher than those of other nucleotides in the motif. We expect that this region will become more ordered upon protein binding due to the formation of additional stabilizing contacts. Complex formation frequently involves disorder-to-order transitions (59).
Figure 4.

Details of a typical bulged-G motif (S1) determined at 1.04 Å resolution. Final refined model of CG-SRL bps superimposed on a 1.04 Å SIGMAA-weighted 2Fo–Fc density map (47) for (a) the C2658•G2663 bp, (b) the A2657•G2664 bp, (c) the G2655•U2656•A2665 base triple, (e) the A2654•C2666 bp and (f) the U2653•C2667 bp, showing hydrogen bond lengths (Å) and standard errors derived from coordinate errors in parentheses (Å). The distance between O2 of 2653 and N4 of 2667 is 2.965 ± 0.019 Å. This interaction is not drawn as a hydrogen bond because the N-H-O angle is not ideal (58). (d) Each atom of the base triple is represented by a thermal ellipsoid (the more elliptical the shape, the greater the anisotropy) with carbon, oxygen, nitrogen and phosphorous atoms colored gray, red, blue and green, respectively; the alternate conformation for G2655 is included.
Conservation of six nucleotides preserves the fold of the motif (Fig. 1). Mutation of any of the six is expected to result in the loss of at least one of the stabilizing contacts, seven of which interact directly with the phosphodiester backbone (Fig. 4). The distortions in the backbone geometry surrounding the bulged G are important for recognition by sarcin (16,21) and EF-G (31). The S-turn encompasses nucleotides U2653, A2654, the bulged G2655 and U2656; the geometry of its backbone reverses direction at A2654 and is restored to A-form at U2656 (Figs 2a and 3a) (19). In contrast to the tetraloop, which reverses chain direction as a result of α and β torsion angle deviation (9), the S-turn has notable torsion angle deviations throughout the backbone of A2654 and G2655 (Table 2). The sugars of the nucleotides at the sharp bends of the S-turn (A2654 and G2655) have C2′-endo puckers, in agreement with NMR data (18,33,35). In the CG-SRL structure, the S-turn geometry places the 2′-hydroxyl groups of the sugars with inverted puckers in the major groove (Fig. 5).
Figure 5.

Sequence-dependent changes on the surfaces of the bulged-G motif (S1). Space-filling presentations of the minor groove (left) and major groove (center) surfaces of the three classes of the motif, with the corresponding sequences (right): (a) AC/YY (CG-SRL), (b) AA/YY/(YY) [from PDB entry 480D (19)] and (c) AA/RR [from PDB entry 1JJ2 (1,10)]. Base hydrogen bond donor (green) and acceptor (yellow) groups are shown, as are base hydrophobic (purple) and 2′-hydroxyl (blue) groups. Underlined and outlined bases in the sequences are labeled in the major and minor grooves, respectively.
The geometry of the strand opposing the S-turn has a common kink and a distinctive feature in which one of two similar conformations is observed (Fig. 3 and Materials and Methods). The backbone kink is common to cross-strand A stacks, including bulged-G motifs. The kink arises from deviations in α, β and γ torsion angles of the adenosine (in this case A2665) that permit interstrand, but disrupt intrastrand, stacking (13,19,20). The WC edges of the adenosines of this motif are presented in the minor groove, providing distinctive surface features. The two distinct conformations of the strand opposing the S-turn arise primarily from an ∼24° rotation about the ε torsion angle of A2665 (Table 2). The difference depends on whether an A•C or A•A trans Hoogsteen/Hoogsteen bp forms on the 5′-side of G2655. The CG-SRL and the two mutant structures (GC-SRL and UA-SRL) present the related but distinct conformations that arise from the pairing of A2654, or its equivalent, with a cytosine or an adenosine base (see below). A•C bps are wider than their A•A counterparts (Fig. 3b), leading to a 3 Å difference in the two structures that affects the placement of the adjacent pyrimidine•pyrimidine (Y•Y) bps (Fig. 3c). The CG-SRL structure represents a class of variants in which the A•C bp (in this case A2654•C2666) stacks on a water-mediated Y•Y bp (in this case U2653•C2667; referred to hereafter as AC/YY). The A2654•C2666 bp has only one direct hydrogen bond (Fig. 4e) and the U2653•C2667 bp has none (Fig. 4f); six solvent-mediated contacts stabilize the former and seven such contacts stabilize the latter. The mutant structures (GC-SRL and UA-SRL) represent the other common class of variants: an A•A bp and one or two water-mediated Y•Y bps stack on one another [referred to hereafter as AA/YY/(YY)]. Both form trans Hoogsteen/Hoogsteen bps.
Virtual torsion angles η and θ
To apply the high-resolution data (Table 2) to the typical RNA structure determined at ∼3 Å resolution, we calculated two virtual torsion angles first proposed by Olson (60): C4′n–1–P–C4′–Pn+1 (η) and P–C4′–Pn+1–C4′n+1 (θ). Analysis of η/θ values is appropriate at this typical resolution because the locations of the backbone phosphate (P) and the sugar moiety (in this case C4′) are sufficiently well determined. Moreover, representing RNA geometry by two virtual angles instead of the six backbone torsion angles is believed to result in minimal loss of structural information (14,54,60). Our analyses of high-resolution structures of bulged-G motifs define the mean and variation of its η and θ torsion angles (Table 3). These values can be used to identify bulged-G motifs in newly determined structures (Materials and Methods) (14). One disadvantage of this type of analysis is that it fails to identify bulged-G motifs in which the S-turn is formed by two strands instead of one (22).
Common hydration
Comparison of bulged-G motif structures suggests 10 common sites of hydration: four lie in the minor groove and contact the WC edges of the invariant adenosines (A2657 and A2665), five lie in the major groove, and one interacts with the backbone of the S-turn (Fig. 3a). The location of each of these solvent molecules is conserved relative to an interacting atom of the RNA molecule and is independent of the lattice environment and of the buffer used during crystallization. The conserved solvent sites coincide with site-specific interactions made by proteins and RNA found in the ribosome. In the structure of the 50S ribosomal subunit (1,10), adenosines equivalent to A2657 and A2665 interact with ribosomal proteins and rRNAs at these conserved hydration sites. Conserved hydration in the major groove marks sites of interaction with protein; a relevant example is the conserved solvent site of A2654 that coincides with a lysine side chain contact made by the sarcin homolog restrictocin (21). In a comparison that includes the 19 structures of bulged-G motifs determined at 2.4 Å resolution or better, metal ions are observed in several examples but do not share a common binding site. In four examples (21), two monovalent metal ions lie in the major groove that is 5′ to the bulged G. In two other examples, a single divalent metal ion contacts the pro-Rp phosphate oxygen of either the bulged G or the 5′ adjacent nucleotide (1,10).
Sequence-dependent surface features
Unlike the six conserved nucleotides that preserve the fold of the motif, the remaining seven to nine nucleotides vary to create a diverse set of sequences, thereby altering the contacts that decorate both grooves. The primary distinctive features are in the minor groove where bases present their WC edges for recognition. Aside from the conserved A2654, sequence variation of the nucleotides located on the 5′-side of the bulged G can be grouped into one of three classes (Fig. 5). In addition to the AA/YY and the AA/YY/(YY) variants, loop E of 5S rRNA from the structure of the 50S ribosomal subunit (1,10) provides an example of the third and less common class where an A•A bp stacks on a purine•purine (R•R) trans Hoogsteen/sugar edge bp (referred to hereafter as AA/RR). To date, G•A bps are the only R•R bps that have been observed. A large number of sequences are possible at the seven to nine variable positions within the bulged-G motif; however, only a few are observed in the ribosome and in the specificity domain of RNase P. Sequence variation is most common in the punctuating WC bps. Each class presents a variety of functional groups to the minor and major grooves for recognition.
Flexible junctions
Comparisons show that the bend angles between the bulged-G motif and its flanking RNA structural elements vary (Fig. 6). We extended our comparisons to include the 19 structures determined at 2.4 Å resolution or better. Several structural elements are observed flanking either side of the bulged-G motif with an A-form helix being the most common. To determine the bend angle between the motif and neighboring elements, each element is represented by a vector. The vector passes through the centroid of the structural element and is parallel to another vector, which is the average of the normals for each base plane in the element. A bulged-G motif with flanking elements is therefore represented by three vectors: two bend angles and an overall torsion angle. One bend angle, between the bulged-G motif and the element on the 5′-side of the bulged G, varies from 20° to 39°. The other bend angle varies from 11° to 29°. The resulting torsion angle varies from 108° to 130°. There is no correlation between the variation in the angles (bend and torsion) and the crystalline environments of the motifs, which include either lattice contacts, RNA contacts, protein contacts or a combination thereof. The variation is independent of sequence and therefore creates a flexible joint where this RNA building block stacks on other RNA structural elements.
Figure 6.

Flexible junctions of the bulged-G motif (S1). Superposition of the 19 bulged-G motif structures determined at 2.4 Å resolution or better (Materials and Methods), showing the corresponding vectors of the motif and its flanking structural elements (left) and the resulting bend and torsion angles (right). The 5′→3′ direction of the bulged strand is shown (arrow).
Variation on the S-turn theme
Previous studies (21) indicated that there are two types of S-turns (Fig. 3): the bulged-G motif and the alternate variety are designated S1 and S2, respectively (14). Both motifs share the characteristic S-shape in the phosphodiester backbone but differ significantly in the positions of equivalent bases, notably that of the bulged nucleotide (21, see figure 4b therein). As a result, the S1 and S2 motifs have distinctive η and θ torsion angles (Table 3). The S1 motif has six nucleotides that preserve its fold, whereas the S2 motif shows no sequence conservation and more structural variability than does the S1 motif (Fig. 3a and d). Significant differences are also observed in the stacking: S1 motifs have a cross-strand A stack and a cross-strand G stack (Fig. 3e) whereas S2 motifs typically have intrastrand stacking, although one variant has one cross-strand interaction (Fig. 3f). Superposition of S1 and S2 S-turns revealed that the two nucleotides located on the 5′-side of the bulged nucleotide share a common geometry and form the first turn of the S-shape, the 5′-turn (Materials and Methods), whereas the 3′-nucleotides contain the features that distinguish the two types of S-turns. Interestingly, this shared 5′-turn is a recurring feature of rRNA, an RNA aptamer and the P4–P6 domain of a group I intron. This turn reverses chain direction and is employed in a variety of different structural roles so that its termini do not superimpose. Visual inspection of these 5′-turns revealed additional S2 motifs (Table 3 and Fig. 3) that were not previously noted (14). The 5′-turn, which is stabilized by a sugar-to-backbone hydrogen bond, may nucleate formation of the second turn of the S-shape. Formation of S-turns is entropically costly because it requires two sequential backbone turns and is therefore expected to form infrequently.
DISCUSSION
The 1.04 Å resolution structure provides an unprecedented view of a typical example of a bulged-G motif (Fig. 4). The structural analyses reported here are the first comprehensive examinations of the common and distinctive features of the geometry and hydration of the bulged-G motif. These features are critical for establishing restraints in the refinement of RNA structure and for building accurate RNA models based on homology modeling that include this ubiquitous motif. Structural studies have identified the motif in the specificity domain of RNase P (17) and in ribosomes from each kingdom of life (1,19,20). The nine bulged-G motifs identified in the ribosome (14,21,22) provide binding sites for protein, RNA and sometimes both. Interestingly, two of the nine motifs appear to be universal structural features of the ribosome. Both are located in regions of conserved secondary structure of the 23S–28S rRNA from bacteria, archaea and eukaryotes, including rRNA from chloroplasts and mitochondria (29,40). One invariant bulged-G motif is in the SRL RNA; the other (G1370) is buried in the central domain of the 50S subunit and interacts with L22, part of which lines the peptide exit tunnel (61).
Common features
The geometry of the S-turn in one strand and a kink in the opposing strand is strikingly similar among bulged-G motifs (Fig. 3). The motif can be identified in newly determined structures by analysis of the characteristic η/θ values of the S-turn (14,54) (Materials and Methods). The backbone shape brings the phosphate groups closer together relative to A-form helices, thereby increasing the concentration of phosphate groups and favoring electrostatic attractions. On the strand opposing the S-turn, a kink inverts the helical curvature of the phosphodiester backbone, thereby straightening its path and reducing the charge density on that strand. This combination of backbone compression and extension appears to make the bulged-G motif amphipathic. One surface is more polar (S-turn side) and either is solvent accessible or forms salt bridges with positive patches on proteins or a divalent metal ion. The other surface is less polar (kinked-strand side) and is usually buried, making RNA–RNA contacts. The conserved residues in the bulged-G motif provide distinctive contacts to both grooves (Fig. 5). Both adenosines of the cross-strand A stack present their distinctive WC edges to the minor groove for recognition. Two of the nine bulged-G motifs from the ribosome form A-minor interactions with these cross-strand adenosines (6,13). In the major groove, the O6 and N7 functional groups of G2664 and the bulged G2655, or the equivalent, are contacted by restrictocin in one case (21) and in other cases by either L15E, L22 or L30 (1,22). The bulged G is also available for stacking interactions; an adenosine base stacks on the 5′-side of the bulged G in the structure of the specificity domain of RNase P (17). The major groove usually lacks the hydroxyl moiety that uniquely provides proton donor and acceptor groups; however, the chain reversals in the S-turn result in the re-positioning of the 2′-hydroxyl groups of A2654 and G2655 from the minor to the major groove.
Distinctive features
Aside from the six conserved nucleotides of the bulged-G motif (Fig. 1), variation of the remaining nucleotides in the motif primarily alters contacts that allow sequence-specific recognition in the minor groove (Fig. 5). Between the WC bps that punctuate the motif, only the distinctive WC edges of the bases are presented to the minor groove for recognition. In addition, the two conformations of the strand opposing the S-turn depend on sequence (Fig. 3). Differences in the geometry of A•C and A•A bps provide changes in the trajectory of the kinked strand in the AC/YY and AA/YY/(YY) variants that may be exploited for recognition. The GC-SRL and UA-SRL structures illustrate distinctive features in the major groove. Two Hoogsteen edge groups (C5 of C2658 and N7 of G2663) are identical in the wild-type and the viable mutation but are different in the lethal mutation, whereas the fold of the three structures is the same (Fig. 2). Both groups are part of the surface that is expected to contact elongation factors, and binding studies indicate that the C2658G•G2663C mutation disrupts binding of EF-G to the SRL RNA (38). The simplest conclusion that can be drawn from these data is that the two Hoogsteen edge groups form energetically significant contacts to EF-G. Alternatively, complex formation with EF-G could induce the SRL RNA structure to adopt a different conformation where differences in stacking interactions may play a role. Viable sequences have a pyrimidine on the 5′-side (C or U at 2658) whereas lethal mutations have a purine (A or G at 2658). Additional data are needed to distinguish between these alternative hypotheses.
Architectural implications
There is a growing consensus that RNA motifs are rigid objects (5–7,9,10,13,19,20). Our studies indicate that the bulged-G motif is no exception. Structural comparisons indicate that the joints between the bulged-G motif and its flanking structural elements are flexible (Fig. 6). The majority of flexibility in RNA structures may arise at junctions between rigid motifs. Apparently, large RNAs are comprised of rigid modules, such as motifs and A-form helices, which stack on one another and bend and twist at their joints to generate a greater variety of folds.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Ira G. Wool for helpful discussions, and for providing the GC-SRL and the UA-SRL RNA; to Kerren Swinger for help with RNA purification and crystal growth of CG-SRL; to Xiaojing Yang and Zhong Ren of Renz Research, Inc. for calculating the bend and torsion angles; and to the staff at both BioCARS and SBC for help with data collection. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707. This work was supported by grants from the National Institutes of Health to C.C.C. (GM59782). Coordinates and structure factors were deposited in the Protein Data Bank [CG-SRL (PDB no. 1Q9A); GC-SRL (PDB no. 1Q93); and UA-SRL (PDB no. 1Q96)].
PDB nos 1Q9A, 1Q93, 1Q96
REFERENCES
- 1.Ban N., Nissen,P., Hansen,J., Moore,P.B. and Steitz,T.A. (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science, 289, 905–920. [DOI] [PubMed] [Google Scholar]
- 2.Wimberly B.T., Brodersen,D.E., Clemons,W.M.,Jr, Morgan-Warren,R.J., Carter,A.P., Vonrhein,C., Hartsch,T. and Ramakrishnan,V. (2000) Structure of the 30S ribosomal subunit. Nature, 407, 327–339. [DOI] [PubMed] [Google Scholar]
- 3.Pioletti M., Schlunzen,F., Harms,J., Zarivach,R., Gluhmann,M., Avila,H., Bashan,A., Bartels,H., Auerbach,T., Jacobi,C. et al. (2001) Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J., 20, 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harms J., Schluenzen,F., Zarivach,R., Bashan,A., Gat,S., Agmon,I., Bartels,H., Franceschi,F. and Yonath,A. (2001) High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell, 107, 679–688. [DOI] [PubMed] [Google Scholar]
- 5.Doherty E.A., Batey,R.T., Masquida,B. and Doudna,J.A. (2001) A universal mode of helix packing in RNA. Nature Struct. Biol., 8, 339–343. [DOI] [PubMed] [Google Scholar]
- 6.Nissen P., Ippolito,J.A., Ban,N., Moore,P.B. and Steitz,T.A. (2001) RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl Acad. Sci. USA, 98, 4899–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krasilnikov A.S. and Mondragon,A. (2003) On the occurrence of the T-loop RNA folding motif in large RNA molecules. RNA, 9, 640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szep S., Wang,J. and Moore,P.B. (2003) The crystal structure of a 26-nucleotide RNA containing a hook-turn. RNA, 9, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Correll C.C. and Swinger,K. (2003) Common and distinctive features of GNRA tetraloops based on a GUAA tetraloop structure at 1.4 Å resolution. RNA, 9, 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein D.J., Schmeing,T.M., Moore,P.B. and Steitz,T.A. (2001) The kink-turn: a new RNA secondary structure motif. EMBO J., 20, 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wimberly B., Varani,G. and Tinoco,I.,Jr (1993) The conformation of loop E of eukaryotic 5S ribosomal RNA. Biochemistry, 32, 1078–1087. [DOI] [PubMed] [Google Scholar]
- 12.Leontis N.B. and Westhof,E. (1998) A common motif organizes the structure of multi-helix loops in 16 S and 23 S ribosomal RNAs. J. Mol. Biol., 283, 571–583. [DOI] [PubMed] [Google Scholar]
- 13.Correll C.C., Freeborn,B., Moore,P.B. and Steitz,T.A. (1997) Metals, motifs and recognition in the crystal structure of a 5S rRNA domain. Cell, 91, 705–712. [DOI] [PubMed] [Google Scholar]
- 14.Duarte C.M., Wadley,L.M. and Pyle,A.M. (2003) RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids Res., 31, 4755–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore P.B. (1999) Structural motifs in RNA. Annu. Rev. Biochem., 68, 287–300. [DOI] [PubMed] [Google Scholar]
- 16.Gluck A. and Wool,I.G. (1996) Determination of the 28 S ribosomal RNA identity element (G4319) for alpha-sarcin and the relationship of recognition to the selection of the catalytic site. J. Mol. Biol., 256, 838–848. [DOI] [PubMed] [Google Scholar]
- 17.Krasilnikov A.S., Yang,X., Pan,T. and Mondragon,A. (2003) Crystal structure of the specificity domain of ribonuclease P. Nature, 421, 760–764. [DOI] [PubMed] [Google Scholar]
- 18.Szewczak A.A. and Moore,P.B. (1995) The sarcin/ricin loop, a modular RNA. J. Mol. Biol., 247, 81–98. [DOI] [PubMed] [Google Scholar]
- 19.Correll C.C., Munishkin,A., Chan,Y.L., Ren,Z., Wool,I.G. and Steitz,T.A. (1998) Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc. Natl Acad. Sci. USA, 95, 13436–13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Correll C.C., Wool,I.G. and Munishkin,A. (1999) The Two Faces of the Escherichia coli 23 S rRNA Sarcin/Ricin Domain: The Structure at 1.11 Å Resolution. J. Mol. Biol., 292, 275–287. [DOI] [PubMed] [Google Scholar]
- 21.Yang X., Gerczei,T., Glover,L. and Correll,C.C. (2001) Crystal structures of restrictocin-inhibitor complexes with implications for RNA recognition and base flipping. Nature Struct. Biol., 8, 968–973. [DOI] [PubMed] [Google Scholar]
- 22.Leontis N.B., Stombaugh,J. and Westhof,E. (2002) Motif prediction in ribosomal RNAs lessons and prospects for automated motif prediction in homologous RNA molecules. Biochimie, 84, 961–973. [DOI] [PubMed] [Google Scholar]
- 23.Montanaro L., Sperti,S., Mattioli,A., Testoni,G. and Stirpe,F. (1975) Inhibition by ricin of protein synthesis in vitro. Inhibition of the binding of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 to ribosomes. Biochem. J., 146, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez-Puentes C. and Vazquez,D. (1977) Effects of some proteins that inactivate the eukaryotic ribosome. FEBS Lett., 78, 143–146. [DOI] [PubMed] [Google Scholar]
- 25.Olmo N., Turnay,J., De Buitrago,G.G., De Silanes,I.L., Gavilanes,J.G. and Lizarbe,M.A. (2001) Cytotoxic mechanism of the ribotoxin alpha-sarcin. Induction of cell death via apoptosis. Eur. J. Biochem., 268, 2113–2123. [DOI] [PubMed] [Google Scholar]
- 26.Hausner T.P., Atmadja,J. and Nierhaus,K.H. (1987) Evidence that the G2661 region of 23S rRNA is located at the ribosomal binding sites of both elongation factors. Biochimie, 69, 911–923. [DOI] [PubMed] [Google Scholar]
- 27.Moazed D., Robertson,J.M. and Noller,H.F. (1988) Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature, 334, 362–364. [DOI] [PubMed] [Google Scholar]
- 28.LaTeana A., Gualerzi,C.O. and Dahlberg,A.E. (2001) Initiation factor IF 2 binds to the alpha-sarcin loop and helix 89 of Escherichia coli 23S ribosomal RNA. RNA, 7, 1173–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cameron D.M., Thompson,J., March,P.E. and Dahlberg,A.E. (2002) Initiation factor IF2, thiostrepton and micrococcin prevent the binding of elongation factor G to the Escherichia coli ribosome. J. Mol. Biol., 319, 27–35. [DOI] [PubMed] [Google Scholar]
- 30.Wool I.G., Gluck,A. and Endo,Y. (1992) Ribotoxin recognition of ribosomal RNA and a proposal for the mechanism of translocation. Trends Biochem. Sci., 17, 266–269. [DOI] [PubMed] [Google Scholar]
- 31.Munishkin A. and Wool,I.G. (1997) The ribosome-in-pieces: binding of elongation factor EF-G to oligoribonucleotides that mimic the sarcin/ricin and thiostrepton domains of 23S ribosomal RNA. Proc. Natl Acad. Sci. USA, 94, 12280–12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wool I.G. (1997) Structure and Mechanism of Action of the Cytotoxic Ribonuclease α-Sarcin. Academic Press, Inc., San Diego, CA. [Google Scholar]
- 33.Szewczak A.A., Moore,P.B., Chan,Y.L. and Wool,I.G. (1993) The conformation of the sarcin/ricin loop from 28S ribosomal RNA. Proc. Natl Acad. Sci. USA, 90, 9581–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rife J.P., Stallings,S.C., Correll,C.C., Dallas,A., Steitz,T.A. and Moore,P.B. (1999) Comparison of the crystal and solution structures of two RNA oligonucleotides. Biophys. J., 76, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seggerson K. and Moore,P.B. (1998) Structure and stability of variants of the sarcin-ricin loop of 28S rRNA: NMR studies of the prokaryotic SRL and a functional mutant. RNA, 4, 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agrawal R.K., Penczek,P., Grassucci,R.A. and Frank,J. (1998) Visualization of elongation factor G on the Escherichia coli 70S ribosome: the mechanism of translocation. Proc. Natl Acad. Sci. USA, 95, 6134–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ban N., Nissen,P., Hansen,J., Capel,M., Moore,P.B. and Steitz,T.A. (1999) Placement of protein and RNA structures into a 5 Å-resolution map of the 50S ribosomal subunit. Nature, 400, 841–847. [DOI] [PubMed] [Google Scholar]
- 38.Chan Y.L., Sitikov,A.S. and Wool,I.G. (2000) The phenotype of mutations of the base-pair C2658.G2663 that closes the tetraloop in the sarcin/ricin domain of Escherichia coli 23 S ribosomal RNA. J. Mol. Biol., 298, 795–805. [DOI] [PubMed] [Google Scholar]
- 39.Cannone J.J., Subramanian,S., Schnare,M.N., Collett,J.R., D’Souza,L.M., Du,Y., Feng,B., Lin,N., Madabusi,L.V., Muller,K.M. et al. (2002) The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron and other RNAs. BMC Bioinformatics, 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannone J.J., Subramanian,S., Schnare,M.N., Collett,J.R., D’Souza,L.M., Du,Y., Feng,B., Lin,N., Madabusi,L.V., Muller,K.M. et al. (2002) The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron and other RNAs: Correction. BMC Bioinformatics, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walter A.E., Wu,M. and Turner,D.H. (1994) The stability and structure of tandem GA mismatches in RNA depend on closing base pairs. Biochemistry, 33, 11349–11354. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 43.Sheldrick G.M. and Schneider,T.R. (1997) SHELXL: high-resolution refinement. Methods Enzymol., 277, 319–343. [PubMed] [Google Scholar]
- 44.Kissinger C.R., Gehlhaar,D.K. and Fogel,D.B. (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr., 55, 484–491. [DOI] [PubMed] [Google Scholar]
- 45.Brunger A.T., Adams,P.D., Clore,G.M., DeLano,W.L., Gros,P., Grosse-Kunstleve,R.W., Jiang,J.S., Kuszewski,J., Nilges,M., Pannu,N.S. et al. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr., 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 46.Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 47.Read R.J. (1986) Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A, 42, 140–149. [Google Scholar]
- 48.Carson M. (1991) Ribbons. Methods Enzymol., 277, 493–505. [PubMed] [Google Scholar]
- 49.Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- 50.Nicholls A., Bharadwaj,R. and Honig,B. (1993) GRASP: graphical representation and analysis of surface properties. Biophys. J., 64, A166. [Google Scholar]
- 51.Malathi R. and Yathindra,N. (1982) Secondary and tertiary structural foldings in tRNA. A diagonal plot analysis using the blocked nucleotide scheme. Biochem. J., 205, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malathi R. and Yathindra,N. (1983) The heminucleotide scheme: an effective probe in the analysis and description of ordered polynucleotide structures. Biopolymers, 22, 2961–2976. [DOI] [PubMed] [Google Scholar]
- 53.Malathi R. and Yathindra,N. (1985) Backbone conformation in nucleic acids: an analysis of local helicity through heminucleotide scheme and a proposal for a unified conformational plot. J. Biomol. Struct. Dyn., 3, 127–144. [DOI] [PubMed] [Google Scholar]
- 54.Duarte C.M. and Pyle,A.M. (1998) Stepping through an RNA structure: a novel approach to conformational analysis. J. Mol. Biol., 284, 1465–1478. [DOI] [PubMed] [Google Scholar]
- 55.Juneau K., Podell,E., Harrington,D.J. and Cech,T.R. (2001) Structural basis of the enhanced stability of a mutant ribozyme domain and a detailed view of RNA–solvent interactions. Structure, 9, 221–231. [DOI] [PubMed] [Google Scholar]
- 56.Sussman D. and Wilson,C. (2000) A water channel in the core of the vitamin B(12) RNA aptamer. Struct. Fold. Des., 8, 719–727. [DOI] [PubMed] [Google Scholar]
- 57.Richards F.M. and Kundrot,C.E. (1988) Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins, 3, 71–84. [DOI] [PubMed] [Google Scholar]
- 58.Llamas Saiz A.L. and Foces-Foces,C. (1990) N-H··Nsp2 hydrogen interactions in organic crystals. J. Mol. Struct., 238, 367–382. [Google Scholar]
- 59.Williamson J.R. (2000) Induced fit in RNA–protein recognition. Nature Struct. Biol., 7, 834–837. [DOI] [PubMed] [Google Scholar]
- 60.Olson W.K. (1980) Configurational statistics of polynucleotide chains. An updated virtual bond model to treat effects of base stacking. Macromolecules, 13, 721–728. [DOI] [PubMed] [Google Scholar]
- 61.Nissen P., Hansen,J., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 62.Leontis N.B. and Westhof,E. (2001) Geometric nomenclature and classification of RNA base pairs. RNA, 7, 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gelbin A., Schneider,B., Clowney,L., Hsieh,S., Olson,W.K. and Berman,H.M. (1996) Geometric parameters in nucleic acids: sugar and phosphate constraints. J. Am. Chem. Soc., 118, 519–529. [Google Scholar]