

Abstract
The aspartate receptor is one of the ligand-specific, homodimeric chemoreceptors that detects extracellular attractants and triggers the chemotaxis pathway of Escherichia coli and Salmonella typhimurium. This receptor regulates the activity of the histidine kinase CheA, which forms a kinetically stable complex with the receptor cytoplasmic domain. An atomic four-helix bundle model has been constructed for this domain, which is functionally subdivided into the signaling and adaptation subdomains. The proposed four-helix bundle structure of the signaling subdomain, which binds CheA, is fully supported by experimental evidence. Much less evidence is available to test the four-helix bundle model of the adaptation subdomain, which possesses covalent adaptation sites and docking surfaces for adaptation enzymes. The present study focuses on a putative helix near the C terminus of the adaptation subdomain. To probe the structural and functional features of positions G467–A494 in this C-terminal region, a cysteine and disulfide scanning approach has been employed. Measurement of the chemical reactivities of scanned cysteines reveals an α-helical periodicity of exposed and buried residues, confirming α-helical secondary structure and mapping out a buried packing face. The effects of cysteine substitutions on activity in vivo and in vitro highlight the functional importance of the helix, especially its buried face. A scan for disulfide bond formation between symmetric pairs of engineered cysteines reveals promiscuous collisions between subunits, indicating the presence of significant thermal dynamics. A scan for functional disulfides reveals lock-on and signal-retaining disulfide bonds formed between symmetric pairs of cysteines at buried positions, indicating that the buried face of the helix lies near the subunit interface of the homodimer in the equilibrium structures of both the apo and aspartate-bound states where it plays a critical role in kinase regulation. These results strongly support the existing four-helix bundle model of the adaptation subdomain structure. A mechanistic model is proposed in which a signal is transmitted through the adaptation subdomain by a change in supercoiling of the four-helix bundle.
The mechanism of signal transduction by which cell-surface receptors regulate cytoplasmic kinases is an issue of central importance in signaling biology. Some receptors activate their appropriate kinases by dimerization, but others trigger kinase activation by transmembrane conformational changes. The latter class includes a large superfamily of cell-surface receptors that regulate cytoplasmic histidine kinases in prokaryotic and eukaryotic two-component signaling pathways (1, 2). An important subfamily of this histidine kinase-coupled receptor superfamily is responsible for regulating the thermo-, photo-, osmo-, redox-, and chemotaxis pathways of a wide variety of prokaryotic organisms (3–5). These taxis receptors, comprising a group of over 2000 homologues (6), exhibit regions of high conservation in their cytoplasmic domains (7, 8). They also appear to have a conserved mechanism of transmembrane signaling and cytoplasmic regulation, because active chimeric receptors have been formed by fusing receptor domains from distantly related pathways (9–12). Even certain unrelated receptors, such as the human insulin receptor, may use a similar mechanism of conformational transmembrane signaling (13, 14).
The chemotaxis pathway of Escherichia coli and Salmonella typhimurium is exquisitely sensitive and can sense the binding of only one or a few molecules of attractant to the cell (15). The aspartate receptor is one of five homodimeric receptors utilized by this pathway to detect specific attractant and repellent molecules in the extracellular environment (4, 5). Aspartate binding to the periplasmic domain of the receptor triggers the propagation of a conformational signal across the cell membrane to the associated cytoplasmic histidine kinase, CheA (4). The receptor–kinase complex, which is stabilized by the coupling protein CheW, exhibits a lifetime on the order of tens of minutes independent of the ligand (16–18). Aspartate binding to the receptor allosterically regulates the autophosphorylation of bound CheA and thereby controls the subsequent phosphotransfer reactions that activate the response regulators CheY or CheB (19, 20). The resulting phospho-CheY controls the swimming activity of the cell by interacting with the flagellar motor switch components, while phospho-CheB regulates the adaptation branch of the chemotaxis pathway (5, 21, 22).
The two identical subunits of the aspartate receptor are each 60 kDa in size, and the receptor can be divided into periplasmic, transmembrane, and cytoplasmic regions as illustrated in Figure 1 (4). The structures of the periplasmic and transmembrane regions are well-characterized, with each dominated by four-helix bundles (23–25). In the periplasmic domain, helices α1–4 of each subunit form a four-helix bundle, and the two symmetric bundles associate at the dimer interface dominated by an α1–α1′ coiled-coil interaction. The two symmetric aspartate-binding sites lie within the interfacial region, where each is composed of residues from both subunits. Two helices from each subunit give rise to the bundle of four membrane-spanning helices α1/TM1, α1′/TM1′, α4/TM2, and α4′/TM2′ that comprise the transmembrane region. The mechanism of the ligand-binding-induced transmembrane signaling is a subtle (~1.6 Å) piston-type displacement of the signaling helix α4/TM2 toward the cytoplasm (4), as observed in the superimposed crystal structures of the apo- and aspartate-occupied periplasmic domain (26). Further support for the piston displacement has been provided by engineered disulfide bonds that lock the membrane-bound receptor in the on and off states, ligand-induced changes in disulfide formation rates, modeling studies of the conformational change induced by docking of the maltose-binding protein, ESR spin-label studies, solid-state NMR studies, and engineered helix displacements driven by substitutions at the membrane–water interface (4, 27–33).
Figure 1.
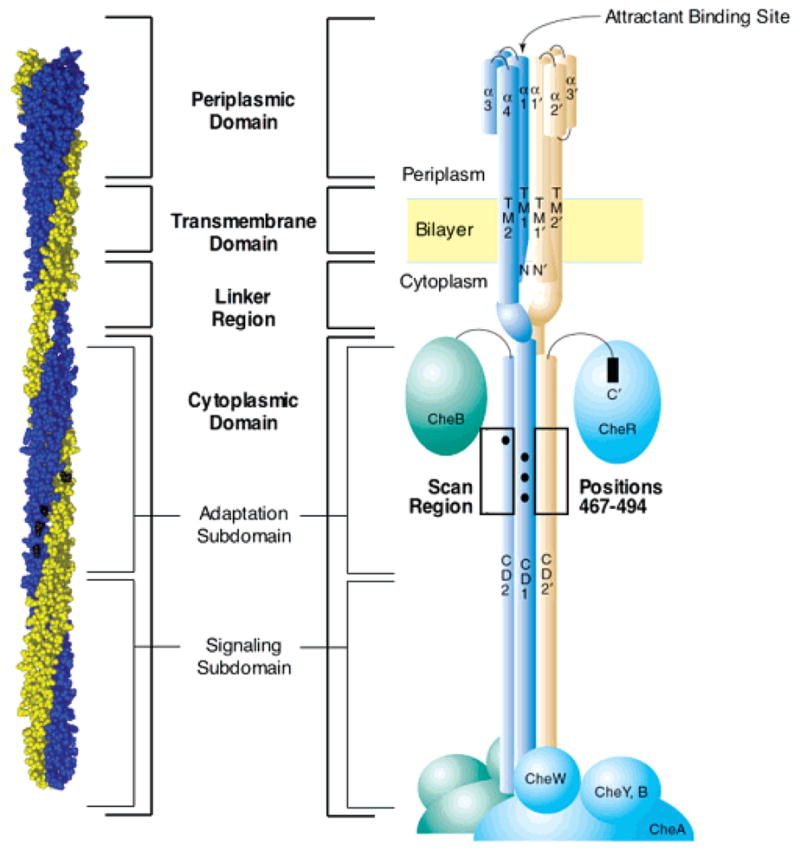
Schematic diagram of the full-length aspartate receptor in the membrane, illustrating its domains and subdomains. The two identical 60-kDa subunits of the homodimer are depicted in blue and gold. The left view is the current atomic model of the full-length, membrane-bound receptor homodimer (39). The right view is a schematic view of the homodimer, in which α helices are depicted as cylinders and associated cytoplasmic proteins are indicated as solid ellipsoids (4). The region of the adaptation subdomain (positions 467–494) examined in the current study by cysteine and disulfide scanning is highlighted. Filled circles represent the covalent adaptation sites within the adaptation subdomain. Note that the homodimer can further assemble into a higher order oligomeric structure proposed to be a trimer of dimers (39, 42, 43).
The receptor cytoplasmic domain can be further subdivided into two functional regions (Figure 1) (4, 34). The signaling subdomain binds the coupling protein CheW and the histidine kinase CheA and is critical for transmitting regulatory signals from the receptor to the kinase (35). The adaptation subdomain, which is the focus of the current study, contains the adaptation sites, lying at positions 295, 302, 309, and 491 in the aspartate receptor, as well as docking surfaces for the adaptation enzymes CheR and CheB (4, 36). The adaptation sites are glutamate side chains that are methyl-esterified by the methyltransferase enzyme CheR and demethylated by phospho-CheB, which hydrolyzes the methyl esters. These adaptation sites play an essential role in the chemotaxis pathway by serving as the chemical memory of the pathway and by enabling the pathway to adapt to a constant background level of stimulus (5, 37). To determine whether swimming is proceeding in a favorable direction, the receptor population constantly compares the current occupancy of the attractant binding site, representing the current attractant concentration, to the level of adaptation site methylation, representing the attractant concentration in the recent past. This comparison ultimately defines the sign of the regulatory signal sent to the CheA kinase. Because the binding of aspartate downregulates the kinase and the methylation of the adaptation sites stimulates the kinase, the signals oppose one another, providing a negative feedback loop as required for adaptation to background stimuli. The adaptation subdomain also plays a central role in thermotaxis (38).
The current model of cytoplasmic domain architecture describes the domain as an extended, antiparallel four-helix bundle (4, 39, 40). This model has been proposed on the basis of evidence from (i) chemical studies, which mapped out the four-helix bundle of the signaling subdomain in the full-length, membrane-bound receptor using a cysteine and disulfide scanning approach (41), and (ii) crystallographic studies, which confirmed the existence of the bundle and provided a high-resolution view of its structure in a water-soluble fragment of the cytoplasmic domain (39). Together, these results have fully defined the signaling subdomain as a four-helix bundle. Furthermore, the crystal structure of the cytoplasmic domain fragment as well as biochemical, genetic, and chemical cross-linking studies of the full-length receptor have shown that interaction surfaces on the signaling subdomain are necessary and sufficient to drive the assembly of receptor dimers into a trimer of dimers that exhibits positive cooperativity in signaling (39, 42–46).
In contrast to the well-defined four-helix bundle structure of the signaling subdomain, the architecture of the adaptation subdomain remains poorly defined. The fragment employed for crystallographic studies of the cytoplasmic domain was truncated in this subdomain and exhibited crystal-packing interactions that perturb helix–helix interactions (39). Efforts to crystallize larger fragments containing the adaptation subdomain have been unsuccessful, perhaps because of the remarkably dynamic nature of the adaptation subdomain in the isolated cytoplasmic domain (47, 48). Cysteine and disulfide scanning studies of the adaptation subdomain have been limited to its N-terminal region and, thus far, have identified only two (CD1 and CD1′) of the four proposed helices in the subdomain (Figure 1) (49). Previous hydrodynamic studies have indicated that the full cytoplasmic domain is highly elongated, and circular dichroism studies revealed a high degree of α helicity (50, 51), but these bulk measurements do not possess the resolution needed to test the four-helix bundle model of the adaptation subdomain structure. In short, further structural and functional analysis of the C-terminal region of the adaptation subdomain is essential to a molecular understanding of the mechanisms of receptor signaling and adaptation.
Cysteine and disulfide scanning has been proven to be an invaluable tool for the determination of protein structure and has the advantage of not being limited by the size of the macromolecule to be studied nor by the lipid environment of membrane proteins (35, 41, 49, 52–56). This technique has been successfully employed to probe the structure and signaling mechanism of bacterial receptors, including studies of the cytoplasmic domain that have revealed large helical regions (4). Figure 1 highlights the C-terminal region (G467–A494) of the adaptation subdomain investigated in the present work. The present cysteine and disulfide scanning studies define the secondary structure of this region as α helical with distinct buried and exposed faces and show that the buried face is critical for allosteric kinase regulation. This helical region exhibits significant dynamics that enable promiscuous collisions with the nearby helices of other receptor subunits. Overall, the results provide further support for the four-helix bundle model of adaptation subdomain structure and support a mechanistic model in which the dynamic packing interactions between the helices of the four-helix bundle directly modulate the activity of the CheA kinase.
EXPERIMENTAL PROCEDURES
Materials
All E. coli strains were provided by Dr. John S. Parkinson, University of Utah (Salt Lake City, UT). Strains used for receptor expression and characterization were RP3808 (Δ(cheA-cheZ)DE2209 tsr-1 leuB6 his-4 eda-50 rpsL136 [thi-1 Δ(gal-attl)DE99 ara-14 lacY1 mtl-1 xyl-5 tonA31 tsx-78]/mks/) and RP8611 (ΔtsrDE7028 Δ(tar-tap)-DE5201 zbd::Tn5 Δ(trg)DE100 leuB6 his-4 rpsL136[thi-1 ara-14 lacY1 mtl-1 xyl-5 tonA31 tsx-78]/CP362 of G. Hazelbauer via F. Dahlquist, pa/) (57), expressing the plasmid pSCF6 previously described (58). CheA (HB101/pMO4) and CheW (HB101/pME5) were made from strains and plasmids provided by Jeff Stock (Princeton). Bob Bourret (University of North Carolina) provided the strain and plasmid used to express CheY (RBB455/pRBB40). Chemical reactivity probe 5-iodoacetamidofluorescein (5-IAF) was purchased from Molecular Probes, Inc. Radiolabeled [γ-32P]-ATP (6000 Ci/mmol), [3H-2,3] l-aspartate (32 Ci/mmol), and s-adenosyl-l-[methyl-3H]methionine (55–85 Ci/mmol) were obtained from Amersham or New England Nuclear. Deoxyoligonucleotides were synthesized by Life Technologies, Inc. Kunkel mutagenesis reagents (T7 DNA polymerase, T4 DNA ligase, and deoxynucleotide triphosphates) were purchased from BioRad. All other reagents were purchased from Sigma unless otherwise noted in the text.
Engineering Cysteine-Containing Receptors
To obtain individual cysteine-containing receptors, site-directed mutagenesis was performed on the plasmid pSCF6 by the Kunkel et al. method as modified by the BioRad Mutagene kit (58, 59). Mutated plasmids were transformed into E. coli RP8611 for amplification and isolated using Qiagen spin columns. PCR plasmid sequencing was carried out using a modification of the Sanger method and the Epicentre Technologies kit; the results verified that receptor genes encoded the intended mutations (35). Plasmids encoding for single-mutant cysteines at positions 490–494 were kindly provided by Randal B. Bass and Mark Danielson (49).
Engineering Dicysteine Receptors
Modified plasmids containing two cysteine mutations (N36C/G467C, N36C/T482C, N36C/N485C, and N36C/S492C) were created by combining fragments of the single-mutant plasmids. The original single-mutation plasmids (of which, for position 36 was provided by Randal B. Bass) were digested with restriction enzymes Pst1 and Mlu1 (New England Biolabs), producing a 0.9-kb fragment containing the N36C mutation or a 4.1-kb fragment containing the G467C, T482C, N485C, or S492C mutation. The digest containing the target 0.9-kb fragment with its N36C mutation was treated with calf intestinal phosphatase (CIP) to prevent self-ligation. The derived fragments were gel-purified and isolated using the Gene Clean Kit (Bio 101). The appropriate fragments were ligated together overnight at 16 °C, using T4 DNA ligase (New England Biolabs). The resulting ligation reactions were then transformed into RP8611 competent cells, and DNA minipreps and membranes were prepared as follows. Under harsh oxidation conditions, a small percentage of the double-cysteine-containing receptor was observed on SDS–poly-acrylamide gels to form higher order products, which could only form if the receptor had two cysteines per subunit.
Expression and Isolation of Engineered Receptors in Membranes
Plasmids encoding each cysteine substitution were transformed into the strain RP3808 for receptor expression. Receptor purification was performed as previously described (49), with the following modifications. Overnight 2 mL of LB cultures were diluted 1:250 into 500 mL of Vogel Bonner Citrate (VBC) minimal growth medium containing 0.75% glycerol, 200 μg/mL MgSO4·7H2O, 2000 μg/mL citric acid·H2O, 10 000 μg/mL K2HPO4, 3500 μg/mL NaNH4·HPO4·4H2O, 40 μg/mL D,L-histidine, 20 μg/mL L-methionine, 20 μg/mL L-leucine, 20 μg/mL L-threonine, 1 μg/mL thiamine, and 150 μg/mL ampicillin (65). Cultures were incubated at 30 °C, shaking at 225 rpm for 17–18 h, and then harvested by centrifugation [Sorvall GS-3 rotor at 6000 rpm (6080g) for 10 min at 4 °C]. Cells were resuspended in 5 mL of ice-cold low-salt buffer containing 20 mM sodium phosphate at pH 7.0 with NaOH, 10% (v/v) glycerol, 10 mM EDTA, 50 mM dithiothreitol (DTT),1 2.5 mM 1,10-phenanthroline, and 0.5 mM PMSF. The cells were then transferred to thin-walled ultracentrifuge tubes (Beckman) in an ice/water bath and lysed by sonication (3 × 17 s bursts with 20 s cooling intervals using a Mysonics model W-385 sonicator with a macrotip). Cell debris was pelleted by centrifugation in a TLA 100.3 rotor (Beckman) at 15 000 rpm (12000g) for 20 min. The supernatant was decanted into fresh tubes, and the membranes were pelleted using a TLA 100.3 rotor at 100 000 rpm (540000g) for 15 min. The pellets were resuspended in 0.6 mL of high-salt buffer containing 20 mM sodium phosphate at pH 7.0 with NaOH, 2 M KCL, 10% (v/v) glycerol, 10 mM EDTA, 5 mM DTT, 2.5 mM 1,10-phenanthroline, and 0.5 mM PMSF. Resuspension was achieved by sonication with a previously mentioned sonicator using a microtip (3 × 17 s bursts with 20 s cooling intervals). Membranes were diluted to 3 mL in high-salt buffer, pelleted again, and resuspended as above, except that DTT and phenanthroline were omitted. The membranes were then resuspended in 0.6 mL of final buffer containing 20 mM sodium phosphate at pH 7.0 with NaOH, 10% (v/v) glycerol, 0.1 mM EDTA, and 0.5 mM PMSF, before diluting to 3 mL in final buffer, pelleting as above, and resuspending in 300 μL of final buffer. These membranes were then snap-frozen in liquid nitrogen and stored at −80 °C.
The total protein yield was determined by BCA assay, calibrated against bovine serum albumin standards. The absorbance measurements were made using a microplate reader (Molecular Devices, Inc.). The fraction of protein consisting of Tar was determined by quantitating the receptor and nonreceptor bands on a Coomassie-stained 10% Laemmli SDS–polyacrylamide gel (acrylamide/bisacrylamide ratio of 40:0.2) using a digital camera (Alpha Innotec).
Chemical Reactivity Assays
This assay was performed as previously described (54) with the following modifications. Receptor-containing membrane samples were diluted to 5 μM Tar in 10 mM sodium phosphate at pH 7.0 with HCl, 50 mM NaCl, 50 mM KCl, and 1 mM EDTA to a 40 μL reaction. The reaction was started with the addition of 5-IAF from a 5 mM stock to a final concentration of 300 μM. The reaction was incubated at 25 °C in a water bath for 15 min, and then one-half the reaction was quenched with 1.25 μL of ·-mercaptoethanol to react with any remaining label. At this time, the second half of the reaction was treated with 1.25 μL of 10% (w/v) SDS and heated to 95 °C, allowed to react for an additional 3 min, and then quenched. A total of 5 μL of 4× Laemmli nonreducing sample buffer [250 mM Tris at pH 6.8 with HCl, 4% SDS, and 40% (v/v) glycerol] was added to each reaction, and they were heated to 95 °C for 2 min before being loaded onto a 10% SDS–PAGE gel with an acrylamide/bisacrylamide ratio of 40:0.2. Fluorescence of the receptor band was visualized on a UV box (Alpha Innotec) and quantitated with a digital camera (Alpha Innotec). The same gels were stained with Coomassie and quantitated with the digital camera to normalize the fluorescence intensity variations because of the fluctuating amount of receptor. Chemical reactivity was defined as the ratio of receptor fluorescence generated by 5-IAF reactions in the folded versus unfolded states.
In Vivo Chemotaxis Assays
Chemotaxis swarm assays were carried out as previously described (49, 60). Briefly, each mutant pSCF6 plasmid was transformed into E. coli RP8611, which is deleted for the wild-type receptor. The vector alone (pBluescript) and vector carrying the wild-type receptor (pSCF6) were used as controls to determine the swarm rates of cells lacking and containing the native receptor. Starter cultures were grown in Luria Broth with 100 μg/mL ampicillin at 37 °C with shaking for 8 h, and then 5 μL was spotted onto 0.23% agar minimal plates containing VBC medium supplemented with 0.1% glycerol, 20 mM lactate, 40 μg/mL D,L-histidine, 20 μg/mL L-leucine, 1 μg/mL thiamine, and 100 μg/mL ampicillin. Cultures were also spotted on minimal media plates, prepared the same way as above but containing 100 μM L-aspartate. Plates were incubated at 30 °C, and colony diameters were measured at 3–4 h intervals approximately 24 h after spotting. The swarm rates were determined by a least-squares linear best fit to the slope of the diameter as a function of time. The aspartate-specific swarm rates were determined by subtracting the (−) aspartate swarm rate from the (+) aspartate swarm rate to correct for pseudotaxis and any other nonaspartate-specific taxis. The resulting rate was normalized to the wild-type rate for comparison.
In Vitro Receptor-Coupled Kinase Assays
A receptor-coupled phosphorylation assay was used to quantitate radiolabeled phosphate from the receptor–CheA–CheW ternary complex to CheY, to directly determine the ability of the modified receptors to regulate the autophosphorylation activity of CheA. This assay was performed as previously described (20, 49, 61), with the following modifications. Isolated membranes were diluted to contain 12 μM Tar monomer and either (1) oxidized by the addition of the redox catalyst CuII(1,10-phenanthroline)3 (400 μM) in the presence of ambient dissolved oxygen for 20 min at 37 °C or (2) reduced by addition of DTT (100 mM) to ensure elimination of disulfides. Oxidized reactions were treated with 0.1 mM sodium persulfate to inactivate oxidation. Aliquots of these membrane samples (to make 6 μM Tar monomer) were combined with purified proteins (kindly provided by Matthew Trammell) CheW (2 μM), CheA (0.25 μM monomer), and CheY (10 μM) in 50 mM Tris at pH 7.5 with HCl, 50 mM KCl, and 5 mM MgCl2, with and without 1 mM aspartate and incubated at room temperature (23 °C) for 30 min to equilibrate the receptor–kinase complex. The addition of [γ-32P]ATP to a final concentration of 0.1 mM initiated the reaction. After 10 s, 5 μL aliquots were quenched with 15 μL of 2× Laemmli sample buffer supplemented with 25 mM EDTA. The [32P] phospho-CheY was resolved on a 15% Laemmli SDS–polyacrylamide gel (acrylamide/bisacrylamide ratio of 40:1.25). The gels were dried and quantitated by phosphorimaging (Molecular Dynamics).
The extent of disulfide formation in the oxidized reactions was determined by analyzing an aliquot of the oxidized membrane on a 10% nonreducing Laemmli SDS–polyacrylamide gel (acrylamide/biacrylamide ratio of 40:0.2). Gels were then Coomassie-stained. The dimer and monomer bands were quantitated with a digital camera (Alpha Innotec), and the percentage of dimer formation was calculated.
In Vitro Aspartate-Binding Assays
Receptors containing lock-on cysteine or disulfide mutations were tested for the ability to bind aspartate in a membrane spin-down assay previously described (27, 62) with the following modifications. Samples of receptor-containing membranes were diluted to contain 6 μM Tar monomer in 150 μL. These samples were either (1) oxidized by addition of the redox catalyst CuII(1,10-phenanthroline)3 (400 μM) in the presence of ambient dissolved oxygen for 20 min at 37 °C or (2) reduced by addition of DTT (100 mM) to ensure elimination of disulfides. Aliquots of 25 μL were then added to 50 μL binding reactions containing 2.5 mM Tris-HCl at pH 7.5, 2.5 mM NaCl, 5.0 mM KCl, and 1 mM EDTA with L-aspartate concentrations ranging from 0.8–9 μM (spiked with [3H-2,3] L-aspartate, 32 Ci/mmol, 1 μCi/μL). These binding reactions were split into two Beckman TLA 100.2 ultracentrifuge tubes. One was incubated with 2.5 μL of glass distilled water, and the other was incubated with 2.5 μL of 200 mM L-aspartate for 10 min at 25 °C. The tubes were spun for 15 min at 22 °C, and 20 μL of the supernatant was removed and added to 50 μL of glass distilled water in scintillation vials. The vials were then filled with Ecoscint H (National Diagnostics), capped, vortexed, and counted for 5 min each. The KD values for each modified receptor were determined by best-fit nonlinear regression analysis of a plot of bound versus free aspartate. Disulfide extents were determined as in the in vitro activity assay.
RESULTS
Engineering a Cysteine Library
To probe the structure and function of the cytoplasmic adaptation subdomain in the full-length, membrane-bound, homodimeric aspartate receptor, we created a library of mutant receptors by cysteine scanning. The wild-type receptor contains no intrinsic cysteines; therefore, each engineered cysteine was unique within each subunit. Briefly, using site-directed mutagenesis, unique single-cysteine substitutions were created in each receptor, from positions G467–A494. Each receptor protein was generated by plasmid overexpression in E. coli strain RP 3808 that lacks receptors and also lacks the methylation and demethylation enzymes CheR and CheB, respectively (57), thereby ensuring that the adaptation state of the receptor population was homogeneous (QEQE or QEQC for the E491C mutant). A total of 28 single-cysteine receptors were generated, and expression of each receptor was similar to that of the wild type.
Chemical Reactivity Analysis of Solvent Exposure and Secondary Structure
Previous studies have shown that the solvent exposure pattern displayed by cysteine residues scanned through a region of primary structure can map out surface-exposed secondary structure elements (35, 49, 54). In the present study, 5-IAF, a bulky, anionic, sulfhydryl-specific probe, was used to quantitate the chemical reactivity of each engineered receptor. Because of its size and charge, the 5-IAF reaction rate is sensitive to the solvent exposure of the cysteine side chain that it alkylates. This probe reacts quickly with solvent-exposed sulfhydryls, but it is largely excluded from the interior of a protein and therefore reacts slowly with buried sulfhydryls. Previous experiments have experimentally confirmed the accuracy of this approach using helix α2, a helix of known structure in the receptor periplasmic domain, as a control (63). This previous analysis scanned a single cysteine from positions T95–H103 on helix α2 in the full-length, membrane-bound receptor and measured the chemical reactivities of each position with three different probes, including 5-IAF. The resulting chemical reactivities were highly correlated with the solvent exposure calculated from the crystal structure (24). Specifically, the α-helical periodicity and buried face observed for helix α2 in the crystal structure were apparent in both the measured chemical reactivity data and the calculated solvent exposure data as expected.
The chemical reactivity assay utilized in the current study incubated mutant receptor membranes with the 5-IAF probe at 25 °C for 15 min. At this time, half of the sample was quenched with β-mercaptoethanol, and the remaining half was denatured with SDS at 95 °C, allowed to react for an additional 3 min to ensure full labeling, and then quenched. Both samples were run on a SDS–polyacrylamide gel, and the receptor fluorescence because of 5-IAF labeling was quantitated. The ratio of receptor 5-IAF labeling in the native versus the denatured state yielded the chemical reactivity, which provides a qualitative measure of solvent exposure. The chemical reactivity parameter ranges from zero for an inaccessible, buried position to unity for a fully exposed position.
The measured chemical reactivities for cysteines G467–A494 are summarized in Figure 2. Two sets of reactivities determined independently under the same conditions illustrate the reproducibility of the experiment. The dashed lines indicate the reactivity levels used to operationally define the three categories of highly exposed, intermediate, and highly buried cysteines. Eight cysteines were found to be highly exposed, having a chemical reactivity ratio exceeding 0.50 (D469, Q470, L473, S476, R480, Q483, E491, and A494). Seven cysteines were determined to be highly buried, with ratios of less than 0.30 (G467, V481, T482, N485, L488, V489, and S492). When the highly exposed and buried positions are mapped onto different secondary structure models, the best segregation to distinct faces is obtained for an α-helical model with a heptad repeat as expected for a coiled-coil four-helix bundle (see Figure 4A in the Discussion).
Figure 2.

Chemical reactivities of cysteine substitutions at positions G467–A494 of the adaptation subdomain. Two independent scans of the region (● and ○) performed under the same conditions are shown to illustrate the reproducibility of the experiment. Chemical reactivity is defined as the ratio of (i) the cysteine reaction rate with 5-iodoacetamido fluorescein in the full-length, membrane-bound receptor to (ii) the corresponding reaction rate in the SDS-denatured state, wherein accessibility is maximal. The reactivity parameter, which is strongly correlated to aqueous solvent exposure in the folded receptor, ranges from zero to unity for cysteines that are inaccessible or fully solvent-exposed, respectively. Experimental conditions: 5 μM receptor and 300 μM 5-iodoacetamido fluorescein in 10 mM sodium phosphate at pH 7.0 with HCl, 50 mM NaCl, 50 mM KCl, and 1 mM EDTA at 25 °C. Positions with reactivities lower than 0.30 in both scans are operationally defined as highly buried. Positions with reactivities above 0.50 in both scans are defined as highly solvent-exposed. Error bars represent the standard deviation of the mean for n ≥ 3 independent measurements.
Figure 4.

Helical wheel model for positions G467–A494 of the adaptation subdomain, depicting the measure chemical reactivities and activity effects of the scanned cysteines and disulfides. (A) Summary of chemical reactivities from Figure 2. White rectangles denote positions with high chemical reactivities, indicating a high degree of solvent exposure; black rectangles denote positions with low reactivities, indicating a high degree of burial. The boxed area identifies the observed buried face, and residues likely to be charged are noted. (B) Black ovals highlight three lock-on disulfides (T482C–T482C′, N485C–N485C′, and S492C–S492C′), which form between a pair of symmetric cysteines within a homodimer and prevent normal attractant-induced kinase inhibition. The white oval indicates one signal-retaining disulfide (G467C–G467C′), which forms within a homodimer and retains normal kinase activation as well as attractant-triggered kinase inhibition. Small black circles and squares indicate cysteine substitutions that inhibit receptor activity in an in vivo chemotaxis assay or in an in vitro receptor-coupled kinase assay, respectively. The black star indicates one cysteine substitution (G467C), which, in the reduced state, exhibits lock-on activity in the in vitro receptor-coupled kinase assay.
Effects of Cysteine Substitutions on Receptor Function in Vivo
Residues crucial for receptor function were determined by testing the engineered receptors in an in vivo assay of cellular chemotaxis. Receptors were overexpressed in E. coli strain RP 8611, lacking both the aspartate and serine chemoreceptors (57). The standard in vivo swarm-plate assay was used to quantitate the ability of each receptor to restore aspartate sensing and chemotaxis to cells swimming in a self-generated aspartate gradient (60). This assay can detect receptor perturbations that block receptor regulation of kinase activity or inhibit normal receptor adaptation (58). Inhibitory substitutions were operationally defined as those that reduced the aspartate-specific swarm rate by a factor of 5-fold or more relative to the rate observed for overexpression of the wild-type receptor. Figure 3A summarizes the relative aspartate-specific swarm rates of the cysteine mutants, including the classification of inhibitory and noninhibitory cysteine substitutions indicated by the dashed line. Altogether, six substitutions were found to be inhibitory (G467C, I468C, N478C, D479C, A486C, and V489C). All six of the inhibitory substitutions are located at buried or interfacial positions on the helix identified by chemical reactivity measurements (see Figure 4B in the Discussion), indicating that the buried face is crucial for receptor function.
Figure 3.

In vivo and in vitro activity of the engineered receptors. (A) In vivo chemotactic swarm plate assay applied to cells expressing cysteine-substituted receptors. For cells expressing a given mutant receptor, the aspartate-specific swarm plate assay measures the difference between the chemotactic swarm rates in the absence and presence of aspartate, normalized to the corresponding difference measured for cells expressing the wild-type receptor. Cysteine substitutions that reduce chemotactic swarming at 37 °C to a rate below 20% of the wild type (below the dashed line) are classified as inhibitory. (B) In vitro receptor-coupled kinase assay applied to cysteine-substituted receptors in their reduced states at 25 °C. Open circles represent normalized receptor-coupled kinase rates in the absence of aspartate, relative to the corresponding rate observed for kinase coupled to the wild-type receptor. This apo state yields maximal kinase activation. Cysteine substitutions that lower the rate to less than 20% of the wild type (below the dashed line) are considered inhibitory. Closed circles indicate rates observed in the presence of saturating aspartate, which triggers kinase inhibition of the wild-type receptor. Cysteine substitutions that prevent normal aspartate-triggered kinase inhibition (above the dashed line) are defined as lock-on cysteines. (C) In vitro receptor-coupled kinase assay applied to cysteine-substituted receptors in their oxidized, disulfide-linked states. Assay conditions and symbols as described for B. Disulfide bonds that retain over 20% wild-type activity (above the dashed line) are considered signal-retaining if they allow aspartate-triggered kinase inhibition (to a level below the dashed line) or lock-on if they prevent aspartate-triggered inhibition (at a level above the dashed line). Error bars represent the standard deviation of the mean for n ≥ 3 independent measurements.
Effects of Cysteine Substitutions and Disulfide Bonds on Receptor Function in Vitro
The in vitro receptor-coupled kinase assay is considerably more sensitive to receptor perturbations than the in vivo swarm assay, because of the absence of the adaptation enzymes that correct many subtle receptor defects in the full pathway (20, 58, 61). Moreover, this in vitro assay can be carried out under oxidizing conditions to study disulfide-bond containing receptors that would be reduced in living cells. The active receptor–kinase signaling complex was reconstituted by adding purified histidine kinase CheA, coupling protein CheW, and phospho-acceptor protein CheY to isolated E. coli RP 3808 membranes containing an overexpressed wild type or cysteine-mutant receptor. The activity of the complex was assayed by observing the rate of phosphotransfer from receptor-bound CheA to a saturating concentration of CheY. Under such conditions, the receptor-stimulated CheA autophosphorylation reaction is the rate-limiting step. The apo state of the receptor stimulates CheA autophosphorylation, while aspartate binding to the complex slows the CheA autophosphorylation rate over 100-fold.
Figure 3B summarizes the effects of cysteine substitutions on receptor-regulated kinase activity in a reducing environment, both in the presence and absence of aspartate. Inhibitory substitutions were defined as those that reduced the kinase activity by a factor of 5-fold or more relative to the wild-type kinase activity in the absence of aspartate. Altogether, seven substitutions were found to be inhibitory (V471C, N478C, D479C, A486C, V489C, A493C, and A494C), six of which fall at buried or interfacial positions on the helix defined by the chemical reactivity data. This spatial distribution further indicates that the buried face is critical for signaling. Interestingly, the buried G467C substitution locked the kinase in the activated state even in the presence of saturating aspartate. The lack of attractant-induced downregulation indicates that this cysteine substitution does not significantly perturb the kinase-activating state of the receptor but instead prevents the aspartate-triggered switch to the kinase-inactivating state. Such a substitution is termed a “lock-on” cysteine because it prevents the normal switching of the receptor from its on state to its off state.
Figure 3C summarizes the effects of intersubunit disulfide bond formation on receptor-regulated kinase activity. Disulfide bond formation was catalyzed by addition of CuII-(1,10-phenanthroline)3 to receptor-containing membranes in the presence of ambient oxygen at 37 °C, yielding a covalent cross-link between a pair of symmetric, engineered cysteines in different subunits (58). Disulfide formation was detected by the appearance of a covalent dimer band in SDS–PAGE and was found to reach between 80 and 95% completion, enabling analysis of the effect of each disulfide on kinase regulation in the in vitro assay. The high efficiency of disulfide formation observed for all 28 cysteine pairs indicates that the region is remarkably dynamic at its physiological temperature. Because 24 of the disulfide bonds completely destroy receptor-coupled kinase activation (Figure 3C), most of these cross-links are likely formed by transient collisions between symmetric cysteines that are far apart in the equilibrium structure. However, four of the disulfide bonds retain receptor activity and thus reveal spatial proximities between pairs of helices in the functional structure. Specifically, intersubunit disulfide bonds were found to retain the ability of the receptor to activate the kinase when incorporated at the G467, T482, N485, and S492 positions. One of these four disulfides at position G467 also retained the ability of the receptor to downregulate the kinase upon binding aspartate and is thus termed a “signal-retaining” disulfide bond. The other three disulfides at the T482, N485, and S492 positions prevented downregulation of the kinase in the presence of saturating aspartate, indicating that these cross-links constitutively lock the receptor in the kinase-activating “on” state. Again, such cross-links are termed “lock-on” disulfides because the disulfide cross-link prevents the receptor from converting from its on state to its off state. Significantly, all of the cysteine substitutions and disulfide bonds with signal-retaining or lock-on character fall on the buried face of the helix defined by the chemical reactivity scan, indicating that this face is located at or near the subunit interface where it plays an important role in transmitting signals from the receptor to the kinase (see Figure 4B in the Discussion).
To test whether the observed lock-on and signal-retaining disulfides were formed between the two subunits within the same dimer or rather between subunits in different dimers, a set of double-cysteine mutants was generated. These double mutants included the cysteine substitution of interest (G467C T482C, N485C, or S492C) and a second substitution, N36C, which has previously been shown to form a disulfide exclusively between the two subunits of the same dimer. Upon oxidation of each double mutant, the products contained the C36–C36′ disulfide, a disulfide at the position of interest, or both disulfides, as resolved by their differing mobilities on SDS–PAGE (41, 53). Using this test, all four of the disulfide bonds that retain significant kinase regulation were found to form between two subunits in the same dimer, rather than between different dimers (data not shown). These findings confirm that the buried face of the helix lies at the subunit interface within a single homodimer of the trimer of dimers (see Figure 4B in the Discussion).
Effects of Lock-On Cysteines and Disulfides on Aspartate Binding
The lock-on mutants were also tested for the ability to bind aspartate in a centrifugation assay (27, 62), because in principle, their inability to downregulate the kinase in response to aspartate could stem from the loss of aspartate binding. The isolated receptor-containing membranes were incubated with various concentrations of radiolabeled aspartate. Then, the membranes were pelleted, and the concentrations of aspartate in the supernatant and membrane fractions were determined. The aspartate-binding curve for each receptor was analyzed by nonlinear least-squares regression to determine the best-fit aspartate KD value, as summarized in Table 1. Each of the locked-on receptors retains a functional aspartate-binding site that is fully loaded with aspartate at the 1 mM aspartate concentration used in the in vitro activity assay. It follows that these receptors successfully bind aspartate at the periplasmic attractant binding site, but the lock-on cysteine substitution or disulfide bond prevents the normal structural transition needed to propagate the inhibitory attractant signal through the adaptation subdomain to the signaling subdomain and its associated CheA kinase.
Table 1.
Aspartate Affinities of Modified Receptors
| receptor mutation | aspartate KD (μM) (mean ± SD) |
|---|---|
| wild type, reduced | 0.9 ± 0.4 |
| wild type, oxidized | 2.5 ± 1.2 |
| G467C, reduced | 1.0 ± 0.2 |
| T482C, oxidized | 5.5 ± 4.6 |
| N485C, oxidized | 4.1 ± 2.6 |
| S492C, oxidized | 0.9 ± 0.7 |
DISCUSSION
The current study extends the use of cysteine and disulfide scanning to characterize the structure and function of positions G467–A494 in the cytoplasmic domain of the transmembrane aspartate receptor, corresponding to the C-terminal region of the adaptation subdomain (Figure 1). The measured chemical reactivities of these positions exhibit a repeating pattern of highly exposed and buried residues with a periodicity consistent with an α-helical secondary structure. A 3.5 residue per turn helix model, characteristic of α helices found in coiled coils or four-helix bundles, provides the best segregation of exposed and buried residues on opposite faces as illustrated in Figure 4. These findings represent the first direct experimental evidence that the putative helix CD2 proposed in the current model of the adaptation subdomain (39, 40) is, in fact, an α helix.
Further examination of the primary structure reveals that the majority of charged or polar residues are located on the exposed helix face, while the nonpolar residues are predominately localized to the buried face. Such segregation of polarity provides additional evidence for the helicity of the C-terminal region of the adaptation subdomain and indicates that this helix is amphiphillic. A sequence alignment of over 60 homologous taxis receptors reveals that the same heptad repeating pattern observed in this helical region of the aspartate receptor is conserved in other members of the taxis receptor superfamily (7, 8), indicating that this amphiphillic helical motif is a conserved structural element. As observed in the aspartate receptor (Figure 4), the heptad repeating pattern of the region typically has nonpolar residues at positions a, d, and g, while polar residues dominate at positions b, c, and f, and interfacial position e exhibits a mixture of nonpolar and polar residues. Additional evidence for the helicity of this region is provided by the helical periodicity of the residues associated with the CheR recognition sequence for adaptation site E491 (64). Residues Q490, E491, and A494 are part of this recognition sequence and, as expected, are located at the solvent-exposed heptad positions in the helical model defined by chemical reactivities.
The activity measurements for the cysteine substitutions in this study highlight the functional importance of the adaptation subdomain C-terminal helix, particular of its buried face. In vivo, 6 of the 28 substitutions tested were found to significantly inhibit the ability of the receptor to restore aspartate-specific chemotaxis. When mapped to the helical secondary structure, 5 of these 6 substitutions fall on the buried face, while the remaining substitution is located at the interfacial position e, highlighting functional importance of the helix-buried face (see Figure 4B). The results of the in vitro assays provide further support for this conclusion, with the 6 of the 7 inhibitory substitutions again mapping to buried or interfacial positions (see Figure 4B). Finally, it is interesting to note that cysteine substitution at the fourth adaptation site (E491C) retains the ability to regulate the kinase in both the in vivo and in vitro studies, showing that this adaptation site is noncritical. The latter observation adds to previous findings that no one site is essential for receptor function and that the fourth site is the least important to receptor adaptation (64, 65).
A scan of the 28 cysteine substitutions for intersubunit disulfide formation reveals promiscuous thermal dynamics that enable each of the symmetric cysteine pairs to collide and form a disulfide bond with high efficiency. Virtually any model for the equilibrium structure of the adaptation subdomain would place most of the cysteine pairs at positions where their β carbons at separations exceeding 4.6 Å, which requires backbone motions to bring them within range for a productive collision between their sufhydryl groups, leading to disulfide bond formation (66). As expected for covalent cross-links that trap structural fluctuations away from the native equilibrium structure, 24 of the 28 disulfide bonds destroy the ability of the receptor to activate CheA kinase in vitro, indicating that they are incapable of binding CheA or are incapable of stimulating bound CheA. It follows that most of the 28 disulfide bonds are formed during extensive backbone motions that yield collisions between subunits in the same homodimer or perhaps even between subunits in different homodimers. The latter interdimer collisions could occur between nearby homodimers in the same trimer of dimers or in adjacent trimers of dimers. Such extensive dynamics are consistent with NMR studies of the isolated cytoplasmic domain (47, 48), and it has previously been noted that such dynamics could play a central role in receptor signaling (67, 68). These findings also illustrate the potential pitfalls of an oversimplified disulfide-mapping approach in which detection of a disulfide bond is considered evidence for close proximity between the cysteine pair. Such an approach can give false positives in a molecule possessing significant thermal motions. Instead, a more reliable disulfide mapping approach is to search for disulfide bonds that retain activity and thus would generally cross-link pairs of positions nearby in the functional, equilibrium structure.
A functional disulfide scan of the 28 C-terminal helix positions reveals four disulfide bonds that retain measurable receptor activity. Three of these disulfides exhibit lock-on behavior (T482–T482′, N485–N485′, and S492–S492′) and are located on the buried face of the helical region. The buried location of these lock-on disulfides further strengthens the conclusion that the buried face is crucial for ligand-induced kinase regulation. The three lock-on disulfides are found to form between two subunits within the same receptor homodimer, indicating that these positions lie in close proximity to their symmetric counterparts at the subunit interface. The remaining functional disulfide bond exhibits signal-retaining behavior (G467–G467′) because it retains both kinase stimulation and downregulation in the absence and presence of aspartate, respectively. Similar to the lock-on disulfides, this signal-retaining disulfide forms between the two subunits in the same homodimer. It follows that this signal-retaining disulfide bond must lie at the native subunit interface in both the on and off states, as demonstrated in Figure 5. Overall, the results of the disulfide scanning analysis support a model in which the C-terminal region of the adaptation subdomain is helical but highly dynamic and that the helix lies at the subunit interface within the dimer. This model further proposes that signal transduction through the adaptation subdomain causes small changes in the packing interactions or the dynamics of the C-terminal helical region that can be trapped in the on state by appropriately placed lock-on disulfides.
Figure 5.
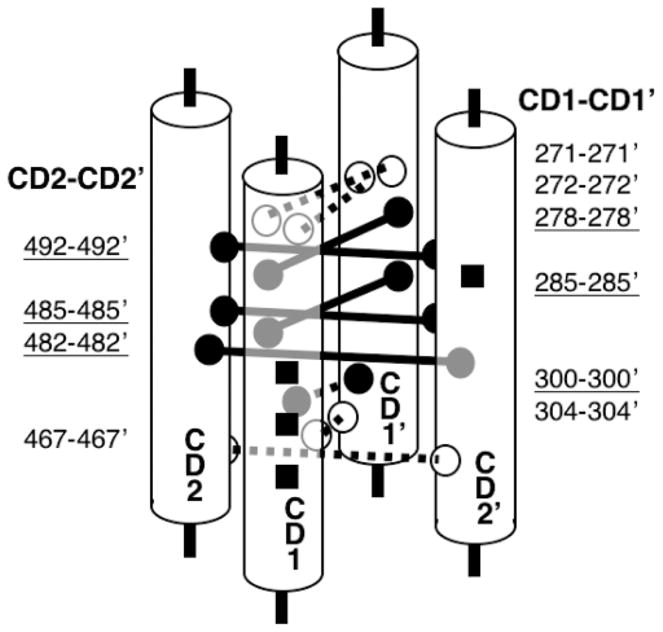
Schematic diagram of adaptation subdomain architecture. Shown are isolated sections of cytoplasmic domain helices CD1, CD1′, CD2, and CD2′ that form the four-helix bundle of the adaptation subdomain. The adaptation sites are shown (black squares). Also shown are lock-on (black/solid) and signal-retaining (white/dashed) disulfide bonds, identified in this and a previous study (49), that form between the two subunits of the homodimer. These disulfide bonds identify functional contacts between symmetric pairs of helices at the core of the four-helix bundle. Additional lines of evidence supporting the four-helix bundle are discussed in the text.
Several lines of evidence argue that the N- and C-terminal helices of the adaptation subdomain pack together to form an antiparallel four-helix bundle in the assembled homodimer. First, the N-terminal helix is known to lie at or near the subunit interface with its buried face packed against the other subunit of the same homodimer, just as found for the C-terminal helix in the present study. Second, the buried faces of both the N- and C-terminal helices are larger than expected for a single pair of coiled-coil helices, wherein only the a and d heptad positions are highly buried. Third, analysis of the sequence alignment for the taxis receptor superfamily has revealed the presence of parallel insertions and deletions in the N- and C-terminal helices that indicate that the lengths of the two helices are constrained to be the same. The simplest explanation for these observations is that the N-and C-terminal helical regions of the adaptation subdomain pack together with their symmetric counterparts in the other subunit to form an antiparallel four-helix bundle. This arrangement is schematically shown in Figure 5 for the adaptation subdomain and in Figure 1, which summarizes the current four-helix bundle model of the cytoplasmic domain in the full-length receptor. The four-helix bundle model for the adaptation subdomain is also consistent with the known four-helix bundle architecture of the signaling subdomain (39–41).
The helix-packing interactions of all four helices within the buried core of the adaptation four-helix bundle have been found to be critical for kinase regulation, as noted above for the two symmetric C-terminal helices (41, 69). A recent study showed that electrostatic repulsion between negative charges on the surface of the subdomain modulates the packing between the adjacent helices of the two subunits within the homodimer, thereby regulating kinase activity (69). Covalent modification of the receptor adaptation sites is believed to modulate this electrostatic interaction in the same way, because methylation or amidation of the adaptation sites decreases the electrostatic repulsion between subunits, thereby stabilizing the helix interactions and activating the kinase. Notably, the previous and present disulfide scanning studies of the cytoplasmic domain reveal that all lock-on disulfides and the majority of signal-retaining disulfides fall within the adaptation subdomain, including disulfides at G271, S272, G278, G285, L300, and A304 from previous studies (49) as well as G467, T482, N485, and S492 from the current study. Together, these findings strongly argue that helix–helix interactions within the four-helix bundle of the adaptation subdomain play a central role in kinase regulation. Stabilization of these helix interactions by neutralization of surface charge repulsion or by engineered lock-on disulfides between helices stimulates kinase activity and can even lock the receptor in the kinase-activating state. However, the discovery of signal-retaining disulfides at these same helix–helix interfaces indicates that the structural rearrangements triggered by signal transduction can be small enough to be accommodated by the intrinsic flexibility of a properly positioned disulfide bond. This observation is reminiscent of the small magnitude (~1.6 Å) of the helix displacement proposed for the transmembrane signal in the periplasmic and membrane-spanning regions of the receptor (4, 26).
A working model consistent with the available evidence proposes that signal transmission through the adaptation subdomain involves a change in the supercoiling of the four-helix bundle. In this model, the attractant signal generated at the periplasmic attractant binding site is carried across the membrane as a piston displacement of the signaling helix TM2 but is converted by the linker to a change in the supercoiling of the cytoplasmic four-helix bundle formed by helices CD1, CD1′, CD2, and CD2′, which are proposed to be continuous throughout the adaptation and signaling subdomains (Figure 1). Similarly, the intersubunit charge repulsion within the bundle between helices CD1 and CD2′ or between CD1′ and CD2, in the vicinity of the adaptation sites triggers a change in the bundle supercoiling that transmits information from the adaptation subdomain to the signaling subdomain. When the signal reaches the signaling subdomain, the changes in supercoiling are transmitted to bound CheA, thereby regulating kinase activity. New evidence consistent with the supercoiling model is the observation that position e of the C-terminal helix in the adaptation subdomain (Figure 4) is generally occupied by a small side chain such as alanine. This position lies at the edge of the buried helix face and is thus partially exposed to the solvent where it exhibits intermediate solvent exposure. Strikingly, however, cysteine substitutions at three of the four position e sites tested strongly perturb receptor activity in vivo or in vitro (Figure 4), and this represents a higher density of perturbing cysteine substitutions than found at any of the other six helix positions. Such an observation suggests that position e is a toggle position at which the attractant and adaptation signals generate a small rotation of this helix relative to the adjacent helices bundle, which would bury side chains at position e in one direction of the toggle and would expose side chains at position e in the other direction of the toggle. Thus, the present results support a model in which small changes in supercoiling carry the signal like a wave through the cytoplasmic four-helix bundle to the bound CheA kinase. Because the signal can be transmitted through the intrinsic flexibility of signal-retaining disulfide bonds at certain locations, the magnitude of this supercoiling change is proposed to be small (no more than a few angstroms). If the displacement is less than 2 Å, then the intrinsic flexibility of side chains involved in interhelix hydrogen bonds and salt bridges would allow the movement without significant disruption of these specific side-chain interactions, ensuring a low-energy transition (73). If, however, the displacement is over 2 Å, many specific contacts would be disrupted, which would mean that an alternative set of contacts would need to be formed; otherwise, the transition would require too much energy to be driven by aspartate binding.
In summary, the results further demonstrate the usefulness of cysteine and disulfide scanning to define secondary structure and functional elements in membrane proteins for which high-resolution structures remain difficult to obtain. The findings represent further progress toward the nearly complete determination of a low-resolution structure for the cytoplasmic domain using chemical methods. The best-defined region of the cytoplasmic domain structure is the signaling subdomain, for which the chemically defined four-helix bundle has been confirmed and elucidated to atomic resolution by the crystal structure of a cytoplasmic domain fragment (39–41). Strong support that this four-helix bundle continues from the signaling subdomain through the adaptation subdomain is defined by the previous (35, 41, 49) and present cysteine and disulfide scanning studies. The structure of the linker region that connects the transmembrane signaling helix to the adaptation subdomain remains unknown, however. This linker plays an essential role in transmembrane signaling (70, 71). A cysteine and disulfide scanning study of this region revealed helical elements and a stable folded core but did not determine its tertiary structure (54). The fact that the linker is a conserved structural element [the HAMP domain (35, 41, 49)] found in a large family of prokaryotic and eukaryotic receptors highlights its structural and functional importance. The current model of the full-length receptor structure proposes that the linker is a simple helical extension of the signaling helix (39), but recent studies of cytoplasmic domain length support a more compact, folded structure for the linker region (72). Overall, with the exception of the linker region, the results provided by cysteine and disulfide scanning studies of the cytoplasmic domain provide strong support for the current four-helix bundle model of the adaptation and signaling subdomains in the full-length, membrane-bound receptor and reveal important mechanistic and functional features of these subdomains.
Footnotes
Abbreviations: DTT, dithiothreitol; 5-IAF, 5-iodoacetamidofluorescein.
Support provided by NIH Grant GM R01-040731.
References
- 1.Swanson RV, Alex LA, Simon MI. Histidine and aspartate phosphorylation: Two-component systems and the limits of homology. Trends Biochem Sci. 1994;19:485–490. doi: 10.1016/0968-0004(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 2.Parkinson JS. Signal transduction schemes of bacteria. Cell. 1993;73:857–871. doi: 10.1016/0092-8674(93)90267-t. [DOI] [PubMed] [Google Scholar]
- 3.Spudich JL, Luecke H. Sensory rhodopsin II: Functional insights from structure. Curr Opin Struct Biol. 2002;12:540–546. doi: 10.1016/s0959-440x(02)00359-7. [DOI] [PubMed] [Google Scholar]
- 4.Falke JJ, Hazelbauer GL. Transmembrane signaling in bacterial chemoreceptors. Trends Biochem Sci. 2001;26:257–265. doi: 10.1016/s0968-0004(00)01770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falke JJ, Bass RB, Butler SL, Chervitz SA, Danielson MA. The two-component signaling pathway of bacterial chemotaxis: A molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu Rev Cell Dev Biol. 1997;13:457–512. doi: 10.1146/annurev.cellbio.13.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhulin IB, Nikolskaya AN, Galperin MY. Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. J Bacteriol. 2003;185:285–294. doi: 10.1128/JB.185.1.285-294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Moual H, Koshland DE., Jr Molecular evolution of the C-terminal cytoplasmic domain of a superfamily of bacterial receptors involved in taxis. J Mol Biol. 1996;261:568–585. doi: 10.1006/jmbi.1996.0483. [DOI] [PubMed] [Google Scholar]
- 8.Danielson MA. Department of Chemistry and Biochemistry. University of Colorado; Boulder, CO: 1997. p. 142. [Google Scholar]
- 9.Trivedi VD, Spudich JL. Photostimulation of a sensory rhodopsin II/HtrII/Tsr fusion chimera activates CheA-autophosphorylation and CheY-phosphotransfer in vitro. Biochemistry. 2003;42:13887–13892. doi: 10.1021/bi034399q. [DOI] [PubMed] [Google Scholar]
- 10.Baumgartner JW, Kim C, Brissette RE, Inouye M, Park C, Hazelbauer GL. Transmembrane signalling by a hybrid protein: Communication from the domain of chemo-receptor Trg that recognizes sugar-binding proteins to the kinase/phosphatase domain of osmosensor EnvZ. J Bacteriol. 1994;176:1157–1163. doi: 10.1128/jb.176.4.1157-1163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krikos A, Conley MP, Boyd A, Berg HC, Simon MI. Chimeric chemosensory transducers of Escherichia coli. Proc Natl Acad Sci USA. 1985;82:1326–1330. doi: 10.1073/pnas.82.5.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Utsumi R, Brissette RE, Rampersaud A, Forst SA, Oosawa K, Inouye M. Activation of bacterial porin gene expression by a chimeric signal transducer in response to aspartate. Science. 1989;245:1246–1249. doi: 10.1126/science.2476847. [DOI] [PubMed] [Google Scholar]
- 13.Moe GR, Bollag GE, Koshland DE., Jr Transmembrane signaling by a chimera of the Escherichia coli aspartate receptor and the human insulin receptor. Proc Natl Acad Sci USA. 1989;86:5683–5687. doi: 10.1073/pnas.86.15.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biemann HP, Harmer SL, Koshland DE., Jr An aspartate/insulin receptor chimera mitogenically activates fibroblasts. J Biol Chem. 1996;271:27927–27930. doi: 10.1074/jbc.271.44.27927. [DOI] [PubMed] [Google Scholar]
- 15.Segall JE, Block SM, Berg HC. Temporal comparisons in bacterial chemotaxis. Proc Natl Acad Sci USA. 1986;83:8987–8991. doi: 10.1073/pnas.83.23.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gegner JA, Graham DR, Roth AF, Dahlquist FW. Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell. 1992;70:975–982. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- 17.Schuster SC, Swanson RV, Alex LA, Bourret RB, Simon MI. Assembly and function of a quaternary signal transduction complex monitored by surface plasmon resonance. Nature. 1993;365:343–347. doi: 10.1038/365343a0. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Matsumura P. Phosphorylating and dephosphorylating protein complexes in bacterial chemotaxis. J Bacteriol. 1997;179:287–289. doi: 10.1128/jb.179.1.287-289.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourret RB, Borkovich KA, Simon MI. Signal transduction pathways involving protein phosphorylation in prokaryotes. Annu Rev Biochem. 1991;60:401–441. doi: 10.1146/annurev.bi.60.070191.002153. [DOI] [PubMed] [Google Scholar]
- 20.Ninfa EG, Stock A, Mowbray S, Stock J. Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J Biol Chem. 1991;266:9764–9770. [PubMed] [Google Scholar]
- 21.Kojima S, Blair DF. The bacterial flagellar motor: Structure and function of a complex molecular machine. Int Rev Cytol. 2004;233:93–134. doi: 10.1016/S0074-7696(04)33003-2. [DOI] [PubMed] [Google Scholar]
- 22.Bren A, Eisenbach M. How signals are heard during bacterial chemotaxis: Protein–protein interactions in sensory signal propagation. J Bacteriol. 2000;182:6865–6873. doi: 10.1128/jb.182.24.6865-6873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bowie JU, Pakula AA, Simon MI. The three-dimensional structure of the aspartate receptor from Escherichia coli. Acta Crystallogr Sect D: Biol Crystallogr. 1995;51:145–154. doi: 10.1107/S0907444994010498. [DOI] [PubMed] [Google Scholar]
- 24.Milburn MV, Prive GG, Milligan DL, Scott WG, Yeh J, Jancarik J, Koshland DE, Jr, Kim SH. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science. 1991;254:1342–1347. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- 25.Pakula AA, Simon MI. Determination of transmembrane protein structure by disulfide cross-linking: The Escherichia coli Tar receptor. Proc Natl Acad Sci USA. 1992;89:4144–4148. doi: 10.1073/pnas.89.9.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chervitz SA, Falke JJ. Molecular mechanism of transmembrane signaling by the aspartate receptor: A model. Proc Natl Acad Sci USA. 1996;93:2545–2550. doi: 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chervitz SA, Falke JJ. Lock on/off disulfides identify the transmembrane signaling helix of the aspartate receptor. J Biol Chem. 1995;270:24043–24053. doi: 10.1074/jbc.270.41.24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughson AG, Hazelbauer GL. Detecting the conformational change of transmembrane signaling in a bacterial chemoreceptor by measuring effects on disulfide cross-linking in vivo. Proc Natl Acad Sci USA. 1996;93:11546–11551. doi: 10.1073/pnas.93.21.11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ottemann KM, Xiao W, Shin YK, Koshland DE., Jr A piston model for transmembrane signaling of the aspartate receptor [see comments] Science. 1999;285:1751–1754. doi: 10.1126/science.285.5434.1751. [DOI] [PubMed] [Google Scholar]
- 30.Isaac B, Gallagher GJ, Balazs YS, Thompson LK. Site-directed rotational resonance solid-state NMR distance measurements probe structure and mechanism in the transmembrane domain of the serine bacterial chemoreceptor. Biochemistry. 2002;41:3025–3036. doi: 10.1021/bi015759h. [DOI] [PubMed] [Google Scholar]
- 31.Draheim RR, Bormans AF, Lai RZ, Manson MD. Tryptophan residues flanking the second transmembrane helix (TM2) set the signaling state of the Tar chemoreceptor. Biochemistry. 2005;44:1268–1277. doi: 10.1021/bi048969d. [DOI] [PubMed] [Google Scholar]
- 32.Gardina PJ, Bormans AF, Manson MD. A mechanism for simultaneous sensing of aspartate and maltose by the Tar chemoreceptor of Escherichia coli. Mol Microbiol. 1998;29:1147–1154. doi: 10.1046/j.1365-2958.1998.00964.x. [DOI] [PubMed] [Google Scholar]
- 33.Miller AS, Falke JJ. Side chains at the membrane–water interface modulate the signaling state of a transmembrane receptor. Biochemistry. 2004;43:1763–1770. doi: 10.1021/bi0360206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ames P, Parkinson JS. Constitutively signaling fragments of Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol. 1994;176:6340–6348. doi: 10.1128/jb.176.20.6340-6348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bass RB, Coleman MD, Falke JJ. Signaling domain of the aspartate receptor is a helical hairpin with a localized kinase docking surface: Cysteine and disulfide scanning studies. Biochemistry. 1999;38:9317–9327. doi: 10.1021/bi9908179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiomi D, Zhulin IB, Homma M, Kawagishi I. Dual recognition of the bacterial chemoreceptor by chemotaxis-specific domains of the CheR methyltransferase. J Biol Chem. 2002;277:42325–42333. doi: 10.1074/jbc.M202001200. [DOI] [PubMed] [Google Scholar]
- 37.Blair DF. How bacteria sense and swim. Annu Rev Microbiol. 1995;49:489–522. doi: 10.1146/annurev.mi.49.100195.002421. [DOI] [PubMed] [Google Scholar]
- 38.Nishiyama SI, Umemura T, Nara T, Homma M, Kawagishi I. Conversion of a bacterial warm sensor to a cold sensor by methylation of a single residue in the presence of an attractant. Mol Microbiol. 1999;32:357–365. doi: 10.1046/j.1365-2958.1999.01355.x. [DOI] [PubMed] [Google Scholar]
- 39.Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 40.Falke JJ, Kim SH. Structure of a conserved receptor domain that regulates kinase activity: The cytoplasmic domain of bacterial taxis receptors. Curr Opin Struct Biol. 2000;10:462–469. doi: 10.1016/s0959-440x(00)00115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bass RB, Falke JJ. The aspartate receptor cytoplasmic domain: In situ chemical analysis of structure, mechanism, and dynamics. Struct Fold Des. 1999;7:829–840. doi: 10.1016/s0969-2126(99)80106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Studdert CA, Parkinson JS. Crosslinking snapshots of bacterial chemoreceptor squads. Proc Natl Acad Sci USA. 2004;101:2117–2122. doi: 10.1073/pnas.0308622100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci USA. 2002;99:7060–7065. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bornhorst JA, Falke JJ. Attractant regulation of the aspartate receptor-kinase complex: Limited cooperative interactions between receptors and effects of the receptor modification state. Biochemistry. 2000;39:9486–9493. doi: 10.1021/bi0002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li G, Weis RM. Covalent modification regulates ligand binding to receptor complexes in the chemosensory system of Escherichia coli. Cell. 2000;100:357–365. doi: 10.1016/s0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]
- 46.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 47.Seeley SK, Weis RM, Thompson LK. The cytoplasmic fragment of the aspartate receptor displays globally dynamic behavior. Biochemistry. 1996;35:5199–5206. doi: 10.1021/bi9524979. [DOI] [PubMed] [Google Scholar]
- 48.Murphy OJ, III, Yi X, Weis RM, Thompson LK. Hydrogen exchange reveals a stable and expandable core within the aspartate receptor cytoplasmic domain. J Biol Chem. 2001;276:43262–43269. doi: 10.1074/jbc.M105585200. [DOI] [PubMed] [Google Scholar]
- 49.Danielson MA, Bass RB, Falke JJ. Cysteine and disulfide scanning reveals a regulatory α-helix in the cytoplasmic domain of the aspartate receptor. J Biol Chem. 1997;272:32878–32888. doi: 10.1074/jbc.272.52.32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long DG, Weis RM. Oligomerization of the cytoplasmic fragment from the aspartate receptor of Escherichia coli. Biochemistry. 1992;31:9904–9911. doi: 10.1021/bi00156a007. [DOI] [PubMed] [Google Scholar]
- 51.Foster DL, Mowbray SL, Jap BK, Koshland DE., Jr Purification and characterization of the aspartate chemoreceptor. J Biol Chem. 1985;260:11706–11710. [PubMed] [Google Scholar]
- 52.Falke JJ, Sternberg DE, Koshland DE., Jr Site-directed sulfhydryl chemistry and spectroscopy: Applications in the aspartate receptor system. Biophys J. 1986;49:20a. [Google Scholar]
- 53.Falke JJ, Koshland DE., Jr Global flexibility in a sensory receptor: A site-directed cross-linking approach. Science. 1987;237:1596–1600. doi: 10.1126/science.2820061. [DOI] [PubMed] [Google Scholar]
- 54.Butler SL, Falke JJ. Cysteine and disulfide scanning reveals two amphiphilic helices in the linker region of the aspartate chemoreceptor. Biochemistry. 1998;37:10746–10756. doi: 10.1021/bi980607g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hughson AG, Lee GF, Hazelbauer GL. Analysis of protein structure in intact cells: Crosslinking in vivo between introduced cysteines in the transmembrane domain of a bacterial chemoreceptor. Protein Sci. 1997;6:315–322. doi: 10.1002/pro.5560060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaback HR, Sahin-Toth M, Weinglass AB. The kamikaze approach to membrane transport. Nat Rev Mol Cell Biol. 2001;2:610–620. doi: 10.1038/35085077. [DOI] [PubMed] [Google Scholar]
- 57.Liu JD, Parkinson JS. Role of CheW protein in coupling membrane receptors to the intracellular signaling system of bacterial chemotaxis. Proc Natl Acad Sci USA. 1989;86:8703–8707. doi: 10.1073/pnas.86.22.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chervitz SA, Lin CM, Falke JJ. Transmembrane signaling by the aspartate receptor: Engineered disulfides reveal static regions of the subunit interface. Biochemistry. 1995;34:9722–9733. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 60.Adler J. Bacterial chemotaxis. Science. 1966;153:708–716. doi: 10.1126/science.153.3737.708. [DOI] [PubMed] [Google Scholar]
- 61.Borkovich KA, Kaplan N, Hess JF, Simon MI. Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc Natl Acad Sci USA. 1989;86:1208–1212. doi: 10.1073/pnas.86.4.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clarke S, Koshland DE., Jr Membrane receptors for aspartate and serine in bacterial chemotaxis. J Biol Chem. 1979;254:9695–9702. [PubMed] [Google Scholar]
- 63.Bass RB, Falke JJ. Detection of a conserved α-helix in the kinase-docking region of the aspartate receptor by cysteine and disulfide scanning. J Biol Chem. 1998;273:25006–25014. doi: 10.1074/jbc.273.39.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shapiro MJ, Koshland DE., Jr Mutagenic studies of the interaction between the aspartate receptor and methyltransferase from Escherichia coli. J Biol Chem. 1994;269:11054–11059. [PubMed] [Google Scholar]
- 65.Bornhorst JA, Falke JJ. Quantitative analysis of aspartate receptor signaling complex reveals that the homogeneous two-state model is inadequate: Development of a heterogeneous two-state model. J Mol Biol. 2003;326:1597–1614. doi: 10.1016/s0022-2836(03)00026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Careaga CL, Falke JJ. Thermal motions of surface α-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J Mol Biol. 1992;226:1219–1235. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim SH. “Frozen” dynamic dimer model for transmembrane signaling in bacterial chemotaxis receptors. Protein Sci. 1994;3:159–165. doi: 10.1002/pro.5560030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim SH, Wang W, Kim KK. Dynamic and clustering model of bacterial chemotaxis receptors: Structural basis for signaling and high sensitivity. Proc Natl Acad Sci USA. 2002;99:11611–11615. doi: 10.1073/pnas.132376499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Starrett DJ, Falke JJ. Adaptation mechanism of the aspartate receptor: Electrostatics of the adaptation subdomain play a key role in modulating kinase activity. Biochemistry. 2005 doi: 10.1021/bi048089z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tatsuno I, Homma M, Oosawa K, Kawagishi I. Signaling by the Escherichia coli aspartate chemoreceptor Tar with a single cytoplasmic domain per dimer. Science. 1996;274:423–425. doi: 10.1126/science.274.5286.423. [DOI] [PubMed] [Google Scholar]
- 71.Gardina PJ, Manson MD. Attractant signaling by an aspartate chemoreceptor dimer with a single cytoplasmic domain. Science. 1996;274:425–426. doi: 10.1126/science.274.5286.425. [DOI] [PubMed] [Google Scholar]
- 72.Lefman J, Zhang P, Hirai T, Weis RM, Juliani J, Bliss D, Kessel M, Bos E, Peters PJ, Subramaniam S. Three-dimensional electron microscopic imaging of membrane invaginations in Escherichia coli overproducing the chemotaxis receptor Tsr. J Bacteriol. 2004;186:5052–5061. doi: 10.1128/JB.186.15.5052-5061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chothia C, Lesk AM. Helix movements in proteins. Trends Biochem Sci. 1985;10:116–118. [Google Scholar]