

Abstract
A strategy for the stereoselective preparation of enantiomerically enriched cis-2,6-disubstituted piperazines from amino acid precursors is described. The target compounds are generated in 95–99% ee with good to excellent levels of diastereoselectivity (usually 14:1 to >20:1) using Pd-catalyzed carboamination reactions between aryl or alkenyl halides and substituted ethylenediamine derivatives to form the heterocyclic rings. The synthesis requires only 4–5 steps from commercially available amino acids, and allows for the modular construction of piperazines bearing different substituents at N1, N4, C2, and C6. The use of this strategy for the construction of 2,3-disubstituted piperazines, fused bicyclic piperazines, and tetrahydroquinoxalines is also reported. In addition, the mechanism of the key carboamination reactions are discussed, and new models that predict and explain the stereochemical outcome of these transformations are presented.
1. Introduction
The N-arylpiperazine scaffold is considered to be a privileged structure in medicinal chemistry,i and is displayed in thousands of biologically active molecules.ii Compounds containing this scaffold have been investigated in clinical studies in over twenty different therapeutic areas, including antidepressants, analgesics, and antibacterials. The biological activity of these compounds is significantly influenced by the degree of ring substitution and the nature of substituents.
One of the most commonly employed methods for the formation of piperazines involves dimerization of amino acids to afford diketopiperazines, which are subsequently reduced.iii This strategy provides straightforward access to 2,5-disubstituted products, and monosubstituted piperazines can also be generated in a similar fashion. However, piperazines that contain other substitution patterns can be much more difficult to prepare. For example, existing routes for the synthesis of enantioenriched 2,6-disubstituted piperazines typically require at least six steps, and are not easily amenable to the rapid construction of libraries of related analogs.iv,v,vi As such, the development of transformations that provide access to substituted piperazines that cannot be easily generated using currently available methods is of significant interest.vii
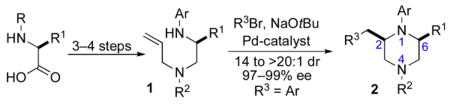 |
(1) |
We recently reported a concise asymmetric synthesis of cis-2,6-disubstituted piperazines (2) that employs a Pd-catalyzed carboamination reaction for the ring-forming step (eq 1).viii,ix,x The substrates (1) for the key transformation can be prepared in enantioenriched form (97–99% ee) from commercially available amino acid precursors using one of two 3–4 step sequences, and the carboamination reactions proceed with good to excellent levels of diastereoselectivity. The reactions generate two bonds and 1–2 stereocenters in a single step with no loss of enantiomeric purity. Significantly, this modular strategy permits the facile variation of the piperazine N1, N4, C2, and C6 substituents. In this article we describe our continued studies on the scope and limitations of this method, and the extension of this strategy to the construction of other substituted piperazines and benzopiperazines. We also report experiments that probe the validity of our initially proposed transition state model for the stereochemical outcome of these reactions, and present a refined hypothesis that is consistent with our current data.
2. Results
2.1. Synthesis of substrates
Retrosynthetic analysis of the substrates (1) required for the piperazine-forming carboamination reactions suggested that these compounds could be prepared from three simple, readily available components: N-substituted allylamines, amino acids, and aryl halides (Scheme 1). This synthetic strategy would allow for facile variation of the substrate Ar, R1, and R2 groups, and a fourth point of variance (R3) could be introduced during the carboamination to generate 2.
Scheme 1.

Retrosynthetic analysis
Our first approach to the construction of 1 is illustrated in Scheme 2, and commenced with a Cu-catalyzed N-phenylation of glycine, (S)-phenylalanine, or (S)-valine to afford 4a–c.xi,xii Subsequent coupling of glycine-derived amino acid 4c with diallylamine provided an acceptable yield of 5c using CDI as the coupling agent. However, amide bond formation proved to be challenging with enantiopure amino acid substrates, as many reagents, including DCC and CDI, provided products with only 85–90% ee. After some experimentation we found that the DEPBT reagent developed by Goodman provided satisfactory results,xiii affording products 5a–b with 98–99% ee. Reduction of 5a–c using LiAlH4 proceeded smoothly to afford the requisite 1,2-diamine substrates 1a–c in overall yields of 36–38% over the three-step sequence.
Scheme 2.

Synthesis of substrates 1a-c
Although the route illustrated in Scheme 2 did provide reasonably efficient access to 1a–c, this approach to substrate synthesis has several limitations. The scope of the Cu-catalyzed N-arylation reactions is somewhat limited, and partial loss of enantiomeric purity is known to be problematic with some amino acids (e.g. alanine).xi In addition, the DEPBT reagent is expensive, as are the materials for its preparation. As such, we sought to develop an alternate route that could be employed for the construction of other piperazine precursors.
A second and more generally applicable set of tactics for the construction of substrates 1 is illustrated in Scheme 3. Treatment of commercially available N-Boc-protected amino acids with DCC and an N-allylamine derivative afforded amides 6a–g. Although separation of these products from the dicyclohexylurea byproduct of the coupling reaction was difficult, LiAlH4 reduction of 6a–g provided 7a–g, which could easily be purified by column chromatography. Cleavage of the Boc-group was effected using HCl, and Pd-catalyzed N-arylationxiv of the resulting primary amines afforded substrates 1d–l in good yields and with near complete retention of enantiomeric purity. Although this sequence requires four steps, the overall yields of the N1-aryl-N4-allyl-1,2-diamine derivatives (35–58%) were comparable to (or greater than) yields obtained using the route shown in Scheme 2. Moreover, all transformations were achieved using inexpensive commercially available reagents.
Scheme 3.

Synthesis of substrates 1d–l
 |
(2) |
 |
(3) |
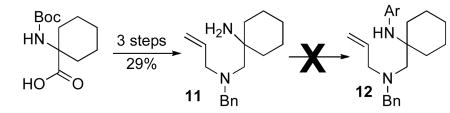 |
(4) |
In addition to providing access to substrates 1d–l bearing a simple N-allyl group, the route outlined in Scheme 3 was also used for the construction of substrate 9, which is substituted at the allylic position, and substrate 10, which contains an internal alkene (eq 2–3). However efforts to prepare a substrate bearing a bulky spirocyclohexane group adjacent to N1 (12) were unsuccessful, as we were unable to achieve N-arylation of the primary amine precursor 11 (eq 4).
2.2. Optimization of reaction conditions
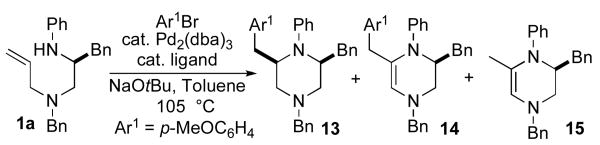
In our initial studies on Pd-catalyzed piperazine-forming reactions, we elected to examine the coupling of 1a with 4-bromoanisole. Our prior experiences in the development of other Pd-catalyzed alkene carboamination reactions suggested that use of NaOtBu as base and toluene as solvent would likely provide good results, and the most important reaction variable to optimize would be the ligand for palladium. As such, several different phosphine ligands were examined for this transformation. As shown below, (Table 1), palladium-catalyzed carboamination reactions of 1a with 4-bromoanisole provided three major products (13–15). The ratio of these products varied depending on the nature of the phosphine ligand, and use of P(2-furyl)3 provided optimal results.xv This finding was rather surprising, given the observation that chelating bis-phosphine ligands (e.g. dppe or dppb) typically provide the best results in related N-arylpyrrolidine-forming transformations.ixa
Table 1.
Ligand effectsa
 | ||||
|---|---|---|---|---|
| Entry | Ligand | Starting material consumed (%) | Product ratiob 13:14:15 |
Isolated yield 13 |
| 1 | dppb | 25 | 0:0:100 | — |
| 2 | dppe | 50 | 68:0:32 | — |
| 3 | Dpe-Phos | 97 | 56:23:21 | — |
| 4 | Xantphos | 82 | 50:35:15 | — |
| 5 | P(o-tol)3 | 87 | 9:17:74 | — |
| 6 | P(2-furyl)3 | 97 | 71:7:21 | 62%c |
Conditions: 1.0 equiv 1a, 1.2 equiv Ar1Br, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % chelating ligand or 8 mol % monodentate ligand, toluene (0.2 M), 105 °C, 8 h.
Determined by 1H NMR analysis of crude reaction mixtures.
This yield was obtained after complete conversion.
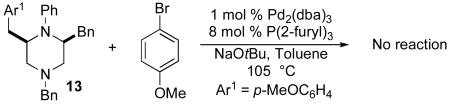
Two experiments were performed to probe the reaction mechanism and the origin of side products 14 and 15. When 15 was resubjected to the reaction conditions used for the transformation shown in Table 1, no reaction was observed (eq 5). Thus, 15 is not an intermediate in the formation of either 13 or 14. Similarly, 13 was not converted to 14 when treated with 4-bromoanisole, NaOtBu, and catalytic Pd2(dba)3/P(2-furyl)3 (eq 6). This indicates that 14 is not generated by way of Pd-catalyzed oxidation of 13.
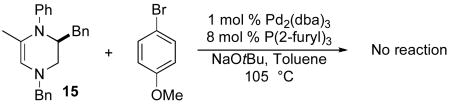 |
(5) |
 |
(6) |
2.3. Synthesis of substituted piperazines
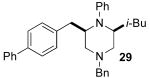
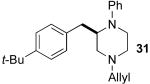
After suitable reaction conditions had been developed, we proceeded to examine the scope of the piperazine-forming carboamination reactions. As shown in Table 2, a number of different N-allyl-1,2-diamines were converted to cis-2,6-disubstituted piperazines in moderate to good yield with excellent diastereoselectivity. In all cases examined, the carboamination reactions proceeded with little or no erosion of enantiomeric purity. The presence of benzyl, allyl, or p-methoxyphenyl (PMP) protecting groups on N4 was tolerated. The reactions were effective with substrates derived from phenylalanine, valine, serine, alanine, leucine, and p-chlorophenylalanine. The synthesis of 2-substituted piperazines from glycine-derived substrates 1c and 7g also proceeded smoothly (entries 17–20).
Table 2.
Synthesis of cis-2,6-disubstituted piperazinesa
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | dr | ee (%) | Yield (%)b |
| 1 | 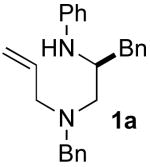 |
 |
>20:1 | 99 | 63 |
| 2 | 1a | 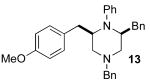 |
>20:1 | 99 | 62 |
| 3 | 1a | 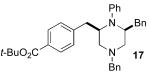 |
>20:1 | 98 | 59 |
| 4 | 1a | 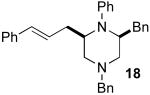 |
>20:1 | 95 | 73c,d |
| 5 | 1a | 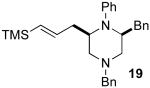 |
>20:1 | 97 | 59c,d |
| 6 | 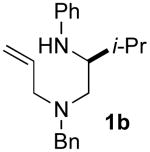 |
 |
>20:1 | 99 | 51c |
| 7 | 1b | 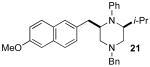 |
>20:1 | 99 | 50 |
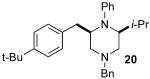
| 8 | 1b | 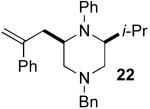 |
>20:1 | 97 | 52c,e |
| 9 | 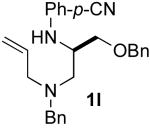 |
 |
14:1 | 97 | 71 |
| 10 | 1l | 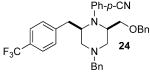 |
14:1 | 98 | 69 |
| 11 | 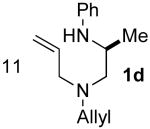 |
 |
>20:1 | 99 | 53 |
| 12 | 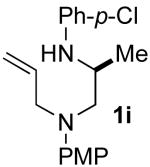 |
 |
>20:1 | 99 | 51 |
| 13 | 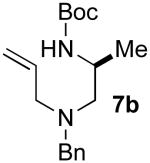 |
 |
1:1 | 99 | 68d,f |
| 14 | 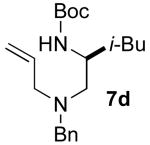 |
 |
2:1 | N/Ag | 63f |
| 15 |  |
 |
>20:1 | 99 | 57 |
| 16 | 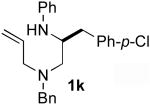 |
 |
>20:1 | N/Ag | 52 |
| 17 | 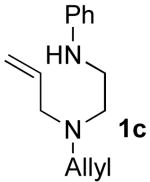 |
 |
— | — | 74h |
| 18 | 1c | 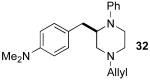 |
— | — | 56 |
| 19 | 1c | 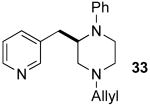 |
— | — | 66h |
| 20 | 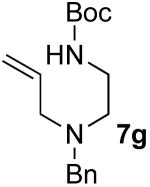 |
 |
— | — | 68f |
Conditions: 1.0 equiv amine, 1.2 equiv ArBr, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 8 mol % P(2-furyl)3, toluene (0.2 M), 105 °C, 8–10 h.
Average isolated yields obtained from two or more experiments.
The reaction was conducted with 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3.
The reaction was conducted at 90 °C using 1.4 equiv of alkenyl halide.
The reaction was conducted at 135 °C in xylenes.
The reaction was conducted using a catalyst composed of 6 mol % Pd(OAc)2 and 8 mol % PPh3.
The reaction was conducted using racemic starting material.
The reaction was conducted using 2 mol % dppb as ligand
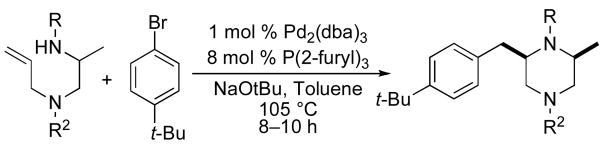
The electronic properties of the aryl bromide electrophile did not have a large influence on yield or stereoselectivity, as transformations of electron-rich, electron-neutral, and electron-poor aryl halides proceeded with similar efficiency. However, our initial efforts to employ alkenyl bromides as coupling partners met with limited success. For example, reactions in which β-bromostyrene was used as the electrophile typically halted at <50% conversion (affording products in ca 25% yield). Fortunately, after further optimization we discovered that acceptable yields could be obtained by increasing the catalyst loading to 2 mol % Pd2(dba)3/16 mol % P(2-furyl)3 and using a slightly larger excess of electrophile (1.4 equiv). Under these conditions, several different 2-allylpiperazine derivatives were prepared with excellent stereocontrol (Table 2, entries 4, 5, and 8).
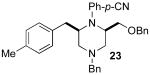
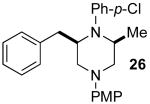
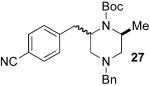
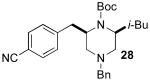
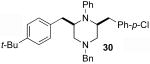
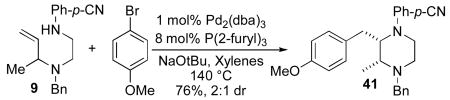
Although most transformations examined proceeded with >20:1 diastereoselectivity, reactions of 1l, which contains an N1-p-cyanophenyl group, afforded piperazines 23 and 24 with slightly lower selectivity (14:1 dr, entries 9–10). In addition, poor stereoselectivity was observed in the conversion of N1-Boc-protected substrates 7b and 7d to piperazines 27 and 28 (entries 13–14). In order to further probe the effect of different N-substituents on stereocontrol, the coupling of 1-bromo-4-tert-butylbenzene with a series of alanine-derived substrates was examined. As shown in Table 3, the nature of the N1-substituent had a large effect on diastereoselectivity. The highest selectivity was obtained when N1 was substituted with an electron-rich p-methoxyphenyl group (Table 3, entry 1). Lower stereoselectivities were observed with substrates bearing electron-withdrawing N1 groups such as p-cyanophenyl or Boc (entries 3–4).xvi Interestingly, the nature of the N4-group also influenced stereocontrol in these reactions. Carboaminations of 1d (N4-allyl) and 1h (N4-p-methoxyphenyl) both proceeded with excellent (>20:1) diastereoselectivity (entries 5–6), but the analogous reaction of 1e (N4-benzyl) gave 36 with only 9:1 dr (entry 2).xvii
Table 3.
N-Substituent effectsa
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R | R2 | Product | drb | Yield (%)c |
| 1 | 1f | PMP | Bn | 35 | >20:1 | 45 |
| 2 | 1e | Ph | Bn | 36 | 9:1 | 54 |
| 3 | 1g | Ph-p-CN | Bn | 37 | 6:1 | 74 |
| 4 | 7b | Boc | Bn | 38 | 1:1 | 34d |
| 5 | 1d | Ph | Allyl | 39 | >20:1 | 45 |
| 6 | 1h | Ph | PMP | 40 | >20:1 | 50 |
Conditions: 1.0 equiv amine, 1.2 equiv ArBr, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 8 mol % P(2-furyl)3, toluene (0.2 M), 105 °C.
Diastereomeric ratio observed in crude reaction mixtures.
Average isolated yields obtained from two or more experiments.
The reaction was conducted using 6 mol % Pd(OAc)2 and 8 mol % PPh3 at 90 °C. Use of 1 mol % Pd2(dba)3 and 8 mol % P(2-furyl)3 provided 38 in 10% yield with 1.3:1 dr.
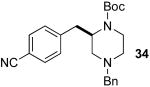
In order to further explore the scope of our palladium-catalyzed piperazine-forming carboamination reactions we examined cyclizations of substrates 9 and 10, which contain substitution on or adjacent to the alkene moiety. Reactions of these substrates were slow at 110 °C, but proceeded to completion in < 24 h when heated to 140 °C in xylenes solvent. As shown in eq 7, diamine 9 bearing an allylic methyl group was converted to piperazine 41 in moderate yield but with low diastereoselectivity. However, cyclopentene-derived substrate 10 was coupled with 4-bromobenzonitrile or 4-bromobenzophenone to afford bicyclic piperazines 42 and 43, which result from syn-addition to the alkene, with excellent diastereoselectivity.
 |
(7) |
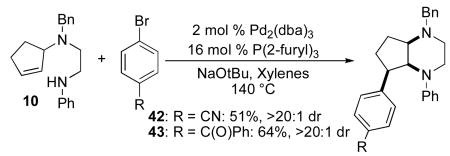 |
(8) |
We also sought to examine the synthesis of 2-substituted tetrahydroquinoxalines via carboamination reactions of N-allylphenylenediamine-derived precursors. We initially envisioned using substrates in which both amino groups were substituted with arenes, which could be installed via Pd-catalyzed N-arylation reactions.xiv As shown below (Table 4, Method A), the N-arylation of 44 proceeded smoothly to afford 45a–c. However, when we attempted to N-arylate the allyl-substituted amino group of 45a–c we were surprised to discover that carboamination had occurred in preference to N-arylation, and tetrahydroquinoxalines 46a–d were formed in moderate to good yield. The conversion of 44 to 46b–c was also achieved using a one-pot procedure (Method B) in which 44 was treated with an aryl bromide in the presence of NaOtBu and a catalyst composed of Pd2(dba)3 and tBu2P(o-biphenyl). After the aryl bromide was consumed, a catalytic amount of (±)-BINAP was added, and the mixture was stirred for 10 min to effect in situ ligand exchange.xviii Addition of a second aryl bromide then afforded the tetrahydroquinoxaline products.
Table 4.
Synthesis of tetrahydroquinoxalines
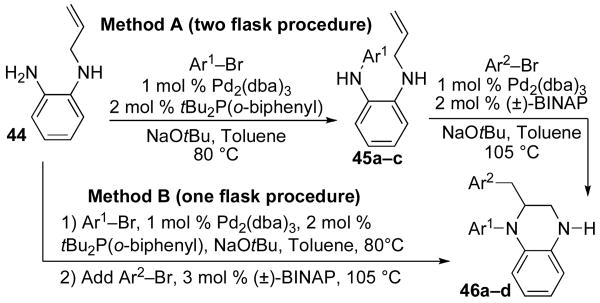 | |||||
|---|---|---|---|---|---|
| Entry | Methoda | Ar1 | Yield 45 (%)b | Ar2 | Yield 46 (%)b |
| 1 | A | Ph-p-OMe | 45a, 75 | Ph-p-OMe | 46a, 48 |
| 2 | A | Ph | 45b, 69 | Ph-p-OMe | 46b, 60 |
| 3 | B | Ph | – | Ph-p-OMe | 46b, 49 |
| 4 | A | Ph | 45b, 69 | 5-indolyl (N-Bn) | 46c, 62 |
| 5 | B | Ph | – | 5-indolyl (N-Bn) | 46c, 41 |
| 6 | A | Ph-p-CN | 45c, 72 | Ph | 46d, 63 |
Conditions for method A: (i) 1.0 equiv 44, 1.0−1.1 equiv Ar1Br, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % tBu2P(o-biphenyl), toluene (0.2 M), 80 °C, 6 h. (ii) 1.0 equiv 45, 1.2 equiv Ar2Br, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % (±)-BINAP, toluene (0.2 M), 105 °C, 10 h. Conditions for method B: 1.0 equiv 44, 1.0 equiv Ar1Br, 2.1 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % tBu2P(o-biphenyl), toluene (0.2 M), 80 °C, 20 min. Then add 3 mol % (±)-BINAP, 85 °C, 10 min. Then add 1.0 equiv Ar2Br, 105 °C, 14h.
Average isolated yields obtained from two or more experiments.
3. Discussion
3.1. Mechanism of piperazine formation
The mechanism of the Pd-catalyzed piperazine-forming reactions described above appears to be similar to that of related carboamination reactions of γ-aminoalkene derivatives that afford pyrrolidines and other nitrogen heterocycles.ix As shown in Scheme 4, the catalytic cycle presumably commences with oxidative addition of the aryl (or alkenyl) bromide to a Pd(0) complex generated in situ from the combination of Pd2(dba)3 and P(2-furyl)3. The resulting Pd(II) intermediate 47 is then transformed to amido complex 48 upon reaction with the amine and NaOtBu.xiv The conversion of 48 to 49 occurs by way of syn-aminopalladation of the pendant alkene,ixa,e,xix,xx and C–C bond-forming reductive elimination from 49 generates the observed piperazine product. The proposed syn-aminopalladation pathway is consistent with the observed selectivity for the conversion of cyclopentene derivative 10 to the bicyclic piperazines 42 and 43 (eq 8), which both result from syn-addition of the aryl group and the amino group to the alkene.
Scheme 4.

Catalytic cycle
The observed formation of side products 14 and 15 (Table 1) is also consistent with the catalytic cycle shown above, and provides further support for a mechanism involving alkene aminopalladation (48 to 49) rather than Heck-type carbopalladation of the substrate alkene. As shown in Scheme 5, 14 and 15 are most likely formed through a competing β-hydride elimination side reaction of intermediate 49 to generate 50, which is a common intermediate leading to both side products. The conversion of 50 to 15 may proceed via displacement of palladium to generate 52, followed by base-mediated isomerization of 52 to the more thermodynamically stable isomer 15. Alternatively, 15 may also be formed from 50 via insertion of the alkene into the Pd–H bond to afford 51 followed by β-hydride elimination and loss of Pd.
Scheme 5.

Formation of side products
Side product 14 may also be formed from 50 via one of two pathways. We currently favor a mechanism involving the conversion of 50 to 52 followed by Heck-arylation of the exo-methylene group to afford 14. However, the generation of 14 through carbopalladation of 50 to provide 53,xxi followed by β-hydride elimination and loss of LnPd(H)2 cannot be ruled out with the data currently on hand.
Interestingly, although side products with the same general structures as 14 and 15 (but with different Ar1 and N–R groups) are formed in ca. 20–30% combined yields in most of the piperazine-forming reactions, analogous unsaturated side products 55 and 56 are typically not observed in pyrrolidine forming reactions. Instead, pyrrolidine regioisomers of the general structure 54 shown below (Scheme 6) are generated in ca. 10% yield. Side products 54 are believed to arise from β-hydride elimination from 57 to afford 58. However, it appears that alkene insertion/reductive elimination processes that lead to 54 are dominant over alkene displacement from Pd (which could, in principle, lead to 55 or 56) in the N-aryl pyrrolidine series. The fact that relatively small amounts of side products that result from β-hydride elimination of 57 are observed also suggests that the conversion of 57 to 58 is less favorable than conversion of 49 to 50. These trends may be due to the fact that rehybridization of either the C2-carbon, the C3-carbon, or both from sp3 to sp2 leads to a greater increase in strain in the five-membered ring series. This would be expected to both decrease the rate of β-hydride elimination and to increase the equilibrium concentration of alkylpalladium intermediates (57) relative to palladium-alkene complexes (58).
Scheme 6.

Side products in pyrrolidine-forming reactions
3.2. Stereochemical models for piperazine formation
In related studies on Pd-catalyzed carboamination reactions that afford pyrrolidine products, we have observed that substrates 59 are converted to cis-2,5-disubstituted pyrrolidines 61 with >20:1 dr, and transformations of 62 afford trans-2,3-disubstituted molecules 64 with ca. 10–20:1 dr (Scheme 7).ix To account for these results, we have suggested that the stereochemistry determining step of pyrrolidine formation involves syn-aminopalladation through transition states 60 and 63, with the alkene π-system and the Pd–N bond eclipsed.xxii Pseudoaxial orientation of R is preferred in transition state 60 to avoid unfavorable A(1,3)-strain interactions with the sp2N-aryl or sp2N-Boc substituent.xxiii There also appears to be a kinetic preference for alkene aminopalladation to occur with orientation of the nonbonding electrons on nitrogen perpendicular to the alkene π-system.xxiv
Scheme 7.

Pyrrolidine stereochemistry
In contrast, carboamination reactions that afford cis-2,6- or cis-2,3-disubstituted piperazine products (Table 2 and eq 7) appear to proceed via transition states that differ significantly from the model illustrated above (Scheme 7). As shown in Scheme 8, an analogous model for piperazine formation would involve transition states 65 and 67. However, neither of these two transition states lead to the observed major stereoisomers.
Scheme 8.

Allylic strain/chair model
In our initial communication on Pd-catalyzed piperazine-forming reactions, we proposed that the cis-2,6-disubstituted products, which are generated with high diastereoselectivity in most cases (14 to >20:1), may result from reaction via transition state 69.viii As shown in Scheme 9, rotation of the N1-aryl group and pyramidalization of the N1 atom would alleviate allylic strain and favor equatorial orientation of the C2-substituent. This model does explain the formation of the observed cis-2,6-disubstituted piperazines. In addition, the observation that substrates bearing electron-poor N1-aryl groups or Boc-groups are transformed with relatively low diastereoselectivity (Table 3) is also consistent with this hypothesis. The presence of either an N-Boc protecting group or a π-electron withdrawing group on the arene would be expected to disfavor pyramidalization of the N1 atom, and might lead to a greater degree of cyclization through transition state 65. However, a chair-like transition state model fails to predict the observed major diastereomer in the reaction of 9, which provides 2,3-disubstituted product 41. Cyclization via 70 should afford trans-2,3-disubstituted piperazine 68, which is generated as the minor isomer. In addition to the inability of this model to account for all observed product stereochemistry, examination of molecular models suggests that the geometry of transition states 69 and 70 leads to relatively poor overlap between the Pd–N σ-bond and the alkene π-system.xxii
Scheme 9.

Pyramidal N1/chair model
Alternatively, a boat-like transition state model (71, Scheme 10) can also be used to explain the observed high stereocontrol in reactions that generate cis-2,6-disubstituted piperazines. In contrast to the transition states shown in Schemes 7–8, the nonbonding electrons on N1 in 71 are directed towards the alkene π-system, and the overlap between the Pd–N bond and the alkene appears to be very good.xxii The favored transition state 71 positions the C2 R-group in a pseudoequatorial position. The high diastereoselectivity can be explained through examination of a second boat-like transition state 72, that would afford the minor diastereomer. This transition state contains a severe steric interaction between the C5 H-substituent and the R-group. In addition, transition state 72 also suffers from significant A(1,3) strain between the C2 R-group and the N-aryl group. As such, cyclization via this transition state should be quite unfavorable.
Scheme 10.

Boat-like transition states for 2,6-disubstituted piperazine formation.
A similar boat-like transition state model can also account for formation of the observed major cis-2,3-disubstituted piperazine stereoisomer in the cyclization of 9. As shown in Scheme 11, pseudoequatorial orientation of the methyl group in transition state 73 would provide 41, whereas axial orientation of the methyl-group (transition state 74) would lead to minor stereoisomer 68. In addition to predicting the correct product stereochemistry, this model also accounts for the modest diastereoselectivity observed in the formation of the 2,3-disubstituted piperazine.xxv In contrast to transition state 72, which is destabilized by two significant steric interactions, transition state 74 suffers from only the interaction between the axial methyl-group and the C2 H-atom. As such the difference in energy between transition states 73 and 74 should be small relative to the difference in energy between 71 and 72.
Scheme 11.

Boat-like transition states for 2,3-disubstituted piperazine formation.
Although the boat-like transition state models illustrated in Schemes 10 and 11 account for the observed major product stereochemistry, the effect of N1 nucleophilicity on diastereoselectivity outlined in Table 3 is more difficult to explain with this model. It is possible that the propensity of a given reaction to proceed through a chair-like vs. boat-like transition state is influenced by either the electronic properties of N1, the substitution pattern of the substrate, or both. Nonetheless, transition state models 69, 71, and 73 serve as useful tools for predicting product stereochemistry.
4. Summary and conclusion
In conclusion, we have developed a concise asymmetric synthesis of cis-2,6-disubstituted piperazines from readily available enantiopure amino acids. The target molecules are generated with good to excellent diastereoselectivity and 95–99% ee in only 4–5 steps. The key Pd-catalyzed carboamination reactions that form the heterocylic ring are effective with a number of different aryl or alkenyl halides as coupling partners. Importantly, this strategy allows the installation of different groups at N1, N4, C2, and C6 in a straightforward manner. This method can also be employed for the synthesis of cis-2,3-disubstituted piperazines or 2-substituted tetrahydroquinoxalines. Further studies directed at adapting this strategy to allow for the synthesis of other six-membered nitrogen heterocycles are currently underway.
5. Experimental
5.1. General
All reactions were carried out under an argon or nitrogen atmosphere in oven- or flame-dried glassware. All catalysts, reagents, and aryl bromides were obtained from commercial sources and were used without further purification. N-phenyl-L-phenylalanine and N-phenyl-L-valine were prepared according to published procedures.xi DEPBT was prepared according to the procedure of Goodmanxiii and was purified by recrystallization from petroleum ether:ethyl acetate (1:1) followed by trituration with ethyl acetate to yield a white solid. Use of pure, colorless, reagent was essential to prevent degradation of enantiomeric purity during amide bond formation. Toluene, THF, ether, and dichloromethane were dried and purified using a GlassContour solvent purification system. Structural and stereochemical assignments were made on the basis of 2-D COSY, HSQC, and NOESY experiments. Ratios of diastereomers were determined by 1H NMR and/or capillary GC analysis of crude reaction mixtures. Yields refer to isolated yields of compounds estimated to 1 be ≥95% pure as determined by H NMR, GC, and/or combustion analysis. The yields reported in Section 5 describe the result of a single experiment, whereas the yields reported in Tables 1–4, Schemes 2–3, eq 2–4 and eq 7–8 are average yields of two or more experiments. Thus, the yields reported in Section 5 may differ from those shown in Tables 1–4, Schemes 2–3, eq 2–4 and eq 7–8.
5.2. General procedure 1: conversion of N-phenyl amino acids to N-allyl-N-alkyl-N′-phenyl amino amides
A flame-dried round-bottomed flask equipped with a magnetic stirbar was cooled under a stream of nitrogen and charged with the N-phenyl amino acid substrate (1.0 equiv). THF was added to provide a 0.5 M solution, which was cooled to 0 °C and stirred. DEPBTxiii (1.2 equiv) was added, followed immediately by N-benzylallylamine (1 equiv), and the resulting mixture was stirred at 0 °C until the starting amine had been consumed as judged by crude 1H NMR analysis of an aliquot (ca. 3– 4 h). Aqueous sodium bicarbonate was then added at 0 °C, and the resulting yellow mixture was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was then purified via flash chromatography on silica gel.
5.2.1. (S)-N-Allyl-N-benzyl-3-phenyl-2-phenylaminopropionamide (5a)
General procedure 1 was employed for the coupling of (S)-3-phenyl-2-phenylaminopropionic acidxi (3.63 g, 15.0 mmol) with N-benzylallylamine (2.21 g, 15.0 mmol). This procedure afforded 3.79 g (68%) of the title compound as a yellow oil. The enantiopurity was judged to be 98% ee by chiral HPLC analysis (chiralcel OJ-H column, 10% isopropanol/hexanes, 1 mL/min, RT = 11.14 min and 18.44 min). This molecule was observed as a 1.5:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.31−7.17 (m, 9 H), 7.14−7.09 (m, 2 H), 6.92−6.91 (m, 1 H), 6.77−6.71 (m, 1 H), 6.66 (d, J = 8.0 Hz, 1 H), 6.54 (d, J = 7.5 Hz, 1 H), 5.73−5.66 (m, 0.4 H), 5.47−5.39 (m, 0.6 H), 5.15−4.97 (m, 2 H), 4.74 (d, J = 14.5 Hz, 0.6 H), 4.56−4.53 (m, 1.4 H), 4.49 (s, 0.5 H), 4.32 (d, J = 14.5 Hz, 0.6 H), 4.22−4.17 (m, 0.7 H), 4.03 (d, J = 17.0 Hz, 0.5 H), 3.65 (dd, J = 6.5, 15.0 Hz, 0.5 H), 3.53 (d, J = 5.5 Hz, 1.2 H), 3.13−3.02 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 172.9, 172.8, 146.6, 146.5, 137.4, 137.3, 136.9, 136.2, 132.5, 132.4, 129.54, 129.50, 129.46, 129.43, 129.0, 128.7, 128.60, 128.56, 128.4, 127.7, 127.5, 126.9, 126.8, 126.5, 118.4, 118.1, 117.6, 114.3, 55.9, 55.6, 49.5, 48.7, 48.6, 46.4, 39.53, 39.49; IR (film) 3326, 3027, 1639 cm−1. Anal. Calcd for C25H26N2O: C, 81.05; H, 7.07; N, 7.56. Found: C, 80.90; H, 7.05; N, 7.50.
5.2.2. (S)-N-Allyl-N-benzyl-3-methyl-2-phenylaminobutyramide (5b)
General procedure 1 was employed for the coupling of (S)-3-methyl-2-phenylaminobutyric acidxi (1.27 g, 6.57 mmol) with N-benzylallylamine (967 mg, 6.57 mmol). This procedure afforded 1.37 g (65%) of the title compound as a tan solid, m.p. 48–55 °C. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 10% isopropanol/hexanes, 1 mL/min, RT = 7.40 min and 10.00 min). This molecule was observed as a 1.5:1 mixture of rotamers. 1H NMR (500 MHz, CDCl3) δ 7.29−7.24 (m, 3 H), 7.16−7.14 (m, 2 H), 7.09−7.06 (m, 2 H), 6.73−6.66 (m, 2 H), 6.54 (d, 1 H), 5.76−5.66 (m, 1 H), 5.21−5.04 (m, 2 H), 4.88 (d, J = 14.5 Hz, 0.6 H), 4.70 (d, J = 17.0 Hz, 0.4 H), 4.46−4.28 (m, 2 H), 4.15 (s, 1 H), 3.97−3.93 (dd, J = 3.0, 17.0 Hz, 0.7 H), 3.84−3.79 (dd, J = 3.0, 17.0 Hz, 0.7 H), 3.60 (dd, J = 6.5, 15.0 Hz, 0.6 H), 2.09−2.08 (m, 1 H), 1.06−1.00 (m, 6 H); 13C NMR (125 MHz, CDCl3) δ 173.6, 173.5, 148.25, 148.21, 137.2, 136.2, 132.7, 132.6, 129.2, 128.9, 128.6, 128.1, 127.7, 127.4, 126.6, 118.1, 117.9, 117.7, 114.5, 114.4, 59.5, 59.2, 49.9, 48.8, 48.1, 48.0, 32.3, 32.2, 20.2, 20.1, 17.72, 17.68; IR (film) 3350, 2962, 1638 cm−1. Anal. Calcd for C21H26N2O: C, 78.22; H, 8.13; N, 8.69. Found: C, 77.96; H, 8.12; N, 8.68.
5.2.3. N,N-diallyl-2-(phenylamino)acetamide (5c)
General procedure 1 was employed for the coupling of N-phenyl glycine (1.32 g, 8.73 mmol)xii with diallylamine (881 mg, 9.07 mmol), except that the reaction was conducted using CDI (1.42 g, 8.73 mmol) in place of DEPBT, and with a reaction concentration of 0.3 M (with respect to N-phenyl glycine). This procedure afforded 1.31 g (65%) of the title compound as a yellow oil. This molecule was observed as a 1.5:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.22−7.18 (m, 2 H), 6.73 (tt, J = 0.8, 7.2 Hz, 1 H), 6.62 (dd, J = 1.2, 8.8 Hz, 2 H), 5.85−5.74 (m, 2 H), 5.28−5.21 (m, 2 H), 5.20−5.15 (m, 2 H), 4.88 (s, 1 H), 4.07 (d, J = 6.0 Hz, 2 H), 3.90−3.89 (m, 4 H); 13C NMR (100 MHz, CDCl3) δ 169.2, 147.5, 132.8, 132.2, 129.4, 118.0, 117.6, 117.4, 113.1, 48.5, 48.3, 45.2; IR (film) 3387, 2919, 1655 cm−1. Anal calcd for C14H18N2O: C, 73.01; H, 7.88; N, 12.16. Found: C, 72.98; H, 7.87; N, 12.07.
5.3. General procedure 2: reduction of N-allyl-N-alkyl-N′-phenyl amino amides to N-allyl-N-alkyl-N′-phenyl-1,2-diamines
A flame-dried flask equipped with a magnetic stirbar was cooled under a stream of nitrogen and charged with the N-allyl-N-alkyl-N′-phenyl amino amide substrate (1.0 equiv). Diethyl ether was added to provide a 0.5 M solution, which was cooled to 0 °C and stirred. A solution of lithium aluminum hydride in diethyl ether (1 M, 2 equiv) was added dropwise, and the resulting mixture was stirred at 0 °C until the starting amide was completely consumed as judged by TLC analysis (ca. 2 h). Water (2 mL) was added dropwise, followed by 10 M aqueous NaOH (5 mL), and additional water (2 mL). The resulting suspension was stirred at rt for 5–10 minutes, then decanted, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel.
5.3.1 (–)-(S)-N1-Allyl-N1-benzyl-3,N2-diphenylpropane-1,2-diamine (1a)
General procedure 2 was conducted using (S)-5a (3.79 g, 10.22 mmol) as substrate. This procedure afforded 3.13 g (86%) of the title compound as a yellow oil that was judged to be 98% ee by chiral HPLC analysis (chiralcel OD-H column, 0.5% isopropanol/hexanes, 1 mL/min, RT = 11.15 min and 12.72 min), [α]23D −43.08° (c 0.26, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.31−7.23 (m, 8 H), 7.21−7.14 (m, 5 H), 6.68 (t, J = 7.6 Hz, 1 H), 6.60 (d, J = 8.4 Hz, 1 H), 5.89−5.79 (m, 1 H), 5.14−5.11 (m, 2 H), 3.93 (s, 1 H), 3.72−3.65 (m, 1 H), 3.62 (d, J = 13.6 Hz, 1 H), 3.51 (d, J = 13.6 Hz, 1 H), 3.14−3.01 (m, 2 H), 2.92 (dd, J = 5.2, 14.0 Hz, 1 H), 2.80 (dd, J = 7.2, 14.4 Hz, 1 H), 2.57−2.47 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 148.1, 139.3, 138.7, 135.5, 129.6, 129.4, 129.2, 128.4, 127.2, 126.3, 118.0, 117.3, 113.4, 58.7, 57.4, 56.9, 52.3, 39.2 (one aromatic carbon signal is absent due to accidental equivalence); IR (film) 3400, 2957, 1600 cm−1. Anal. Calcd for C25H28N2: C, 84.23; H, 7.92; N, 7.86. Found: C, 84.23; H, 7.98; N, 7.87.
5.3.2. (–)-(S)-N1-allyl-N1-benzyl-3-methyl-N2-phenylbutane-1,2-diamine (1b)
General procedure 2 was conducted using (S)-5b (1.34 g, 4.16 mmol) as substrate. This procedure afforded 1.13 g (88%) of the title compound as a yellow oil that was judged to be 99% ee by chiral HPLC analysis (chiralcel OD-H column, 0.1% isopropanol/hexanes, 0.2 mL/min, RT = 31.65 min and 35.18 min), [α]23D −59.5° (c 0.98, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.31−7.23 (m, 5 H), 7.22−7.12 (m, 2 H), 6.65 (t, J = 7.5 Hz, 1 H), 6.56 (d, J = 8.5 Hz, 2 H), 5.88 (ddt, J = 6.0, 10.5, 16.5 Hz, 1 H), 5.18−5.13 (m, 2 H), 3.72 (s, 1 H), 3.63 (d, J = 13.5 Hz, 1 H), 3.50 (d, J = 13.5Hz, 1 H), 3.37−3.33 (m, 1 H), 3.11 (dd, J = 6.0, 14.5 Hz, 1 H), 3.04 (dd, J = 7.0, 13.5 Hz, 1 H), 2.52−2.44 (m, 2 H), 2.13−2.06 (m, 1 H), 0.87 (dd, J = 4.5, 7.0 Hz, 6 H); 13C NMR (100 MHz, CDCl3) δ 149.1, 139.7, 136.0, 129.4, 129.2, 128.4, 127.1, 117.8, 116.8, 113.2, 58.9, 57.5, 55.9, 54.3, 29.5, 18.6, 17.5; IR (film) 3400, 3025, 1601 cm−1. Anal. Calcd for C21H28N2: C, 81.77; H, 9.15; N, 9.08. Found: C, 81.73; H, 9.21; N, 9.09.
5.3.3 N1,N1-diallyl-N2-phenylethane-1,2-diamine (1c)
General procedure 2 was conducted using 5c (1.01 g, 4.39 mmol) as substrate. This procedure afforded 805 mg (85%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.20−7.16 (m, 2 H), 6.70 (t, J = 7.2 Hz, 1 H), 6.62 (dd, J = 1.2, 8.8 Hz, 2 H), 5.90−5.80 (m, 2 H), 5.21−5.13 (m, 4 H), 4.26 (s, 1 H), 3.16−3.12 (m, 6 H), 2.72 (t, J = 6.4 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ 148.7, 135.5, 129.3, 117.8, 117.3, 113.1, 56.7, 51.8, 41.2; IR (film) 3379, 2811, 1602 cm−1. Anal calcd for C14H20N2: C, 77.73; H, 9.32; N, 12.95. Found: C, 77.50; H, 9.32; N, 13.02.
5.4. General procedure 3: conversion of N-Boc amino acids to N-Boc-N′-allyl-1,2-diamines
A flame-dried flask equipped with a magnetic stirbar was cooled under a stream of nitrogen and charged with the N-Boc amino acid (1.0 equiv), DCC (2.0 equiv) and a sufficient volume of dichloromethane to provide a solution with a 0.5 M amine concentration. The solution was stirred for 30 min at rt then the appropriate allylamine derivative (1.0 equiv) was added. The resulting mixture was stirred at room temperature for 12 h then filtered to remove the dicyclohexylurea byproduct. The resulting solution was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The crude N-Boc-N′-allyl amino amide product was purified by flash chromatography on silica gel. In some cases the purified product was contaminated with small amounts of dicyclohexylurea. This material was carried on without further purification.
A flame-dried flask equipped with a magnetic stirbar was cooled under a stream of nitrogen and charged with the N-Boc-N′-allyl amino amide (1.0 equiv). Diethyl ether was added to provide a 0.5 M solution, which was cooled to 0 °C. A solution of lithium aluminum hydride in diethyl ether (1 M, 2.0 equiv) was added dropwise and the reaction was stirred at 0 °C until the starting amide was consumed as judged by TLC analysis (ca. 3 h). Water (2 mL) was added dropwise, followed by 10 M aqueous NaOH (5 mL), and additional water (2 mL). The resulting suspension was stirred at rt for 5–10 min, then decanted, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel.
5.4.1. (S)-tert-Butyl-1-(diallylamino)propan-2-ylcarbamate (7a)
General procedure 3 was used for the coupling of (S)-2-tert-butoxycarbonylaminopropionic acid (1.16 g, 6.13 mmol), with diallylamine (754 mL, 6.13 mmol). This procedure afforded 1.64 g (100%) of (S)-tert-butyl 1-(diallylamino)-1-oxopropan-2-ylcarbamate (6a) as a white solid, which was contaminated with 5% dicyclohexylurea. This material was carried on without further purification. This molecule was observed as a 5:1 mixture of rotamers. 1H NMR (500 MHz, CDCl3) δ 5.83−5.70 (m, 2 H), 5.39 (d, J = 7.5 Hz, 0.8 H), 5.25−5.11 (m, 4 H), 4.99−4.96 (m, 0.2 H), 4.59−4.57 (m, 0.8 H), 4.43−4.41 (m, 0.2 H), 4.18−4.10 (m, 0.2 H), 4.06−3.88 (m, 3.6 H), 3.70−3.60 (m, 0.2 H), 1.43 (s, 9 H), 1.31 (d, J = 6.5 Hz, 3 H). MS (ESI) 291.1690 (291.1685 calcd for C14H24N2O3, M + Na+).
Amide (S)-6a (1.64 g, 6.16 mmol) was reduced following general procedure 3. This procedure afforded 1.13 g (72%) of the title compound as a white solid, m.p. 45–47 °C. 1H NMR (500 MHz, CDCl3) δ 5.85−5.77 (m, 2 H), 5.18−5.11 (m, 4 H), 4.68 (s, 1 H), 3.67−3.63 (m, 1 H), 3.12 (dd, J = 6.0, 14.0 Hz, 2 H), 3.04 (dd, J = 6.5, 14.0 Hz, 2 H), 2.40−2.30 (m, 2 H), 1.44 (s, 9 H), 1.12 (d, J = 6.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 156.0, 135.9, 117.7, 79.1, 58.7, 57.3, 44.8, 28.7, 19.7; IR (film) 3326, 2930, 1690 cm−1; MS (EI) 254.2003 (254.1994 calcd for C14H26N2O2, M + H+).
5.4.2.(±)-tert-Butyl-1-[allyl(benzyl)amino]propan-2-ylcarbamate (7b)
General procedure 3 was used for the coupling of 2-(tert-butoxycarbonylamino)propanoic acid (4.5 g, 23.78 mmol) and N-benzylallylamine (3.50 g, 23.78 mmol). This procedure afforded 7.57 g (100%) of tert-butyl 1-[allyl(benzyl)amino]-1-oxopropan-2-ylcarbamate (6b) as a white solid, which was contaminated with ca. 15% of dicyclohexylurea. This molecule was observed as a 1.3:1 mixture of rotamers. 1H NMR (300 MHz, CDCl3) δ 7.38−7.28 (m, 3 H), 7.20−7.18 (m, 2 H), 5.83−5.68 (m, 1 H), 5.43 (s, 1 H), 5.26−5.06 (m, 2 H), 4.70−4.52 (m, 3 H), 4.03−3.91 (m, 1 H), 3.87−3.80 (m, 1 H), 1.44 (s, 5 H), 1.42 (s, 4 H), 1.34 (d, J = 6.9 Hz, 1.8 H), 1.27 (d, J = 6.9 Hz, 1.2 H); 13C NMR (100 MHz, CDCl3) δ 173.8, 155.3, 137.2, 136.4, 132.7, 132.5, 129.1, 128.8, 128.1, 128.0, 127.6, 127.0, 117.9, 79.8, 50.2, 49.2, 48.4, 48.0, 46.5, 28.6, 19.8; IR (film) 3306, 2978, 1703, 1648 cm−1. MS (ESI) 341.1834 (341.1841 calcd for C18H26N2O3, M + Na+).
Amide (S)-6b (1.52 g, 4.77 mmol) was reduced following general procedure 3. This procedure afforded 900 mg (62%) of the title compound as a white solid, m.p. 37–39 °C. 1H NMR (500 MHz, CDCl3) δ 7.31−7.29 (m, 3 H), 7.25−7.22 (m, 2 H), 5.89−5.81 (m, 1 H), 5.19−5.13 (m, 2 H), 4.58 (s, 1 H), 3.78−3.68 (m, 1 H), 3.64 (d, J = 14.0 Hz, 1 H), 3.51 (d, J = 14.0 Hz, 1 H), 3.14−3.10 (m, 1 H), 3.06−3.02 (m, 1 H), 2.41−2.31 (m, 2 H), 1.46 (s, 9 H), 1.10 (d, J = 6.0 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ 156.0, 139.6, 135.8, 129.1, 128.4, 127.2, 117.9, 79.2, 59.0, 58.5, 57.2, 44.7, 28.7, 19.7; IR (film) 3350, 2360, 1702 cm−1. Anal. Calcd for C18H28N2O2: C, 71.02; H, 9.27; N, 9.20. Found: C, 71.12; H, 9.55; N, 9.42.
5.4.3. (S)-tert-Butyl-1-[allyl(4-methoxyphenyl)amino]-propan-2-ylcarbamate (7c)
General procedure 3 was used for the coupling of (S)-2-tert-butoxycarbonylaminopropionic acid (828 mg, 4.38 mmol) and N-(p-methoxyphenyl)allylamine (710 mg, 4.38 mmol). This procedure afforded 1.28 g (88%) of (S)-tert-butyl 1-[allyl(4-methoxyphenyl)amino]-1-oxopropan-2-ylcarbamate (6c) as a white solid, which was contaminated with ca. 15% of dicyclohexylurea. 1H NMR (500 MHz, CDCl3) δ 7.13 (d, J = 8.5 Hz, 2 H), 6.92 (d, J = 9.5 Hz, 2 H), 5.82 (ddt, J = 6.0, 11.0, 17.0 Hz, 1 H), 5.28 (d, J = 8.0 Hz, 1 H), 5.12 (dd, J = 1.5, 11.0 Hz, 1 H), 5.07 (dd, J = 1.5, 17.0 Hz, 1 H), 4.34−4.27 (m, 2 H), 4.18−4.11 (m, 1 H), 3.82 (s, 3 H), 1.41 (s, 9 H), 1.11 (d, J = 6.5 Hz, 3 H). MS (ESI) 357.1785 (357.1790 calcd for C18H26N2O4, M + Na+).
Amide (S)-6c (1.28 g, 3.83 mmol) was reduced following general procedure 3. This procedure afforded 4.83 g (65%) of the title compound as a white solid, m.p. 99–102 °C. 1H NMR (400 MHz, CDCl3) δ 6.84−6.75 (m, 4 H), 5.85−5.76 (m, 1 H), 5.14−5.10 (m, 2 H), 4.46 (s, br, 1 H), 3.95−3.76 (m, 3 H), 3.74 (s, 3 H), 3.36 (dd, J = 6.0, 14.4 Hz, 1 H), 3.05 (dd, J = 5.6, 14.4 Hz, 1 H), 1.43 (s, 9 H), 1.16 (d, J = 6.8 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 155.6, 152.0, 143.5, 134.5, 116.7, 115.2, 114.9, 57.3, 55.9, 55.3, 45.7, 29.9, 28.6, 19.2; IR (film) 3357, 1676 cm−1; MS (EI) 320.2095 (320.2100 calcd for C18H28N2O3).
5.4.4. (S)-tert-butyl 1-[allyl(benzyl)amino]-4-methylpentan-2-ylcarbamate (7d)
General procedure 3 was used for the coupling of 2-(tert-butoxycarbonylamino)-4-methylpentanoic acid (1.70 g, 7.35 mmol) with N-benzylallylamine (1.08 g, 7.35 mmol). This procedure afforded 2.65 g (100%) of (S)-tert-butyl 1-[allyl(benzyl)amino]-3-(benzyloxy)-1-oxopropan-2-ylcarbamate (6d) as a white solid, which was contaminated with ca. 15% dicyclohexylurea. This material was carried on without further purification. This molecule was observed as a 1.2:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.36−7.28 (m, 2.72 H), 7.22– 7.19 (m, 2.27 H), 5.84−5.70 (m, 1 H), 5.26−5.09 (m, 3 H), 4.70−4.62 (m, 2 H), 4.60−4.51 (m, 1 H), 4.07−3.98 (m, 1 H), 3.87−3.80 (m, 1 H), 1.78−1.71 (m, 2 H), 1.44 (s, 4.9 H), 1.42 (s, 4.1 H), 1.36−1.27 (m, 1 H), 0.97−0.88 (m, 6 H), 0.84 (d, J = 6.8 Hz, 0.55 H), 0.77 (d, J = 6.4 Hz, 0.45 H). MS (ESI) 383.2310 (383.2311 calcd for C21H32N2O3, M + Na+).
Amide (S)-6d (2.62 g, 7.26 mmol) was reduced following general procedure 3. This procedure afforded 1.57 g (62%) of the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.34−7.28 (m, 4 H), 7.24−7.21 (m, 1 H), 5.90−5.80 (m, 1 H), 5.19−5.12 (m, 2 H), 4.29 (s, 1 H), 3.80−3.70 (m, 1 H), 3.65 (dd, J = 13.6 Hz, 1 H), 3.53 (dd, J = 13.6 Hz, 1 H), 3.16−3.02 (m, 2 H), 2.37 (d, J = 6.8 Hz, 2 H), 1.72−1.62 (m, 1 H), 1.46 (s, 9 H), 1.34−1.29 (m, 1H), 1.22−1.15 (m, 1 H), 0.90 (d, J = 6.8 Hz, 6 H); 13C NMR (100 MHz, CDCl3) δ 156.0, 139.8, 136.1, 129.1, 128.3, 127.0, 117.6, 79.0, 58.7, 58.5, 57.4, 47.3, 43.4, 28.7, 25.0, 23.5, 22.5; IR (film) 3359, 2956, 1703 cm−1. MS (ESI) 347.2702 (347.2699 calcd for C21H34N2O2, M + H+).
5.4.5. (±)–tert-Butyl 1-[allyl(benzyl)amino]-3-(4-chlorophenyl)propan-2-ylcarbamate (7e)
General procedure 3 was employed for the coupling of (±)-2-(tert-butoxycarbonylamino)-3-(4-chlorophenyl)propanoic acid (1.24 g, 4.13 mmol) with N-allylbenzylamine (0.608 g, 4.13 mmol). This procedure afforded 1.56 g (88%) of (±)-tert-butyl 1-[allyl(benzyl)amino]-3-(4-chlorophenyl)-1-oxopropan-2-ylcarbamate (6e) as a white solid which was contaminated with 15% dicyclohexylurea. This material was carried on without further purification. This molecule was observed as a complex mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.25−6.92 (m, 9 H), 5.69−4.99 (m, 3 H), 4.79−3.62 (m, 5 H), 3.01−2.75 (m, 2 H), 1.35 (m, 9 H). MS (ESI) 451.1765 (451.1764 calcd for C24H29ClN2O3, M + Na+).
Amide (±)-6e (1.56 g, 3.64 mmol) was reduced following general procedure 3. This procedure afforded 1.16 g (63%) of the title compound as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.32−7.24 (m, 5 H), 7.22−7.19 (d, 2 H), 7.05−7.03 (d, J = 8.0 Hz, 2 H), 5.84−5.79 (m, 1 H), 5.17– 5.12 (dd, J = 1.2, 9.6 Hz, 2 H), 4.53 (s, 1 H), 3.91 (s, 1 H), 3.61 (d, J = 12.0 Hz, 1 H), 3.52 (d, J = 12.0 Hz, 1 H), 3.13−3.01 (m, 2 H), 2.76−2.75 (d, J = 4.8 Hz, 2 H), 2.39−2.37 (d, J = 6.8 Hz, 2 H), 1.42 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 155.6, 139.0, 136.7, 135.3, 131.9, 130.7, 128.9, 128.2, 128.1, 126.9, 117.8, 79.0, 58.2, 57.0, 56.1, 49.6, 38.5, 28.3; IR (film) 2360, 1705, 1492, 910, 735 cm−1; MS (ESI) 415.2168 (415.2152 calcd for C24H31ClN2O2, M + H+).
5.4.6. (R)-tert-butyl 1-[allyl(benzyl)amino]-3-(benzyloxy)propan-2-ylcarbamate (7f)
General procedure 3 was used for the coupling of (S)-3-(benzyloxy)-2-(tert-butoxycarbonylamino)propanoic acid (5.99 g, 20.28 mmol) with N-benzylallylamine (2.98 g, 20.28 mmol). This procedure afforded 7.67 g (89%) of (S)-tert-butyl 1-[allyl(benzyl)amino]-3-(benzyloxy)-1-oxopropan-2-ylcarbamate (6f) as a white solid, which was contaminated with ca. 15% dicyclohexylurea. This material was carried on without further purification. The enantiopurity was judged to be 98% ee by chiral HPLC analysis (chiralcel OD column, 5% isopropanol/hexanes, 1 mL/min, RT = 7.19 min and 9.13 min). This molecule was observed as a 4.6:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.38−7.28 (m, 6 H), 7.20−7.17 (m, 4 H), 5.82−5.69 (m, 1 H), 5.43 (m, 0.8 H), 5.26−5.07 (m, 2 H), 4.99−4.97 (m, 0.2 H), 4.71−4.53 (m, 3.8 H), 4.44−4.40 (m, 0.2 H), 4.17−4.07 (m, 0.2 H), 4.02−3.96 (m, 1.8 H), 3.86−3.79 (m, 2.8 H), 3.71−3.66 (m, 0.2 H), 1.43 (m, 9 H). MS (ESI) 447.2264 (447.2260 calcd for C25H32N2O4, M + Na+).
Amide (S)-6f (7.65 g, 18.0 mmol) was reduced following general procedure 3. This procedure afforded 4.83 g (65%) of the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.31−7.17 (m, 10 H), 5.80 (dt, J = 6.8, 10.4 Hz, 1 H), 5.15−5.08 (m, 2 H), 4.81 (s, 1 H), 4.42−4.35 (m, 2 H), 3.80 (s, 1 H), 3.65−3.58 (m, 2 H), 3.51−3.44 (m, 2 H), 3.11 (dd, J = 8.0, 14.4 Hz, 1 H), 3.01 (dd, J = 6.4, 14.4 Hz, 1 H), 2.64 (dd, J = 7.6, 12.8 Hz, 1 H), 2.47 (dd, J = 6.0, 12.4 Hz, 1 H), 1.41 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 155.9, 139.7, 138.5, 136.0, 129.1, 128.5, 128.4, 127.80, 127.77, 127.1, 117.7, 79.3, 73.4, 70.2, 58.6, 57.3, 54.5, 48.9, 28.6; IR (film) 3436, 2976, 1713 cm−1; MS (ESI) 411.2637 (411.2648 calcd for C25H34N2O3, M + H+).
5.4.7. tert-Butyl-2-(diallylamino)ethylcarbamate (7g)
General procedure 3 was employed for the coupling of N-Boc glycine (3.12 g, 17.81 mmol) with N-benzylallyl amine (2.62 g, 17.81 mmol). The procedure afforded 4.72 g (87%) of tert-butyl-2-[allyl(benzyl)amino]-2-oxoethylcarbamate (6g) as a white solid, which was contaminated with ca. 15% dicyclohexylurea. This material was carried on without further purification. This molecule was observed as a 1.5:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.38−7.27 (m, 3 H), 7.24−7.22 (m, 1 H), 7.13 (d, J = 7.6 Hz, 1 H) 5.79−5.65 (m, 1 H), 5.55 (s, 1 H), 5.30−5.09 (m, 2 H), 4.71−4.58 (m, 1.2 H), 4.50−4.45 (m, 0.8 H), 4.04−4.03 (m, 2 H), 3.84−3.82 (m, 0.4 H), 3.80−3.77 (m, 1.2 H), 3.69−3.67 (m, 0.4 H), 1.44 (s, 5.4 H), 1.39 (s, 3.6 H). MS (ESI) 327.1681 (327.1685 calcd for C17H24N2O3, M + Na+).
Amide 6g (4.47 g, 14.7 mmol) was reduced following general procedure 3. This procedure afforded 2.04 g (48%) of the title compound as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.30−7.27 (m, 4 H), 7.25−7.19 (m, 1 H), 5.87−5.77 (m, 1 H) 5.17−5.10 (m, 2 H), 4.84 (s, 1 H), 3.54 (s, 2 H), 3.18−3.10 (m, 2 H), 3.05 (d, J = 6.4 Hz, 2 H), 2.51 (t, J = 6 Hz, 2 H), 1.40 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 156.2, 139.3, 135.6, 129.0, 128.4, 127.2, 118.0, 79.1, 58.2, 56.8, 52.6, 38.2, 28.6; IR (film) 3361, 2977, 1715 cm−1. Anal calcd for C17H26N2O2: C, 70.31; H, 9.02; N, 9.65. Found: C, 70.23; H, 9.09; N, 9.60.
5.4.8. (±)–tert-Butyl 2-[benzyl(but-3-en-2-yl)amino]ethylcarbamate (7h)
General procedure 3 was used for the coupling of 2-(tert-butoxycarbonylamino)acetic acid (6.95 g, 39.7 mmol) with N-benzyl-but-3-en-2-ylamine (6.40 g, 39.7 mmol) except only 1.4 equiv of DCC was employed. This procedure afforded 10.7 g (85%) of (±)-tert-butyl 2-[benzyl(but-3-en-2-yl)amino]-2-oxoethylcarbamate (6h) as a white solid, which was contaminated with ca. 15% of dicyclohexylurea. This material was carried on without further purification. This molecule was observed as a 1.2:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.37−7.27 (m, 2.27 H), 7.23−7.16 (m, 2.73 H), 5.89−5.67 (m, 1 H), 5.65−5.58 (m, 1 H), 5.54−5.44 (m, 1 H), 5.31−5.06 (m, 2 H), 4.78 (d, J = 15.6 Hz, 0.4 H), 4.41 (d, J = 12.4 Hz, 1.1 H), 4.28 (d, J = 15.6 Hz, 0.5 H), 4.19−4.00 (m, 1 H), 3.96−3.66 (m, 1 H), 1.45 (s, 4.9 H), 1.41 (s, 4.1 H), 1.23 (d, J = 7.2 Hz, 3 H). MS (ESI) 341.1828 (341.1841 calcd for C18H26N2O3, M + Na+).
Amide (±)-6h (5.14 g, 16.1 mmol) was reduced following general procedure 3. This procedure afforded 2.50 g (51%) of the title compound as a yellow solid, m.p. 38–40 °C. 1H NMR (400 MHz, CDCl3) δ 7.31−7.24 (m, 4 H), 7.24−7.21 (m, 1 H), 5.90−5.81 (m, 1 H), 5.17−5.05 (m, 2 H), 4.84 (s, 1 H), 3.63−3.52 (m, 2 H), 3.30 (t, J = 6.4 Hz, 1 H), 3.12−3.06 (m, 2 H), 2.64−2.57 (m, 1 H), 2.54−2.48 (m, 1 H), 1.44 (s, 9 H), 1.14 (d, J = 6.4 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ 156.1, 140.5, 139.5, 128.6, 128.4, 126.9, 115.9, 78.8, 56.7, 54.6, 49.0, 38.7, 28.5, 15.4; IR (film) 3425, 3365, 2974, 1714 cm−1. MS (ESI) 327.2035 (327.2048 calcd for C18H28N2O2, M + Na+).
5.4.9. (±)-tert-Butyl 2-[benzyl(cyclopent-2-enyl)amino]ethylcarbamate (7i)
General procedure 3 was used for the coupling of N-Boc glycine (1.25 g, 7.16 mmol) with N-benzylcyclopent-2-enylamine (1.25 g, 7.16 mmol). This procedure afforded 1.81 g (77%) of (±)-tert-butyl 2-[benzyl(cyclopent-2-enyl)amino]-2-oxoethylcarbamate (6i) as a white solid, which was contaminated with ca. 15% dicyclohexylurea. This material was carried on without further purification. This molecule was observed as a 1.4:1 mixture of rotamers. 1H NMR (500 MHz, CDCl3) δ 7.34−7.31 (m, 1 H), 7.29−7.27 (m, 1 H), 7.22−7.13 (m, 3 H), 5.98−5.95 (m, 1 H), 5.80−5.74 (m, 0.58 H), 5.65 (s, 0.42 H), 5.54−5.52 (m, 0.42 H), 5.51−5.49 (m, 1 H), 4.96−4.93 (m, 0.58 H), 4.58−4.50 (m, 1 H), 4.42−4.38 (m, 1 H), 4.22−4.09 (m, 1 H), 4.02−4.01 (m, 0.5 H), 3.91−3.87 (m, 0.5 H), 3.77−3.73 (m, 1 H), 2.41−2.22 (m, 3 H), 1.45 (s, 5.2 H), 1.41 (s, 3.8 H). MS (ESI) 353.1840 (353.1841 calcd for C19H26N2O3, M + Na+).
Amide (±)-6i (1.81 g, 5.48 mmol) was reduced following general procedure 3. This procedure afforded 1.21 g (70%) of the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.32−7.26 (m, 4 H), 7.25−7.21 (m, 1 H), 5.90−5.88 (m, 1 H) 5.71−5.68 (m, 1 H), 4.84 (s, 1 H), 4.06−4.05 (m, 1 H), 3.64 (d, J = 13.6 Hz, 1 H), 3.44 (d, J = 14.0 Hz, 1 H), 3.16−3.07 (m, 2 H), 2.60−2.50 (m, 1 H), 2.49−2.44 (m, 1 H), 2.40−2.22 (m, 2 H), 1.97−1.88 (m, 1 H), 1.74−1.59 (m, 1 H), 1.43 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 156.2, 140.5, 133.7, 131.9, 128.8, 128.4, 127.0, 79.0, 67.3, 55.4, 49.6, 38.6, 31.8, 28.6, 23.8; IR (film) 3367, 2975, 1715 cm−1. MS (ESI) 317.2220 (317.2229 calcd for C19H28N2O2, M + H+).
5.4.10. tert-butyl 1-[allyl(benzyl)carbamoyl]-cyclohexylcarbamate (6j)
General procedure 3 was employed for the coupling of 1-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid (0.391 g, 1.61 mmol) with N-allylbenzylamine (0.236 g, 1.61 mmol). After purification, 0.469 g (78%) of tert-butyl 1-(allyl(benzyl)carbamoyl)cyclohexylcarbamate was obtained as a white solid contaminated with 37% of the dicyclohexylurea side product. This material was carried on without further purification. This molecule was observed as a complex mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.29−7.183 (m, 5 H), 5.78−5.14 (m, 1 H), 5.10 (dd, J = 10.0, 17.2 Hz, 2 H), 4.72 (s, 3 H), 4.21 (t, 1 H), 4.06 (s, 2 H), 3.62−3.59 (m, 0.83 H), 2.24 (d, 1.33 H), 2.04−1.93 (s, 6.5 H), 1.93−1.74 (m, 7 H), 1.74−1.56 (m, 12 H), 1.44−1.08 (m, 34 H).
5.5. General procedure 4: conversion of N-Boc-N′-allyl-1,2-diamines) to N-aryl-N′-allyl-1,2-diamines
A flask equipped with magnetic stirbar was charged with the appropriate N-Boc-N′-allyl-1,2-diamine (1.0 equiv) and a sufficient volume of dioxane to provide a 0.1 M solution. A solution of 4 M aqueous HCl (33 equiv) was added and the reaction mixture was heated to 50 °C for 2 h. The reaction mixture was cooled to room temperature and NH4OH was added dropwise until the solution pH was >11. The resulting mixture was extracted with ethyl acetate, and the combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to afford the corresponding primary amine product. The crude product was immediately carried on without further purification.
A flame dried Schlenk tube equipped with a magnetic stirbar was charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), (±)-BINAP (2 mol %), sodium tert-butoxide (1.2 equiv), the appropriate aryl bromide (1.0 equiv), and a 0.5 M solution of the primary amine (1.0 equiv) in toluene. The reaction mixture was heated to 80 °C with stirring until the starting material had been consumed as judged by TLC analysis (ca. 6 h). The mixture was cooled to rt and a solution of aqueous ammonium chloride was added (4 mL). The resulting mixture was extracted with ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel.
5.5.1. (–)-(S)-N1,N1-Diallyl-N2-phenylpropane-1,2-diamine (1d)
General procedure 4 was used for the deprotection of 1.08 g (4.24 mmol) of (S)-7a (1.08 g, 4.24 mmol). This procedure afforded 654 mg (85%) of (S)-N1,N1-diallylpropane-1,2-diamine (8a) as a yellow oil. This material was used without further purification. 1H NMR (500 MHz, CDCl3) δ 5.95−5.87 (m, 2 H), 5.19−5.15 (m, 4 H), 3.70 (s, br, 2 H), 3.36−3.30 (m, 1 H), 3.24−3.20 (m, 2 H), 3.13−3.10 (m, 2 H), 2.66 (dd, J = 10.5, 15.0 Hz, 1 H), 2.53 (dd, J = 4.5, 14 Hz, 1 H). 1.40 (d, J = 6.5 Hz, 3 H).
General procedure 4 was used for the N-arylation of (S)-8a (379 mg, 2.46 mmol) with bromobenzene (386 mg, 2.46 mmol). This procedure afforded 390 mg (69%) of the title compound as a yellow oil. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 1% IPA/hexanes, 0.2 mL/min, RT = 38.71 min and 43.68 min), [α]23D −4.61° (c 0.23, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.18−7.15 (m, 2 H), 6.69 (t, J = 7.5 Hz, 1 H), 6.64 (d, J = 8.5 Hz, 2 H), 5.83 (ddt, J = 6.5, 7.0, 10 Hz, 2 H), 5.18−5.12 (m, 4 H), 4.20 (s, 1 H), 3.50−3.43 (m, 1 H), 3.17 (dd, J = 6.0, 14.0 Hz, 2 H), 3.04 (dd, J = 7.0, 14.5 Hz, 2 H), 2.54 (dd, J = 8.5, 13.0 Hz, 1 H), 2.42 (dd, J = 6.0, 13.0 Hz, 1 H), 1.19 (d, J = 6.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 148.4, 135.7, 129.3, 117.7, 117.3, 113.7, 59.0, 57.2, 46.6, 19.9; IR (film) 3350, 2924, 1602 cm−1; MS (EI) 230.1787 (230.1783 calcd for C15H22N2).
5.5.2. (±)-N1-Allyl-N1-benzyl-N2-phenylpropane-1,2-diamine (1e)
General procedure 4 was used for the deprotection of (±)-7b (890 mg, 2.93 mmol). This procedure afforded 526 mg (88%) of (±)-N1-allyl-N1-benzylpropane-1,2-diamine (8b) as a yellow oil. This material was used without further purification. 1H NMR (500 MHz, CDCl3) δ 7.33−7.29 (m, 3 H), 7.25−7.22 (m, 2 H), 5.91−5.83 (m, 1 H), 5.18−5.13 (m, 2 H), 3.75−3.70 (m, 1 H), 3.43 (d, J = 13.5 Hz, 1 H), 3.21−3.17 (m, 1 H), 3.04−2.99 (m, 1 H), 2.98−2.94 (m, 1 H), 2.33−2.30 (m, 1 H), 2.26−2.22 (m, 1 H), 1.47 (s, 2 H), 0.99 (d, J = 6.0 Hz, 3 H).
General procedure 4 was used for the N-arylation of (±)-8b (278 mg, 1.36 mmol) with bromobenzene (144 μl, 1.36 mmol). This procedure afforded 270 mg (71%) of the title compound as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.37−7.31 (m, 4 H), 7.30−7.27 (m, 1 H), 7.23−7.20 (m, 2 H), 6.73 (tt, J = 1.0, 7.5 Hz, 1 H), 6.64 (dd, J = 1.0, 8.5 Hz, 2 H), 5.96−5.88 (m, 1 H), 5.25−5.19 (m, 2 H), 4.14 (s, 1 H), 3.73 (d, J = 13.0 Hz, 1 H), 3.57−3.50 (m, 2 H), 3.23−3.19 (m, 1 H), 3.12−3.08 (m, 1 H), 2.64−2.60 (m, 1 H), 2.50−2.46 (m, 1 H), 1.22 (d, J = 6.5 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ 148.4, 139.5, 135.7, 129.4, 129.2. 128.5, 127.2, 118.0, 117.3, 113.6, 59.4, 58.7, 57.3, 46.7, 19.9; IR (film) 3360, 2806, 1602 cm−1. MS (ESI) 281.2015 (281.2018 calcd for C19H24N2, M + H+).
5.5.3. (±)-N1-Allyl-N1-benzyl-N2-(4-methoxyphenyl)-propane-1,2-diamine (1f)
General procedure 4 was used for the N-arylation of (±)-8b (185 mg, 0.91 mmol) with 4-bromoanisole (114 mL, 0.905 mmol) using 2 mol % of 2-(di-tert-butylphosphino)biphenyl in place of BINAP as the ligand. This procedure afforded 228 mg (81%) of the title compound as an orange oil. 1H NMR (500 MHz, CDCl3) δ 7.36−7.32 (m, 4 H), 7.30−7.26 (m, 1 H), 6.82−6.79 (m, 2 H), 6.64−6.60 (m, 2 H), 5.94−5.86 (m, 1 H), 5.23−5.18 (m, 2 H), 3.97 (s, 1 H), 3.78 (s, 3 H), 3.72 (d, J = 13.5 Hz, 1 H), 3.54 (d, J = 13.0 Hz, 1 H), 3.45−3.39 (m, 1 H), 3.22−3.18 (m, 1 H), 3.09−3.05 (m, 1 H), 2.62−2.58 (m, 1 H), 2.47−2.44 (m, 1 H), 1.19 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 152.2, 142.6, 139.4, 135.7, 129.1, 128.4, 127.2, 117.9, 115.2, 115.0, 59.6, 58.7, 57.3, 55.9, 47.6, 20.0; IR (film) 3350, 2928, 2360, 1510 cm−1. MS (ESI) 311.2126 (311.2123 calcd for C20H26N2O, M + H+).
5.5.4. (±)-4-{1-[allyl(benzyl)amino]propan-2-ylamino}-benzonitrile (1g)
General procedure 4 was used for the N-arylation of (±)-8b (278 mg, 1.36 mmol) with 4-bromobenzonitrile (1.36 mmol). This procedure afforded 290 mg (70%) of the title compound as an orange oil. 1H NMR (500 MHz, CDCl3) δ 7.39−7.37 (m, 2 H), 7.32−7.24 (m, 5 H), 6.46 (d, J = 7.5 Hz, 2 H), 5.90−5.82 (m, 1 H), 5.21−5.17 (m, 2 H), 4.49 (d, J = 4.5 Hz, 1 H), 3.68 (d, J = 13.5 Hz, 1 H), 3.50−3.44 (m, 2 H), 3.21−3.17 (m, 1 H), 3.08−3.04 (m, 1 H), 2.57−2.52 (m, 1 H), 2.50−2.46 (m, 1 H), 1.14 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 151.4, 139.3, 135.4, 133.8, 129.1, 128.5, 127.4, 120.8, 118.2, 112.7, 98.3, 59.1, 59.0, 57.8, 46.5, 19.2; IR (film) 3361, 2807, 2211, 1607 cm−1. MS (ESI) 306.1962 (306.1970 calcd for C20H23N3, M + H+).
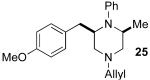
5.5.5. (+)-(S)-N1-Allyl-N2-(4-chlorophenyl)-N1-(4-methoxyphenyl)propane-1,2-diamine (1i)
General procedure 4 was used for the deprotection of (S)-7c (513 mg, 1.60 mmol). This procedure afforded 318 mg (91%) of (S)-N1-allyl-N1-(4-methoxyphenyl)propane-1,2-diamine (8c) as a yellow oil. This material was used without further purification. 1H NMR (500 MHz, CDCl3) δ 6.81 (d, J = 9.0 Hz, 2 H), 6.75 (d, J = 9.0 Hz, 2 H), 5.86−5.79 (m, 1 H), 5.14−5.11 (m, 2 H), 3.90 (dd, J = 1.5, 5.0 Hz, 2 H), 3.75 (s, 3 H), 3.26−3.21 (m, 1 H), 3.20 (d, J = 4.5 Hz, 1 H), 3.00−2.96 (m, 1 H), 1.60 (s, br, 2 H), 1.09 (d, J = 6.0 Hz, 3 H).
General procedure 4 was used for the N-arylation of (S)-8c (64 mg, 0.29 mmol) with 4-bromochlorobenzene (55 mg, 0.29 mmol). This procedure afforded 65 mg (69%) of the title compound as a yellow oil. The enantiopurity was judged to be 98% ee by chiral HPLC analysis (chiralcel OD column, 1% isopropanol/hexanes, 0.2 mL/min, RT = 59.14 min and 63.37 min), [α]23D +21.46° (c 0.11, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.8 Hz, 2 H), 6.83 (d, J = 9.2 Hz, 2 H), 6.75 (d, J = 9.2 Hz, 2 H), 6.48 (d, J = 8.8 Hz, 2 H), 5.86−5.77 (m, 1 H), 5.16– 5.11 (m, 2 H), 3.95−3.79 (m, 2 H), 3.77 (s, 3 H), 3.74−3.63 (m, 2 H), 3.32−3.22 (m, 2 H), 1.22 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 152.5, 146.4, 143.6, 134.6, 129.2, 121.9, 116.9, 116.2, 114.9, 114.6, 57.8, 55.9, 55.7, 48.0, 19.5; IR (film) 3391, 2929, 1598 cm−1; MS (EI) 330.1490 (330.1500 calcd for C19H23ClN2).
5.5.6. (±)-N1-Allyl-N1-(4-methoxyphenyl)-N2-phenylpropane-1,2-diamine (1h)
Diamine (±)-7c was prepared from racemic alanine using a sequence of transformations identical to that described above for the synthesis of (S)-7c. General procedure 4 was then used for the N-arylation of (±)-7c (350 mg, 1.59 mmol) with bromobenzene (168 μL, 1.36 mmol). This procedure afforded 308 mg (65%) of the title compound as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.19 (t, J = 7.5 Hz, 2 H), 6.88−6.85 (m, 2 H), 6.82−6.78 (m, 2 H), 6.72 (t, J = 7 Hz, 1 H), 6.62 (d, J = 8.0 Hz, 2 H), 5.90−5.82 (m, 1 H), 5.20−5.16 (m, 2 H), 3.98−3.93 (m, 1 H), 3.89−3.85 (m, 1 H), 3.80−3.72 (m, 5 H), 3.39−3.35 (m, 1 H), 3.29−3.25 (m, 1 H), 1.27 (d, J = 6.5 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 152.3, 147.8, 143.7, 134.6, 129.4, 117.5, 116.8, 115.9, 114.8, 113.5, 57.8, 55.9, 55.6, 47.8, 19.7; IR (film) 2929, 2341, 1601 cm−1. MS (ESI) 297.1964 (297.1967 calcd for C19H24N2O, M + H+).
5.5.7. (–)-(S)-N1-Allyl-N1-benzyl-N2-phenyl-4-methylpentane-1,2-diamine (1j)
General procedure 4 was used for the deprotection of (S)-7d (1.49 g, 4.30 mmol). This procedure afforded 1.06 g (100%) of (S)-N1-allyl-N1-benzyl-4-methylpentane-1,2-diamine (8d) as a yellow oil. This material was used without further purification. 1H NMR (500 MHz, CDCl3) δ 7.31−7.28 (m, 4 H), 7.25−7.22 (m, 1 H), 5.91−5.83 (m, 1 H), 5.18−5.13 (m, 2 H), 3.77 (d, J = 13.5 Hz, 1 H), 3.40 (d, J = 13.5 Hz, 1 H), 3.23−3.19 (m, 1 H), 2.96−2.91 (m, 2 H), 2.34−2.31 (m, 1 H), 2.27−2.23 (m, 1 H), 1.75−1.68 (m, 1 H), 1.53 (s, 2 H), 1.15−1.06 (m, 2 H), 0.89 (d, J = 6.5 Hz, 3 H), 0.86 (d, J = 7.0 Hz, 3 H).
General procedure 4 was used for the N-arylation of (S)-8d (1.06 g, 4.30 mmol) with bromobenzene (453 μl, 4.30 mmol). This procedure afforded 1.09 g (79%) of the title compound as an yellow oil. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 5% isopropanol/hexanes, 1 mL/min, RT = 6.16 min and 8.90 min), [α]23D−44.10° (c 3.2, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.32−7.27 (m, 4 H), 7.25−7.22 (m, 1 H), 7.13 (t, J = 7.5 Hz, 2 H), 6.65 (t, J = 7.0 Hz, 1 H), 6.54 (d, J = 8.0 Hz, 2 H), 5.90−5.82 (m, 1 H), 5.18−5.13 (m, 2 H), 3.72 (d, J = 6.0 Hz, 1 H), 3.63 (d, J = 13.5 Hz, 1 H), 3.56 (d, J = 13.5 Hz, 1 H), 3.48−3.41 (m, 1 H), 3.14−3.06 (m, 2 H), 2.55−2.45 (m, 2 H), 1.77−1.69 (m, 1 H), 1.50−1.45 (m, 1 H), 1.38−1.32 (m, 1 H), 0.96 (d, J = 7.0 Hz, 3 H), 0.88 (d, J = 6.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 148.6, 139.8, 135.9, 129.4, 129.2, 128.4, 127.2, 117.8, 117.0, 113.2, 59.2, 58.3, 57.8, 49.8, 43.6, 25.1, 23.3, 23.1; IR (film) 3400, 2954, 1601 cm−1. MS (ESI) 323.2471 (323.2487 calcd for: C22H30N2, M + H+).
5.5.8. (±)-N1-Allyl-N1-benzyl-N2-phenyl-3-(4-chlorophenyl)propane-1,2-diamine (1k)
General procedure 4 was used to deprotect (±)-7e (1.08 g, 2.61 mmol). This procedure afforded 0.81 g (98%) of (±)-N1-allyl-N1-benzyl-3-(4-chlorophenyl)propane-1,2-diamine (8e) as a clear oil. This material was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.22−7.19 (m, 4 H), 7.16−7.13 (m, 3 H), 7.05 (d, J = 8.4 Hz, 2 H), 5.82−5.72 (m, 1 H), 5.12 (t, J = 9.8 Hz, 2 H), 3.66 (d, J = 13.2 Hz, 1 H), 3.43 (d, J = 13.4 Hz, 1 H), 3.14 (dd, J = 5.8, 14.0 Hz, 1 H), 3.10−3.03 (m, 1 H), 2.95 (dd, J = 7.6, 14.4 Hz, 1 H), 2.65 (dd, J = 4.8, 13.6 Hz, 1 H), 2.39−2.30 (m, 3 H).
General procedure 4 was used for the N-arylation of (±)-8e (0.99 g, 3.14 mmol) with bromobenzene (0.49 g, 3.14 mmol). This procedure afforded 1.00 g (81%) of the title compound as an orange oil. 1H NMR (400 MHz, CDCl3) δ 7.33−7.26 (m, 5 H), 7.23−7.16 (m, 4 H), 7.06 (d, J = 8.8 Hz, 2 H), 6.71 (t, J = 7.2 Hz, 1 H), 6.58 (d, J = 8.4 Hz, 2 H), 5.91−5.80 (m, 1 H), 5.16 (d, J = 15.6 Hz, 2 H), 3.85 (s, 1 H), 3.70−3.65 (m, 1 H), 3.61 (d, J = 13.2 Hz, 1 H), 3.53 (d, J = 13.2 Hz, 1 H), 3.15−3.04 (m, 2 H), 2.88−2.79 (m, 2 H), 2.54−2.44 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 147.7, 139.1, 137.0, 135.3, 132.0, 130.8, 129.3, 129.0, 128.3, 127.1, 118.0, 117.4, 113.3, 58.6, 57.4, 56.7, 52.0, 38.2; IR (film) 2254, 1493, 912, 742 cm−1; MS (ESI) 391.1928 (391.1941 calcd for C25H27ClN2, M + H+).
5.5.9. (–)-(R)-4-{1-[Allyl(benzyl)amino]-3-(benzyloxy)-propan-2-ylamino}benzonitrile (1l)
General procedure 4 was used for the deprotection of (R)-7f (3.9 g, 9.5 mmol). This procedure afforded 3.1 g (100%) of (S)-N1-allyl-N1-benzyl-3-benzyloxypropane-1,2-diamine (8f) as a yellow oil. This material was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.37−7.21 (m, 10 H), 5.86 (dt, J = 6.0, 11.0 Hz, 1 H), 5.17−5.12 (m, 2 H), 4.50 (s, 2 H), 3.67 (d, J = 13.5 Hz, 1 H), 3.51−3.48 (m, 2 H), 3.30−3.26 (m, 1 H), 3.20−3.12 (m, 2 H), 3.00 (dd, J = 5.4, 7.5 Hz, 1 H), 2.46−2.38 (m, 2 H), 1.60 (s, 2 H); MS (ESI) 311.2116 (311.2123 calcd for C20H26N2O, M + H+).
General procedure 4 was used for the N-arylation of (S)-8f (900 mg, 2.89 mmol) with 4-bromobenzonitrile (527 mg, 2.89 mmol). This procedure afforded 864 mg (73%) of the title compound as an orange oil. The enantiopurity was judged to be 98% ee by chiral HPLC analysis (chiralcel AD column, 0.7% isopropanol/hexanes, 1 mL/min, RT = 24.75 min and 28.10 min), [α]23D −25.59° (c 0.79, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.35−7.28 (m, 12 H), 6.40 (d, J = 8.5 Hz, 2 H), 5.85 (dt, J = 6.5, 10.5 Hz, 1 H), 5.22−5.16 (m, 2 H), 4.54 (d, J = 6.5 Hz, 1 H), 4.49−4.44 (m, 2 H), 3.67 (dd, J = 3.0, 9.0 Hz, 1 H), 3.65−3.56 (m, 2 H), 3.53−3.46 (m, 2 H), 3.17−3.10 (m, 2 H), 2.74 (dd, J = 7.5, 13.5 Hz, 1 H), 2.60 (dd, J = 6.5, 13.5 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 151.0, 139.5, 138.0, 135.6, 133.8, 129.1, 128.6, 128.5, 127.99, 127.90, 127.4, 120.7, 118.1, 112.7, 98.6, 73.5, 69.9, 59.3, 58.3, 54.3, 51.4; IR (film) 3365, 2211, 1606 cm−1; MS (ESI) 412.2383 (412.2389 calcd for C27H29N3O, M + H+).
5.5.10. (±)-4-{2-[benzyl(but-3-en-2-yl)amino]-ethylamino}benzonitrile (9)
General procedure 4 was used for the deprotection of (±)-7h (1.35 g, 4.44 mmol). This procedure afforded 842 mg (93%) of (±)-N1-benzyl-N1-(but-3-en-2-yl)ethane-1,2-diamine (8h) as a yellow oil. This material was used without further purification. 1H NMR (300 MHz, CDCl3) δ 7.39−7.19 (m, 5 H), 5.94−5.82 (m, 1 H), 5.16−5.03 (m, 2 H), 3.65−3.53 (m, 2 H), 3.34– 3.25 (m, 1 H), 2.72−2.61 (m, 2 H), 2.60−2.44 (m, 2 H), 1.21 (s, br, 2 H), 1.14 (d, J = 6.9 Hz, 3 H).
General procedure 4 was used for the N-arylation of (±)-8h (681 mg, 3.33 mmol) with 4-bromobenzonitrile (606 mg, 3.33 mmol). This procedure afforded 905 mg (89%) of the title compound as an orange oil. 1H NMR (300 MHz, CDCl3) δ 7.43−7.21 (m, 7 H), 6.41 (d, J = 9.0 Hz, 2 H), 5.94−5.83 (m, 1 H), 5.21−5.08 (m, 2 H), 4.65 (s, 1 H), 3.66−3.52 (m, 2 H), 3.39−3.30 (m, 1 H), 3.08−2.97 (m, 2 H), 2.82−2.63 (m, 2 H), 1.20 (d, J = 6.6 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 151.5, 140.2, 139.2, 133.5, 128.6, 128.4, 127.1, 120.7, 116.2, 112.2, 97.8, 56.8, 54.3, 47.8, 40.7, 15.3; IR (film) 3380, 2968, 2212, 1608 cm−1. MS (ESI) 306.1975 (306.1970 calcd for C20H23N3, M + H+).
5.5.11. (±)-N1-benzyl-N1-(cyclopent-2-enyl)-N2-phenylethane-1,2-diamine (10)
General procedure 4 was used for the deprotection of (±)-7i (1.20 g, 3.79 mmol). This procedure afforded 754 mg (92%) of (±)-N1-benzyl-N1-(cyclopent-2-enyl)ethane-1,2-diamine (8i) as a yellow oil. This material was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.35−7.28 (m, 4 H), 7.25−7.21 (m, 1 H), 5.90−5.87 (m, 1 H) 5.73−5.70 (m, 1 H), 4.08−4.04 (m, 1 H), 3.66 (d, J = 14.0 Hz, 1 H), 3.46 (d, J = 14.0 Hz, 1 H), 2.73−2.61 (m, 2 H), 2.56−2.42 (m, 2 H), 2.41−2.23 (m, 2 H), 1.97−1.88 (m, 1 H), 1.78−1.69 (m, 1 H), 1.31 (s, 2 H).
General procedure 4 was used for the N-arylation of (±)-8i (754 mg, 3.48 mmol) with bromobenzene (367μL, 3.48 mmol). This procedure afforded 507 mg (50%) of the title compound as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.34−7.29 (m, 4 H), 7.25−7.21 (m, 1 H), 7.16−7.12 (m, 2 H), 6.67 (td, J = 1.0, 7.5 Hz, 1 H), 6.54 (dd, J = 1.0, 8.5 Hz, 2 H), 5.91−5.88 (m, 1 H), 5.73−5.71 (m, 1 H), 4.16 (s, 1 H), 4.11−4.08 (m, 1 H), 3.67 (d, J = 13.5 Hz, 1 H), 3.46 (d, J = 13.5 Hz, 1 H), 3.12−3.04 (m, 2 H), 2.78−2.73 (m, 1 H), 2.67−2.63 (m, 1 H), 2.41−2.33 (m, 1 H), 2.32−2.24 (m, 1 H), 1.98−1.91 (m, 1 H), 1.79−1.72 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 148.8, 140.6, 133.8, 131.9, 129.3, 128.9, 128.5, 127.1, 117.2, 113.1, 67.3, 55.3, 49.0, 41.8, 31.9, 23.9; IR (film) 3392, 2943, 1602 cm−1. MS (ES) 293.2012 (293.2018 calcd for C20H24N2, M + H+).
5.5.12. 1-{[allyl(benzyl)amino]methyl}-cyclohexanamine (11)
A slightly modified general procedure 4 was followed for the deprotection of 6j (0.475 g, 1.27 mmol). The crude reaction mixture was diluted with H2O and washed with CH2Cl2 (3 × 20 mL) before being made basic (pH >10) with 10 M NaOH. The aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the organic layers were combined, washed once with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel to afford 0.284 g (82%) of N-allyl-1-amino-N-benzylcyclohexanecarboxamide as a white solid, m.p. 58–59 °C. 1H NMR (400 MHz, CDCl3) δ 7.32−7.18 (m, 5 H), 5.85−5.74 (m, 1 H), 5.12 (d, J = 11.6 Hz, 1 H), 5.08 (d, 15.6 Hz, 1 H), 4.86 (s, 2 H), 4.22 (s, 2 H), 2.08 (t, J = 10.0 Hz, 2 H), 1.56 (m, 2 H), 1.48−1.41 (m, 3 H), 1.36−1.25 (m, 3 H).
A slightly modified general procedure 3 was used for the reduction of 1-{[allyl(benzyl)amino]methyl}cyclohexanamine (0.937 g, 3.44 mmol) with lithium aluminum hydride. The crude reaction mixture was diluted with CH2Cl2 and extracted with 1 M HCl (3 × 25 mL). The aqueous extracts were combined and made basic (pH > 10) with 10 M NaOH before being extracted with CH2Cl2 (4 × 20 mL). The organic layers were combined, washed once with brine, dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude to product was purified by flash chromatography on silica gel to give 0.412 g (46%) of the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.35−7.27 (m, 4 H), 7.23−7.21 (m, 1 H), 5.89 (m, 1 H), 5.12 (dd, J = 4.8, 12.8 Hz, 2 H), 3.70 (s, 2 H), 3.11 (d, J = 6.8 Hz, 2 H), 2.44 (s, 2 H), 1.50−1.43 (m, 6 H), 1.37−1.33 (m, 4 H), 1.34−1.23 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 139.8, 135.5, 128.6, 128.0, 126.7, 117.3, 64.6, 60.5, 58.7, 52.4, 36.8, 25.9, 21.8; IR (film) 4197, 3054, 2306, 1421, 1265, 896, 705 cm−1; MS (ESI) 259.2167 (259.2174 calcd for C17H26N2, M + H+).
5.6. General Procedure 5: N-arylation of N-allyl phenylenediamine
A flame-dried Schlenk tube equipped with a magnetic stirbar was charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), 2-(di-tert-butylphosphino)biphenyl (2 mol %), sodium tert-butoxide (1.2 equiv), the appropriate aryl bromide (1.0−1.1 equiv), and a 0.5 M solution of N-allyl phenylenediaminexxvi (1.0 equiv) in toluene. The reaction mixture was heated to 80 °C with stirring until the starting material had been consumed as judged by TLC analysis (ca. 6 h). The mixture was cooled to rt and a solution of aqueous ammonium chloride was added (4 mL). The resulting mixture was extracted with ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel.
5.6.1. N1-Allyl-N2-(4-methoxyphenyl)benzene-1,2-diamine (45a)
General procedure 5 was used for the N-arylation of N-allylphenylenediamine (44) (300 mg, 2.02 mmol) with 4-bromoanisole (254 μl, 2.02 mmol). This procedure afforded 387 mg (75%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.07 (d, J = 7.6 Hz, 2 H), 6.86−6.82 (m, 2 H), 6.80−6.71 (m, 4 H), 6.03−5.93 (m, 1 H), 5.31−5.26 (m, 1 H), 5.20−5.17 (m, 1 H), 4.95 (s, 1 H), 4.26 (s, 1 H), 3.82 (d, J = 4.8 Hz, 2 H), 3.80 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 153.8, 142.8, 139.0, 135.6, 130.4, 125.0, 122.8, 118.0, 117.7, 116.2, 114.9, 111.6, 55.8, 46.5; IR (film) 3367, 1508 cm−1. MS (ESI) 255.1487 (255.1497 calcd for C16H18N2O, M + H+).
5.6.2. N1-Allyl-N2-phenylbenzene-1,2-diamine (45b)
General procedure 5 was used for the N-arylation of N-allylphenylenediamine (44) (300 mg, 2.02 mmol) with bromobenzene (232 μl, 2.20 mmol). This procedure afforded 313 mg (69%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.23−7.19 (m, 2 H), 7.14−7.09 (m, 2 H), 6.82 (t, J = 7.2 Hz, 1 H), 6.74−6.69 (m, 4 H), 5.96−5.90 (m, 1 H), 5.26−5.21 (m, 1 H), 5.16−5.12 (m, 1 H), 5.09 (s, 1 H), 4.33 (s, 1 H), 3.81−3.78 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 146.0, 144.1, 135.6, 129.5, 128.3, 126.4, 125.2, 119.4, 117.5, 116.2, 115.4, 111.5, 46.4; IR (film) 3370, 3045, 1598 cm−1. MS (ESI) 225.1383 (225.1392 calcd for C15H16N2, M + H+).
5.6.3. 4-[2-(allylamino)phenylamino]benzonitrile (45c)
General procedure 5 was used for the N-arylation of N-allylphenylenediamine (44) (300 mg, 2.02 mmol) with 4-bromobenzonitrile (368 mg, 2.02 mmol). This procedure afforded 362 mg (72%) of the title compound as an orange oil. 1H NMR (300 MHz, CDCl3) δ 7.44 (d, J = 8.4 Hz, 2 H), 7.19 (t, J = 8.4 Hz, 1 H), 7.10 (d, J = 7.2 Hz, 1 H), 6.74 (d, J = 7.5 Hz, 2 H), 6.66 (d, J = 9.0 Hz, 2 H), 5.96−5.84 (m, 1 H), 5.50 (s, 1 H), 5.26−5.15 (m, 2 H), 4.18 (s, 1 H), 3.78 (t, J = 12.0 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ 150.0, 144.7, 135.2, 133.9, 128.3, 127.1, 125.0, 120.3, 117.6, 116.5, 114.3, 111.9, 100.9, 46.2; IR (film) 3338, 1609 cm−1. MS (ESI) 250.1335 (250.1344 calcd for C16H15N3, M + H+).
5.7. General procedure 6: Pd-catalyzed synthesis of N-aryl piperazines
A flame-dried Schlenk tube equipped with a magnetic stirbar was cooled under a stream of nitrogen and charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), P(2-furyl)3 (8 mol %), sodium tert-butoxide (1.2–1.4 equiv), and the aryl bromide (1.2–1.4 equiv). The Schlenk tube was purged with nitrogen and the amine was added as a solution in toluene (2.5 mL/0.5 mmol substrate). The Schlenk tube was then heated to 105 °C with stirring until the starting material has been consumed as judged by 1H NMR analysis of an aliquot taken from the reaction mixture. The reaction mixture was then cooled to room temperature, saturated aqueous ammonium chloride (2–3 mL) was added, and the resulting mixture was extracted with ethyl acetate (3 × 8 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was then purified by flash chromatography on silica gel.
5.7.1. (+)-(2S,6R)-2,4-Dibenzyl-6-(4-methoxybenzyl)-1-phenylpiperazine (13)
The reaction of (S)-1a (150 mg, 0.42 mmol) with 4-bromoanisole (95 mg, 0.51 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 120 mg (62%) of the title compound was obtained as a white solid, m.p. 119–122 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 10% isopropanol/hexanes, 0.5 mL/min, RT = 15.12 min and 30.50 min), [α]23D +13.51° (c 0.48, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.47−7.38 (m, 7 H), 7.26−7.18 (m, 3 H), 7.06 (d, J = 8.0 Hz, 4 H), 6.95 (d, J = 8.4 Hz, 3 H), 6.84 (t, J = 6.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, 1 H), 3.83– 3.74 (m, 5 H), 3.52−3.45 (m, 2 H), 3.11−2.99 (m, 2 H), 2.89 (d, J = 11.6 Hz, 2 H), 2.83-2.73 (m, 2 H), 2.13−2.10 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 158.1, 147.0, 140.3, 138.9, 132.4, 130.4, 130.1, 129.9, 129.5, 128.7, 128.6, 127.4, 126.2, 117.4, 114.1, 113.5, 63.2, 55.8, 55.6, 55.4, 54.3, 54.1, 37.5, 36.5; IR (film) 3026, 2812, 1597 cm−1; MS (ESI) 463.2744 (463.2749 calcd for C32H34N2O, M + H+).
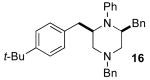
5.7.2. (+)-(2S,6R)-2,4-Dibenzyl-6-(4-tert-butylbenzyl)-1-phenylpiperazine (16)
The reaction of (S)-1a (150 mg, 0.42 mmol) with 4-bromo-tert-butylbenzene (108 mg, 0.51 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 132 mg (64%) of the title compound was obtained as a white solid, m.p. 74–77 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 2% isopropanol/hexanes, 0.1 mL/min, RT = 55.13 min and 101.14 min), [α]23D + 5.48° (c 0.27, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.47−7.38 (m, 7 H), 7.24−7.17 (m, 5 H), 7.07−6.99 (m, 6 H), 6.83 (t, J = 7.2 Hz, 1 H), 3.80 (d, J = 6.4 Hz, 2 H), 3.54−3.44 (m, 2 H), 3.07 (t, J = 12.8 Hz, 2 H), 2.94−2.87 (m, 2 H), 2.79 (t, J = 11.2 Hz, 2 H), 2.15−2.07 (m, 2 H), 1.31 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 149.0, 147.1, 140.4, 139.0, 137.3, 130.1, 129.9, 129.5, 129.1, 128.7, 128.6, 127.5, 126.2, 125.6, 117.4, 113.5, 63.2, 55.6, 55.5, 54.5, 54.1, 37.5, 36.9, 34.6, 31.6; IR (film) 2960, 1597 cm−1; MS (ESI) 489.3272 (489.3270 calcd for C35H40N2, M + H+).
5.7.3. (+)-(2R,6S)-tert-butyl 4-[(4,6-dibenzyl-1-phenylpiperazin-2-yl)methyl]benzoate (17)
The reaction of (S)−1a (150 mg, 0.42 mmol) with 4-bromo-tert-butylbenzoate (130 mg, 0.51 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 132 mg (59%) of the title compound was obtained as a white solid, m.p. 75–77 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OD-H column, 5% isopropanol/hexanes, 2 mL/min, RT = 1.40 min and 1.94 min), [α]23D + 35.1° (c 0.30, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 1.5, 6.5 Hz, 2 H), 7.47−7.40 (m, 7 H), 7.27−7.25 (m, 2 H), 7.24−7.18 (m, 1 H), 7.08−7.06 (m, 6 H), 6.87 (t, J = 7.0 Hz, 1 H), 3.82 (t, J = 11.0 Hz, 2 H), 3.49 (m, 2 H), 3.16−3.06 (m, 2 H), 2.91−2.81 (m, 4 H), 2.18−2.11 (m, 2 H), 1.61 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 165.9, 146.9, 145.2, 140.2, 138.7, 130.10, 130.06, 129.96, 129.86, 129.4, 129.3, 128.7, 128.6, 127.6, 126.3, 117.8, 113.7, 81.0, 63.1, 55.7, 55.5, 54.3, 54.1, 37.6, 37.4, 28.4; IR (film) 2975, 1711, 1598 cm−1; MS (ESI) 533.3166 (533.3168 calcd for C36H40N2O2, M + H+).
5.7.4. (+)-(E)-(2S,6R)-2,4-dibenzyl-6-cinnamyl-1-phenylpiperazine (18)
The reaction of (S)-1a (98 mg, 0.274 mmol) with (E)-β-bromostyrene (71 mg, 0.384 mmol) was conducted for 10 h according to general procedure 6 except with 1.4 equiv of NaOtBu, a catalyst loading of 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3, and a reaction temperature of 90 °C. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 92 mg (73%) of the title compound was obtained as a pale yellow solid, m.p. 43–48 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 95% ee by chiral HPLC analysis (chiralcel OJ-H column, 3% isopropanol/hexanes, 0.2 mL/min, RT = 65.89 min and 106.91 min), [α]23D + 101.30° (c 0.75, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.48−7.46 (m, 2 H), 7.44−7.36 (m, 5 H), 7.34−7.30 (m, 4 H), 7.25−7.17 (m, 4 H), 7.05−7.00 (m, 4 H), 6.87 (t, J = 7.0 Hz, 1 H), 6.29 (d, J = 16.0 Hz, 1 H), 6.21−6.15 (m, 1 H), 3.81 (d, J = 9.5 Hz, 1 H), 3.71 (d, J = 9.0 Hz, 1 H), 3.57−3.51 (m, 2 H), 3.00−2.93 (m, 2 H), 2.83−2.74 (m, 3 H), 2.42 (dd, J = 6.0, 13.5 Hz, 1 H), 2.33 (dd, J = 3.0, 11.0 Hz, 1 H), 2.20 (dd, J = 3.0, 11.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 147.3, 140.3, 138.9, 137.7, 132.6, 129.8, 129.7, 129.5, 128.7, 128.6, 128.6, 128.0, 127.4, 127.3, 126.24, 126.21, 118.1, 114.6, 63.1, 56.1, 55.3, 54.4, 54.0, 37.6, 35.2; IR (film) 3025, 2923, 2811, 1596 cm−1. MS (ESI) 459.2789 (459.2800 calcd for C33H34N2, M + H+).
5.7.5. (–)-(E)-(2S,6R)-2,4-dibenzyl-1-phenyl-6-[3-(trimethylsilyl)allyl]piperazine (19)
The reaction of (S)-1a (100 mg, 0.281 mmol) with (E)-(2-bromovinyl)trimethylsilane (61 μL, 0.393 mmol) was conducted for 10 h according to general procedure 6 except with 1.4 equiv of NaOtBu, a catalyst loading of 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3, and a reaction temperature of 90 °C. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 76 mg (59%) of the title compound was obtained as a yellow viscous oil. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 97% ee by chiral HPLC analysis (chiralcel OD-H column, 0.1% isopropanol/hexanes, 0.5 mL/min, RT = 8.68 min and 9.82 min), [α]23D − 50.60° (c 4.9, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.48−7.34 (m, 7 H), 7.27−7.18 (m, 3 H), 7.05 (d, J = 6.8 Hz, 2 H), 7.01 (d, J = 8.4 Hz, 2 H), 6.89 (t, J = 7.2 Hz, 1 H), 6.01−5.93 (m, 1 H), 5.62 (d, J = 18.4 Hz, 1 H), 3.82−3.79 (m, 1 H), 3.67−3.65 (m, 1 H), 3.58−3.50 (m, 2 H), 2.95 (t, J = 12.4 Hz, 1 H), 2.85−2.74 (m, 3 H), 2.69−2.61 (m, 1 H), 2.41−2.31 (m, 2 H), 2.22 (dd, J = 3.2, 11.2 Hz, 1 H), 0.06 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 147.4, 144.1, 140.3, 138.8, 133.6, 129.74, 129.68, 129.5, 128.6, 128.5, 127.4, 126.2, 118.3, 115.1, 63.2, 56.3, 55.3, 54.7, 53.7, 39.0, 37.6, −1.0; IR (film) 3026, 2953, 1597, 1500 cm−1. MS (ESI) 455.2885 (455.2883 calcd for C30H38N2Si, M + H+).
5.7.6. (+)-(2R,6S)-4-benzyl-2-(4-tert-butylbenzyl)-6-isopropyl-1-phenylpiperazine (20)
The reaction of (S)-1b (150 mg, 0.49 mmol) with 4-bromo-tert-butylbenzene (145 mg, 0.68 mmol) was conducted for 10 h according to general procedure 6 using 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 110 mg (51%) of the title compound was obtained as a white solid, m.p. 83–86 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OD-H column, 100% hexanes, 1 mL/min, RT = 6.29 min and 7.06 min), [α]23D + 3.13° (c 0.17, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.36−7.29 (m, 7 H), 7.24−7.18 (m, 4 H), 7.01 (t, J = 7.2 Hz, 1 H), 6.95 (d, J = 8.0 Hz, 2 H), 3.57 (d, J = 13.2 Hz, 1 H), 3.49−3.47 (m, 1 H), 3.44 (d, J = 16.0 Hz, 1 H), 3.30−3.27 (m, 1 H), 2.74−2.62 (m, 2 H), 2.57−2.54 (m, 1 H), 2.49−2.45 (m, 2 H), 2.37−2.32 (m, 1 H), 1.94−1.89 (m, 1 H), 1.32 (9 H), 0.90 (d, J = 6.8 Hz, 3 H), 0.81 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 150.3, 148.8, 138.5, 136.8, 129.4, 129.3, 129.0, 128.3, 127.1, 125.2, 121.8, 63.4, 63.0, 60.7, 57.2, 53.7, 38.3, 34.5, 31.6, 29.8, 20.8, 18.2 (one aromatic carbon signal is absent due to accidental equivalence); IR (film) 2915, 1604 cm−1. MS (ESI) 441.3269 (441.3270 calcd for C31H40N2, M + H+).
5.7.7. (+)-(2S,6R)-4-Benzyl-2-isopropyl-6-(6-methoxynaphthalen-2-ylmethyl)-1-phenylpiperazine (21)
The reaction of (S)-1b (100 mg, 0.32 mmol) with 2-bromo-6-methoxynapthalene (108 mg, 0.45 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 77 mg (51%) of the title compound was obtained as a white solid, m.p. 114–119 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel AD column, 5% isopropanol/hexanes, 0.9 mL/min, RT = 4.22 min and 5.78 min), [α]23D + 33.5° (c 0.34, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 6.4, 8.8 Hz, 2 H), 7.36−7.26 (m, 7 H), 7.21−7.19 (m, 3 H), 7.13−7.07 (m, 3 H), 7.01 (t, J = 7.2 Hz, 1 H), 3.91 (s, 3 H), 3.56 (d, J = 12.8 Hz, 1 H), 3.54−3.49 (m, 1 H), 3.35 (d, J = 13.2 Hz, 1 H), 3.29−3.25 (m, 1 H), 2.86−2.73 (m, 2 H), 2.55 (dd, J = 3.2, 10.8 Hz, 1 H), 2.47−2.41 (m, 2 H), 2.35 (dd, J = 6.4, 10.8 Hz, 1 H), 1.93−1.87 (m, 1 H), 0.89 (d, J = 6.8 Hz, 3 H), 0.79 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 157.4, 150.4, 138.5, 135.1, 133.2, 129.44, 129.36, 129.17, 129.16, 128.5, 128.4, 127.6, 127.2, 126.8, 122.0, 118.8, 105.8, 63.5, 63.2, 60.8, 57.3, 55.5, 53.7, 39.0, 29.8, 20.8, 18.2 (one aromatic carbon signal is absent due to accidental equivalence); IR (film) 2956, 1604 cm−1; MS (ESI) 465.2899 (465.2906 calcd for C32H36N2O, M + H+).
5.7.8. (+)-(2S,6R)-4-benzyl-2-isopropyl-1-phenyl-6-(2-phenylallyl)piperazine (22)
The reaction of (S)-1b (150 mg, 0.487 mmol) with α-bromostyrene (125 mg, 0.682 mmol) was conducted for 10 h according to general procedure 6 except with 1.4 equiv of NaOtBu, a catalyst loading of 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3, xylenes solvent, and a reaction temperature of 135 °C. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 104 mg (52%) of the title compound was obtained as a viscous yellow oil. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 97% ee by chiral HPLC analysis (chiralcel OD-H column, 0.1% isopropanol/hexanes, 0.5 mL/min, RT = 7.44 min and 8.68 min), [α]23D + 24.60° (c 3.5, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.32−7.27 (m, 6 H), 7.24−7.22 (m, 4 H), 7.10−7.03 (m, 5 H), 5.10 (s, 1 H), 4.92 (s, 1 H), 3.52−3.43 (m, 2 H), 3.12−3.03 (m, 2 H), 2.68−2.56 (m, 3 H), 2.39– 2.32 (m, 1 H), 2.26−2.22 (m, 1 H), 2.10−2.05 (m, 1 H), 1.67−1.60 (m, 1 H), 0.82 (d, J = 6.8 Hz, 3 H), 0.73 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 150.1, 146.6, 141.0, 138.3, 129.3, 129.1, 128.3, 127.5, 127.1, 126.5, 124.6, 123.7, 114.8, 64.2, 63.4, 58.3, 57.9, 53.4, 39.1, 28.9, 20.5, 17.0; IR (film) 2958, 2360, 1596 cm−1. MS (ESI) 411.2794 (411.2800 calcd for C29H34N2, M + Na+).
5.7.9. (+)-(2R,6R)-4-[4-Benzyl-2-benzyloxymethyl-6-(4-methylbenzyl)piperazin-1yl]benzonitrile (23)
The reaction of (R)-1l (150 mg, 0.37 mmol) with 4-bromotoluene (75 mg, 0.48 mmol) was conducted for 10 h according to general procedure 6. The product was formed with 14:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 128 mg (70%) of the title compound was obtained as a white solid, m.p. 125–133 °C. This material was determined to contain a 14:1 mixture of diastereomers as judged by 1H NMR analysis. The enantiopurity of the major diastereomer was judged to be 97% ee by chiral HPLC analysis (chiralcel AD column, 10 % isopropanol/hexanes, 1 mL/min, RT = 5.32 min and 7.41 min), [α]23D + 168.99° (c 0.17, CH2Cl2). Data are for the major diastereomer: 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 9.0 Hz, 2 H), 7.40−7.33 (m, 7 H), 7.32−7.28 (m, 3 H), 6.96 (d, J = 8.0 Hz, 2 H), 6.84 (d, J = 9.0 Hz, 2 H), 6.69 (d, J = 8.0 Hz, 2 H), 4.55−4.48 (m, 2 H), 3.95−3.89 (m, 2 H), 3.72−3.70 (m, 1 H), 3.61 (d, J = 12.0 Hz, 1 H), 3.39−3.35 (m, 2 H), 3.26 (dd, J = 1.5, 11.0 Hz, 1 H), 2.83 (d, J = 11.5 Hz, 1 H), 2.75 (t, J = 12.0 Hz, 1 H), 2.44 (d, J = 12.5 Hz, 1 H), 2.31−2.28 (m, 1 H), 2.28 (s, 3 H), 1.96 (dd, J = 3.0, 11.0 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 150.2, 138.6, 138.2, 136.2, 135.8, 134.1, 129.8, 129.5, 129.2, 128.7, 128.6, 128.13, 128.06, 127.6, 120.6, 112.1, 99.6, 73.7, 68.5, 62.8, 55.0, 54.4, 53.1, 52.6, 37.0, 21.2; IR (film) 2919, 1603 cm−1; MS (ESI) 524.2679 (524.2678 calcd for C34H35N3O, M + Na+).
5.7.10. (+)-(2R,6R)-4-[4-Benzyl-2-benzyloxymethyl-6-(4-trifluoromethylbenzyl)piperazin-1-yl]benzonitrile (24)
The reaction of (R)-1l (150 mg, 0.37 mmol) with 4-bromobenzotrifluoride (98 mg, 0.44 mmol) was conducted for 10 h according to general procedure 6. The product was formed with 14:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 131 mg (65%) of the title compound was obtained as a white solid, m.p. 154–157 °C. This material was determined to contain a 14:1 mixture of diastereomers as judged by 1H NMR analysis. The diastereomers were subsequently separated by careful flash chromatography on silica gel. The enantiopurity of the major diastereomer was judged to be 99% ee by chiral HPLC analysis (chiralcel AD column, 10 % isopropanol/hexanes, 1 mL/min, RT = 5.99 min and 8.68 min), [α]23D + 155.6° (c 0.13, CH2Cl2).
Major (cis) diastereomer (24): 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.0 Hz, 2 H), 7.41−7.31(m, 12 H), 6.82 (t, J = 8.8 Hz, 4 H), 4.59−4.51 (m, 2 H), 3.97−3.89 (m, 2 H), 3.74−3.71 (m, 1 H), 3.66 (d, J = 12.4 Hz, 1 H), 3.38 (d, J = 6.8 Hz, 1 H), 3.32−3.27 (m, 2 H), 2.80 (t, J = 12.4 Hz, 1 H), 2.71 (d, J = 11.6 Hz, 1 H), 2.51 (d, J = 12.4 Hz, 1 H), 2.37 (dd, J = 2.4, 11.6 Hz, 1 H), 1.95 (dd, J = 3.2, 12.0 Hz, 1 H); 13C NMR (500 MHz, CDCl3) δ 150.0, 142.8, 138.4, 138.1, 134.1, 130.0, 129.6, 129.0, 128.74, 128.69, 128.2, 128.1, 127.7, 125.6 (q, J = 14.5 Hz), 124.3 (q, J = 271 Hz), 120.4, 112.1, 99.1, 73.6, 68.4, 62.8, 54.7, 54.5, 53.2, 52.1, 37.3; IR (film) 2817, 1603 cm−1; MS (ESI) 578.2402 (578.2395 calcd for C34H32F3N3O, M + Na+).
Minor (trans) diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.4 Hz, 2 H), 7.37−7.26 (m, 11 H), 7.19−7.17 (m, 2 H), 7.10 (d, J = 8.4 Hz, 2 H), 6.79 (d, J = 8.0 Hz, 2 H), 4.38 (s, 2 H), 3.80−3.76 (m, 1 H), 3.68−3.62 (m, 2 H), 3.49 (dd, J = 2.4, 9.6 Hz, 1 H), 3.34−3.31 (m, 2 H), 3.19 (dd, J = 2.4, 11.2 Hz, 1 H), 3.03−2.97 (m, 1 H), 2.53−2.49 (m, 2 H), 2.46−2.41 (m, 1 H), 2.30 (dd, J = 3.2, 11.2 Hz, 1 H).
5.7.11. (+)-(2R,6S)-4-Allyl-2-(4-methoxybenzyl)-6-methyl-1-phenylpiperazine (25)
The reaction of (S)-1d (124 mg, 0.54 mmol) with 4-bromoanisole (121 mg, 0.65 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 91 mg (50%) of the title compound was obtained as a yellow oil. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (OD-H column, 100% hexanes, 1 mL/min, RT = 19.75 min and 22.39 min), [α]23D + 112.18° (c 0.26, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.35 (t, J = 8.5 Hz, 2 H), 7.17 (d, J = 8.0 Hz, 2 H), 7.07 (t, J = 7.5 Hz, 1 H), 6.98 (d, J = 9.0 Hz, 2 H), 6.79−6.76 (m, 2 H), 5.90−5.82 (m, 1 H), 5.20−5.12 (m, 2 H), 3.77 (s, 3 H), 3.45−3.40 (m, 1 H), 3.38−3.35 (m, 1 H), 3.06−3.01 (m, 1 H), 2.92−2.88 (m, 1 H), 2.71 (dd, J = 2.0, 11.0 Hz, 1 H), 2.57 (dd, J = 3.5, 13.5 Hz, 1 H), 2.51 (d, J = 10.0 Hz, 1 H), 2.44−2.40 (m, 1 H), 2.26−2.18 (m, 2 H), 0.94 (d, J = 6.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 158.1, 149.2, 135.2, 131.8, 130.2, 129.4, 123.2, 118.1, 113.8, 61.9, 60.5, 60.4, 57.4, 55.4, 54.2, 37.9, 18.8 (one carbon signal is absent due to accidental equivalence); IR (film) 3400, 2929, 1597 cm−1; MS (ESI) 337.2263 (337.2280 calcd for C22H28N2O, M + H+).
5.7.12. (+)-(2R,6S)-2-Benzyl-1-(4-chlorophenyl)-4-(4-methoxyphenyl)-6-methylpiperazine (26)
The reaction of (S)-1i (47 mg, 0.14 mmol) with bromobenzene (22 mg, 0.17 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 29 mg (51%) of the title compound was obtained as a yellow oil. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OD-H column, 1% isopropanol/hexanes, 0.5 mL/min, RT = 9.27 min and 10.30 min), [α]23D + 58.87° (c 0.55, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 7.34−7.27 (m, 4 H), 7.21−7.18 (m, 1 H), 7.12−7.09 (m, 4 H), 6.86−6.81 (m, 4 H), 3.76 (s, 3 H), 3.59−3.55 (m, 1 H), 3.52−3.49 (m, 1 H), 3.25 (dd, J = 3.5, 12.0 Hz, 1 H), 3.07 (dd, J = 3.0, 12.0 Hz, 1 H), 2.92−2.88 (m, 2 H), 2.68 (dd, J = 3.5, 13.5 Hz, 1 H), 2.58 (dd, J = 10.5, 13.5 Hz, 1 H), 1.05 (d, J = 6.5 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 154.2, 147.5, 146.0, 139.4, 129.5, 129.3, 128.7, 128.0, 126.5, 123.6, 118.9, 114.7, 60.2, 58.3, 55.8, 54.7, 53.8, 38.6, 18.5; IR (film) 2930, 1510 cm−1; MS (ESI) 407.1895 (407.1890 calcd for C25H27ClN2O, M + H+).
5.7.13. (+)-tert-Butyl-4-benzyl-2-(4-cyanobenzyl)-6-methylpiperazine-1-carboxylate (27)
The reaction of (S)-7b (150 mg, 0.493 mmol) of with 4-bromobenzonitrile (117 mg, 0.641 mmol) was conducted for 10 h according to general procedure 6 except using a catalyst composed of Pd(OAc)2 (6 mol %) and PPh3 (8 mol %) with a reaction temperature of 90 °C. The product was formed with 1:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 135 mg (68%) of the title compound was obtained as a 1:1 mixture of diastereomers. The stereoisomers were subsequently separated by careful column chromatography.
(2R,6S)-Cis-Diastereomer (27): pale yellow solid, m.p. 180–185 °C. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 5% isopropanol/hexanes, 1.0 mL/min, RT = 7.40 min and 8.78 min), [α]23D + 8.90° (c 0.8, CH2Cl2). 1H. 1H NMR (500 MHz, CDCl3) δ 7.38−7.35 (m, 7 H), 6.92 (d, J = 6.5 Hz, 2 H), 4.18−4.12 (m, 1 H), 3.89 (d, J =10.5 Hz, 1 H), 3.63 (d, J =13.0 Hz, 1 H), 3.24 (d, J = 13.0 Hz, 1 H), 3.07 (t, J = 11.5 Hz, 1 H), 2.77−2.73 (m, 2 H), 2.46 (d, J = 12.0 Hz, 1 H), 2.32 (dd, J = 4.5, 11.5 Hz, 1 H), 1.78 (dd, J = 4.0, 11.5 Hz, 1 H), 1.50 (s, 9 H), 1.37 (d, J = 7.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 154.9, 145.8, 138.8, 132.2, 130.4, 129.7, 128.6, 127.5, 119.3, 110.0, 80.0, 63.0, 58.9, 53.9, 52.3, 47.4, 40.6, 28.8, 21.1; IR (film) 2969, 2226, 1686 cm−1. MS (ESI) 406.2496 (406.2495 calcd for C25H31N3O2, M + H+).
(+)-(2S,6S)-Trans-Diastereomer (27b): pale yellow solid, m.p. 109–114 °C. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OJ-H column, 5% isopropanol/hexanes, 0.2 mL/min, RT = 39.29 min and 42.49 min). 1H NMR (500 MHz, CDCl3) δ 7.40−7.31 (m, 7 H), 7.05 (d, J = 8.0 Hz, 2 H), 4.09−4.05 (m, 1 H), 3.72−3.65 (m, 1 H), 3.58 (d, J = 13.0 Hz, 1 H), 3.25 (d, J = 12.5 Hz, 1 H), 3.19−3.14 (m, 1 H), 2.86−2.78 (m, 2 H), 2.44−2.41 (m, 1 H), 2.04−2.00 (m, 2 H), 1.48 (s, 9 H), 1.39 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 156.4, 145.3, 138.3, 132.2, 130.3, 129.6, 128.5, 127.5, 119.3, 110.0, 80.2, 62.9, 61.6, 55.9, 52.7, 48.5, 37.4, 28.7, 20.7; IR (film) 2972, 2226, 1704 cm−1. MS (ESI) 406.2498 (406.2495 calcd for C25H31N3O2, M + H+).
5.7.14. tert-butyl 4-benzyl-2-(4-cyanobenzyl)-6-isobutylpiperazine-1-carboxylate (28)
The reaction of (±)-7d (54 mg, 0.156 mmol) with 4-bromobenzonitrile (35 mg, 0.187 mmol) was conducted for 10 h according to general procedure 6 except using a catalyst composed of Pd(OAc)2 (6 mol %) and PPh3 (8 mol %) with a reaction temperature of 90 °C. The product was formed with 2:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 50 mg (72%) of the title compound was obtained as a 12:1 mixture of diastereomers. The stereoisomers were subsequently separated by careful column chromatography.
(±)-(2R,6S)-Major (cis) diastereomer (observed as a 1:1 mixture of rotamers) (28): pale yellow solid, m.p. 125–134 °C; 1H NMR (400 MHz, CDCl3) δ 7.40−7.30 (m, 7 H), 7.08−6.92 (m, 2 H), 4.02−3.88 (m, 2 H), 3.56 (d, J = 12.9 Hz, 1 H), 3.30 (d, J =12.6 Hz, 1 H), 3.06 (t, J = 11.7 Hz, 1 H), 2.78 (t, J = 14.1 Hz, 2 H), 2.49−2.41 (m, 1 H), 2.17 (dd, J = 3.9, 11.1 Hz, 1 H), 2.04−1.92 (m, 1 H), 1.83 (dd, J = 4.2, 11.4 Hz, 1 H), 1.50−1.40 (m, 10 H), 0.97−0.92 (m, 7 H); 13C NMR (100 MHz, CDCl3) δ 155.0, 145.8, 138.9, 132.2, 130.4, 129.5, 129.1, 129.0, 128.6, 127.5, 119.3, 110.0, 80.1, 63.0, 55.7, 53.6, 52.8, 50.6, 43.8, 40.5, 28.8, 28.7, 28.5, 25.9, 24.2, 22.1; IR (film) 2957, 2226, 1686 cm−1. MS (ESI) 448.2957 (448.2964 calcd for C28H37N3O2, M + H+).
(±)-(2S,6S)-Minor (trans) diastereomer (28b): yellow solid, m.p. 91–96 °C; 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.4 Hz, 2 H), 7.39−7.27 (m, 5 H), 7.22 (d, J = 8.0 Hz, 2 H), 3.95−3.91 (m, 1 H), 3.85−3.80 (m, 1 H), 3.64 (d, J = 12.8 Hz, 1 H), 3.37 (d, J = 13.2 Hz, 1 H), 3.28 (dd, J = 5.2, 12.8 Hz, 1 H), 3.03−2.97 (m, 1 H), 2.67 (dd, J = 3.6, 11.6 Hz, 1 H), 2.45−2.40 (m, 2 H), 2.37−2.33 (m, 1 H), 1.78−1.72 (m, 1 H), 1.47 (m, 10 H), 1.25−1.10 (m, 1 H), 0.87 (t, J = 6.0 Hz, 6 H). 13C NMR (100 MHz, CDCl3) δ 156.1, 145.5, 138.6, 132.2, 130.3, 129.1, 128.5, 127.4, 119.3, 110.1, 80.1, 63.0, 55.6, 55.2, 53.9, 52.6, 41.6, 38.5, 28.7, 25.4, 23.3, 22.5; IR (film) 2956, 2227, 1694 cm−1. MS (ESI) 448.2972 (448.2964 calcd for C28H37N3O2, M + H+).
5.7.15. (+)-(2R,6S)-4-Benzyl-2-(biphenyl-4-ylmethyl)-6-isobutyl-1-phenylpiperazine (29)
The reaction of (S)-1j (150 mg, 0.465 mmol) with 4-bromobiphenyl (130.1 mg, 0.558 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 126 mg (57%) of the title compound was obtained as a pale yellow viscous oil. This material was judged to be of >20:1 dr by 1H NMR analysis. The enantiopurity was judged to be 99% ee by chiral HPLC analysis (chiralcel OD-H column, 2 % isopropanol/hexanes, 1 mL/min, RT = 3.54 min and 4.23 min), [α]23D + 157.20° (c 4.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.6 Hz, 2 H), 7.45−7.38 (m, 6 H), 7.36−7.31 (m, 6 H), 7.06 (d, J = 8.4 Hz, 2 H), 6.99 (d, J = 8.4 Hz, 2 H), 6.84 (t, J = 7.2 Hz, 1 H), 3.76– 3.66 (m, 2 H), 3.57 (d, J = 13.2 Hz, 1 H), 3.48 (d, J = 12.8 Hz, 1 H), 2.95 (t, J = 12.8 Hz, 1 H), 2.82−2.73 (m, 3 H), 2.39 (dd, J = 3.6, 11.2 Hz, 1 H), 2.20 (dd, J = 3.2, 11.2 Hz, 1 H), 1.94−1.84 (m, 1 H), 1.53−1.44 (m, 1 H), 1.28−1.22 (m, 1 H), 0.95 (d, J = 6.4 Hz, 3 H), 0.89 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 147.7, 141.2, 139.5, 139.1, 138.9, 129.9, 129.6, 129.5, 128.9, 128.5, 127.4, 127.3, 127.26, 127.2, 118.3, 115.4, 63.1, 56.6, 56.2, 54.8, 52.8, 40.3, 37.6, 26.2, 24.4, 21.8; IR (film) 2954, 2360, 1595 cm−1. MS (ESI) 475.3114 (475.3113 calcd for C34H38N2, M + H+).
5.7.16. (±)-4-Benzyl-2-(4-tert-butylbenzyl)-6-(4-chlorobenzyl)-1-phenylpiperazine (30)
The reaction of (±)-1k (200 mg, 0.511 mmol) with 1-bromo-4-tert-butylbenzene (131 mg, 0.614 mmol) was conducted according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 0.140 g (52%) of the title compound was obtained as a yellow, low-melting, solid. This material was judged to be of >20:1 dr by 1H NMR analysis. 1H NMR (400 MHz, CDCl3) δ 7.42−7.36 (m, 7 H), 7.26 (d, J = 8.4 Hz, 2 H), 7.13 (d, J = 8.0 Hz, 2 H), 7.04−7.01 (m, 4 H), 6.86−6.81 (m, 3 H), 3.80 (d, J = 10.8 Hz, 1 H), 3.72 (d, J = 10.8 Hz, 1 H), 3.55 (d, J =12.4 Hz, 1 H), 3.38 (d, J = 12.4 Hz, 1 H), 3.07−2.90 (m, 3 H), 2.79−2.71 (m, 3 H), 2.15 (d, J = 14.4 Hz, 1 H), 2.06 (d, J = 14.4 Hz, 1 H), 1.30 (s, 9 H); 13C NMR (400 MHz, CDCl3) δ 148.9, 146.7, 138.7, 138.5, 136.9, 131.7, 130.6, 129.9, 129.7, 128.9, 128.5, 128.4, 127.3, 125.4, 117.4, 113.4, 62.9, 55.3, 54.5, 53.5, 36.7, 36.6, 34.3, 31.4, 31.2; IR (film) 2953, 1597 cm−1; MS (ESI) 523.2875 (523.2880 calcd for C35H39ClN2, M + H+).
5.7.17. (±)-4-Allyl-2-(4-tert-butylbenzyl)-1-phenylpiperazine (31)
The reaction of 1c (100 mg, 0.462 mmol) with 4-bromo-tert-butylbenzene (96.2 μl, 0.554 mmol) was conducted for 10 h according to general procedure 6 except dppb (2 mol %) was used as the ligand. Upon purification, 120 mg (74%) of the title compound was obtained as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.37−7.33 (m, 4 H), 7.14 (d, J = 8.4 Hz, 2 H), 7.02 (d, J = 8.0 Hz, 2 H), 6.89 (t, J = 7.2 Hz, 1 H), 6.03−5.93 (m, 1 H), 5.27−5.19 (m, 2 H), 3.98−3.94 (m, 1 H), 3.39 (dt, J = 2.8, 11.6 Hz, 1 H), 3.31−3.25 (m, 1 H), 3.20−3.11 (m, 2 H), 3.03−2.95 (m, 2 H), 2.88 (dt, J = 2.4, 9.2 Hz, 1 H), 2.60 (dd, J = 2.8, 12.8 Hz, 1 H), 2.32 (td, J = 3.6, 11.2 Hz, 1 H), 2.16 (dd, J = 3.2, 11.6 Hz, 1 H), 1.34 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 149.9, 148.9, 137.2, 135.4, 129.5, 129.2, 125.4, 119.0, 118.0, 115.9, 61.9, 58.2, 54.0, 53.6, 43.6, 34.5, 32.1, 31.6; IR (film) 2960, 1598 cm−1. Anal calcd for C24H32N2: C, 82.71; H, 9.25; N, 8.04. Found: C, 82.72; H, 9.32; N, 8.01.
5.7.18. (±)-4-[(4-Allyl-1-phenylpiperazin-2-yl)methyl]-N,N-dimethylaniline (32)
The reaction of 1c (100 mg. 0.462 mmol) with 4-bromo-N,N-dimethylaniline (111 mg, 0.554 mmol) of was conducted for 10 h according to general procedure 6 except only 4 mol % P(2-furyl)3 was employed. Upon purification, 87 mg (56%) of the title compound was obtained as an orange solid, m.p. 59–61 °C. 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.5 Hz, 2 H), 7.04 (d, J = 8.5 Hz, 2 H), 6.98 (d, J = 8.0 Hz, 2 H), 6.84 (t, J = 7.5 Hz, 1 H), 6.69 (d, J = 8.5 Hz, 2 H), 5.98−5.89 (m, 1 H), 5.22−5.15 (m, 2 H), 3.87−3.84 (m, 1 H), 3.35 (dt, J = 2.5, 12.0 Hz, 1 H), 3.25−3.16 (m, 1 H), 3.11−3.01 (m, 2 H), 3.01−2.90 (m, 8 H), 2.86−2.83 (m, 1 H), 2.49 (dd, J = 2.5, 13.0 Hz, 1 H), 2.27 (td, J = 3.5, 11.0 Hz, 1 H), 2.10 (dd, J = 3.0, 11.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 149.9, 149.2, 135.4, 130.1, 129.4, 128.3, 118.8, 118.0, 115.8, 113.1, 61.9, 58.4, 53.8, 53.6, 43.6, 41.0, 31.5; IR (film) 2952, 2798, 1615 cm−1. Anal calcd for C22H29N3: C, 78.76; H, 8.71; N, 12.53. Found: C, 78.59; H, 8.59; N, 12.27.
5.7.19. (±)-4-Allyl-1-phenyl-2-(pyridin-3-ylmethyl)-piperazine (33)
The reaction of 1c (100 mg, 0.462 mmol) with 3-bromopyridine (54.4 μl, 0.554 mmol) was conducted for 10 h according to general procedure 6 except dppb (2 mol %) was used as the ligand. Upon purification, 90 mg (66 %) of the title compound was obtained as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 8.44−8.42 (m, 2 H), 7.44 (d, J = 7.5 Hz, 1 H), 7.31 (t, J = 7.0 Hz, 2 H), 7.20−7.17 (m, 1 H), 6.96 (d, J = 9.0 Hz, 2 H), 6.86 (dt, J = 1.0, 7.5 Hz, 1 H), 5.93−5.85 (m, 1 H), 5.21−5.15 (m, 2 H), 3.93−3.90 (m, 1 H), 3.34 (dt, J = 3.5, 12.0 Hz, 1 H), 3.27−3.22 (m, 1 H), 3.18−3.13 (m, 1 H), 3.11−3.07 (m, 1 H), 2.99−2.96 (m, 1 H), 2.92−2.88 (m, 1 H), 2.71 (dt, J = 2.0, 11.5 Hz, 1 H), 2.59 (dd, J = 3.0, 12.5 Hz, 1 H), 2.29 (td, J = 3.5, 11.0 Hz, 1 H), 2.14 (dd, J = 3.2, 11.5 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 150.9, 149.8, 147.7, 136.8, 135.7, 135.1, 129.6, 123.5, 119.5, 118.3, 116.2, 61.9, 58.3, 53.7, 53.5, 43.6, 29.9; IR (film) 3392, 2948, 2801, 1597 cm−1. Anal calcd for C19H23N3: C, 77.78; H, 7.90; N, 14.32. Found: C, 77.48; H, 7.85; N, 14.36.
5.7.20. (±)-tert-Butyl-4-benzyl-2-(4-cyanobenzyl)-piperazine-1-carboxylate (34)
The reaction of 7g (250 mg, 0.861 mmol) with 4-bromobenzonitrile (188 mg, 1.03 mmol) was conducted for 12 h according to general procedure 6 except using a catalyst composed of Pd(OAc)2 (6 mol %) and PPh3 (8 mol %) with a reaction temperature of 90 °C. Upon purification, 229 mg (68%) of the title compound was obtained as a white solid, m.p. 129–133 °C. This molecule was observed as a 2:1 mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.53−7.50 (m, 0.4 H), 7.42 (d, J = 7.6 Hz, 1.6 H), 7.37−7.29 (m, 5 H), 7.14−7.00 (m, 2 H), 4.30−3.70 (m, 2 H), 3.60 (d, J = 12.8 Hz, 1 H), 3.28 (d, J = 12.4 Hz, 1 H), 3.18 (td, J = 2.8, 12.8 Hz, 1 H), 3.10−3.00 (m, 1 H), 2.96−2.80 (m, 2 H), 2.53 (d, J = 11.6 Hz, 1 H), 2.11 (td, J = 3.2, 12 Hz, 1 H), 1.96 (d, J = 9.6, 1 H), 1.37 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 154.4, 144.9, 138.0, 134.6, 132.0, 130.1, 129.3, 127.3, 119.0, 109.8, 79.8, 62.8, 53.5, 52.1, 40.3, 39.2, 36.2, 28.3; IR (film) 3400, 2916, 1691 cm−1. MS (ESI) 392.2338 (392.2338 calcd for C24H29N3O2, M + H+).
5.7.21. (±)-4-Benzyl-2-(4-tert-butylbenzyl)-1-(4-methoxyphenyl)-6-methylpiperazine (35)
The reaction of (±)-1f (65 mg, 0.209 mmol) with 4-bromo-tert-butylbenzene (44 μl, 0.251 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 45 mg (49%) of the title compound was obtained as a yellow oil. This material was judged to be of >20:1 dr by 1H NMR analysis. 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 4.0 Hz, 4 H), 7.25−7.24 (m, 1 H), 7.21 (d, J = 8.5 Hz, 2 H), 7.17 (d, J = 8.5 Hz, 2 H), 6.92−6.89 (m, 4 H), 3.82 (s, 3 H), 3.64 (d, J = 13.0 Hz, 1 H), 3.35−3.29 (m, 2 H), 3.16−3.06 (m, 1 H), 2.76 (t, J = 11.0 Hz, 2 H), 2.58 (dd, J = 3.5, 13.5 Hz, 1 H), 2.31−2.26 (m, 1 H), 2.16 (t, J = 10.0 Hz, 1 H), 2.02 (t, J = 9.5 Hz, 1 H), 1.29 (s, 9 H), 0.78 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 156.9, 148.8, 142.6, 138.2, 136.5, 129.4, 129.1, 128.9, 128.3, 127.2, 125.2, 114.4, 63.2, 62.2, 60.5, 58.9, 56.3, 55.6, 38.6, 34.5, 31.6, 19.0; IR (film) 2961, 2360, 1508 cm−1. MS (ESI) 443.3060 (443.3062 calcd for C30H38N2O, M + H+).
5.7.22. (±)-(2R,6S)-4-Benzyl-2-(4-tert-butylbenzyl)-6-methyl-1-phenylpiperazine (36)
The reaction of (±)-1e (100 mg, 0.357 mmol) with 4-bromo-tert-butylbenzene (75 ml, 0.428 mmol) was conducted for 10 h according to general procedure 6. The product was formed with 9:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 81 mg (55%) of the title compound was obtained as a yellow oil. This material was judged to be of 9:1 dr by 1H NMR analysis. The diastereomers were subsequently separated by careful column chromatography.
Major (cis) diastereomer (36): 1H NMR (400 MHz, CDCl3) δ 7.43−7.31 (m, 7 H), 7.23 (d, J = 8.0 Hz, 2 H), 7.10 (d, J = 7.6 Hz, 2 H), 6.99−6.92 (m, 3 H), 3.66−3.63 (m, 3 H), 3.43 (d, J = 12.8 Hz, 1 H), 2.76−2.66 (m, 2 H), 2.60−2.58 (m, 2 H), 2.52−2.48 (m, 1 H), 2.38 (d, J = 9.6 Hz, 1 H), 1.32 (s, 9 H), 1.14 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 148.8, 148.3, 138.6, 137.1, 129.53, 129.48, 129.0, 128.4, 127.3, 125.3, 120.4, 118.9, 63.2, 59.8, 58.2, 56.1, 51.5, 37.7, 34.5, 31.6, 18.5; IR (film) 2962, 2360, 1596 cm−1. MS (ESI) 413.2957 (413.2957 calcd for C29H36N2, M + H+).
Minor (trans) diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.38−7.30 (m, 7 H), 7.10−7.02 (m, 5 H), 6.70 (d, J = 7.6 Hz, 2 H), 3.68−3.60 (m, 1 H), 3.57−3.50 (m, 2 H), 3.40– 3.34 (m, 1 H), 2.98 (t, J = 11.6 Hz, 1 H), 2.89 (d, J = 11.2 Hz, 1 H), 2.64 (d, J = 10.4 Hz, 1 H), 2.53 (d, J = 11.2 Hz, 1 H), 2.32 (d, J = 9.6 Hz, 1 H), 2.08 (t, J = 10.4 Hz, 1 H), 1.23 (s, 9 H), 0.99 (d, J = 6.0 Hz, 3 H).
5.7.23. (±)-4-[4-benzyl-2-(4-tert-butylbenzyl)-6-methylpiperazin-1-yl]benzonitrile (37)
The reaction of (±)-1g (142 mg, 0.466 mmol) with 4-bromo-tert-butylbenzene (98 μl, 0.559 mmol) was conducted for 10 h according to general procedure 6. The product was formed with 6:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 156 mg (76%) of the title compound was isolated as a yellow solid. This material was judged to be of 6:1 dr by 1H NMR analysis. The diastereomers were subsequently separated by careful column chromatography.
Major (cis) diastereomer (37): m.p. 59–66 °C. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.8 Hz, 2 H), 7.47−7.38 (m, 5 H), 7.21 (d, J = 6.8 Hz, 2 H), 6.88−6.84 (m, 4 H), 4.04−3.99 (m, 1 H), 3.84−3.78 (m, 1 H), 3.62 (d, J = 12.4 Hz, 1 H), 3.46 (d, J = 12.4 Hz, 1 H), 3.09 (t, J = 12.4 Hz, 1 H), 2.94−2.86 (m, 2 H), 2.61 (d, J = 12.8 Hz, 1 H), 2.40 (dd, J = 4.4, 10.8 Hz, 1 H), 1.99 (dd, J = 3.2, 11.6 Hz, 1 H), 1.40 (d, J = 6.8 Hz, 3 H), 1.31 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 150.2, 149.3, 138.6, 136.4, 134.0, 129.6, 129.0, 128.6, 127.5, 125.6, 120.7, 112.3, 98.0, 63.0, 58.8, 55.2, 53.2, 48.2, 37.3, 34.5, 31.5, 18.3; IR (film) 2962, 2360, 1603 cm−1. MS (ESI) 438.2905 (438.2909 calcd for C30H35N3, M + H+).
Minor (trans) diastereomer (isolated as 2:1 mixture of trans:cis diastereomers) : 1H NMR (400 MHz, CDCl3) δ 7.59−7.52 (m, 2 H), 7.44−7.30 (m, 5 H), 7.18 (d, J = 8.4 Hz, 1 H), 7.11−7.08 (m, 2 H), 6.86−6.82 (m, 2 H), 6.70 (d, J = 8.0 Hz, 1 H), 3.70−3.66 (m, 2 H), 3.62−3.54 (m, 1 H), 3.42 (t, J = 14.0 Hz, 1 H), 3.11−2.99 (m, 1 H), 2.95−2.84 (m, 2 H), 2.44−2.36 (m, 2 H), 2.09−2.03 (m, 1 H), 1.24 (s, 9 H), 1.02 (d, J = 5.6 Hz, 3 H).
5.7.24. (±)-tert-Butyl 4-benzyl-2-(4-tert-butylbenzyl)-6-methylpiperazine-1-carboxylate (38)
The reaction of (±)-7b (100 mg, 0.329 mmol) with 4-bromo-tert-butylbenzene (75 mL, 0.428 mmol) was conducted for 10 h according to general procedure 6 except using a catalyst composed of Pd(OAc)2 (6 mol %) and PPh3 (8 mol %) with a reaction temperature of 90 °C. The product was formed with 1:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 50 mg (35%) of the title compound was obtained as a yellow oil. This material was judged to be of 1:1 dr by 1H NMR analysis. Data are for the mixture. 1H NMR (500 MHz, CDCl3) δ 7.40−7.28 (m, 6 H), 7.20−7.14 (m, 2 H), 6.91 (d, J = 8.0 Hz, 1 H), 4.18−4.10 (m, 1 H), 3.98−3.93 (m, 1 H), 3.51 (d, J = 12.8 Hz, 1 H), 3.39 (d, J = 13.2 Hz, 1 H), 3.06 (t, J = 12.0 Hz, 1 H), 2.73−2.63 (m, 3 H), 2.21 (dd, J = 4.4, 10.8 Hz, 1 H), 1.83 (dd, J = 4.0, 11.6 Hz, 1 H), 1.51 (s, 9 H), 1.37 (d, J = 6.8 Hz, 3 H), 1.27 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 155.0, 148.8, 139.0, 137.1, 129.4, 129.3, 129.2, 128.5, 127.3, 125.4, 125.3, 79.7, 63.1, 58.4, 63.1, 58.4, 54.3, 53.2, 40.2, 34.5, 31.6, 28.8, 28.7, 21.0; IR (film) 2965, 1690 cm−1. MS (ESI) 437.3162 (437.3168 calcd for C28H40N2O2, M + H+).
5.7.25. (±)-4-Allyl-2-(4-tert-butylbenzyl)-6-methyl-1-phenylpiperazine (39)
The reaction of (±)-1d (50 mg, 0.217 mmol) with 4-bromo-tert-butylbenzene (46 ml, 0.260 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 35 mg (46%) of the title compound was obtained as a yellow oil. This material was judged to be of >20:1 dr by 1H NMR analysis. 1H NMR (400 MHz, CDCl3) δ 7.36−7.32 (m, 2 H), 7.27−7.24 (m, 2 H), 7.16 (d, J = 7.6 Hz, 2 H), 7.04 (t, J = 7.2 Hz, 1 H), 7.00 (d, J = 8.4 Hz, 2 H), 5.93−5.82 (m, 1 H), 5.21−5.13 (m, 2 H), 3.51−3.46 (m, 1 H), 3.44−3.36 (m, 1 H), 3.08−3.03 (m, 1 H), 2.93−2.88 (m, 1 H), 2.69 (d, J = 8.8 Hz, 1 H), 2.62−2.58 (m, 1 H), 2.53−2.46 (m, 2 H), 2.34−2.30 (m, 1 H), 2.25−2.21 (m, 1 H), 1.29 (s, 9 H), 0.96 (d, J = 6.4 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 149.0, 148.9, 136.7, 135.3, 129.4, 129.0, 125.3, 122.8, 118.1, 114.6, 61.9, 60.2, 60.0, 57.4, 53.9, 38.1, 34.5, 31.6, 18.8; IR (film) 2962, 1596 cm−1. MS (ESI) 363.2798 (363.2800 calcd for C25H34N2, M + H+).
5.7.26. (±)-2-(4-tert-Butylbenzyl)-4-(4-methoxyphenyl)-6-methyl-1-phenylpiperazine (40)
The reaction of (±)-1h (68 mg, 0.229 mmol) with 4-bromo-tert-butylbenzene (48 μl, 0.275 mmol) was conducted for 10 h according to general procedure 6. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 49 mg (50%) of the title compound was obtained as a yellow oil. This material was judged to be of >20:1 dr by 1H NMR analysis. 1H NMR (400 MHz, CDCl3) δ 7.38 (t, J = 8.4 Hz, 2 H), 7.30 (d, J = 8.0 Hz, 2 H), 7.19 (d, J = 7.6 Hz, 2 H), 7.07 (d, J = 8.0 Hz, 3 H), 6.92−6.89 (m, 2 H), 6.87−6.84 (m, 2 H), 3.78 (s, 3 H), 3.68−3.57 (m, 2 H), 3.26 (dd, J = 3.2, 11.2 Hz, 1 H), 3.09 (dd, J = 3.2, 12.0 Hz, 1 H), 3.00−2.93 (m, 2 H), 2.71−2.58 (m, 2 H), 1.31 (s, 9 H), 1.09 (d, J = 6.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 154.2, 149.1, 148.7, 146.2, 136.6, 129.5, 129.0, 125.5, 122.5, 121.7, 119.0, 114.6, 59.8, 58.4, 55.8, 54.6, 53.4, 38.0, 34.6, 31.6, 18.6; IR (film) 2961, 1596, 1510 cm−1. MS (ESI) 429.2901 (429.2906 calcd for C29H36N2O, M + H+).
5.7.27. (±)-(2S,3R)-4-[4-Benzyl-2-(4-methoxybenzyl)-3-methylpiperazin-1-yl]benzonitrile (41)
The reaction of (±)-9 (100 mg, 0.327 mmol) with 4-bromoanisole (50 ml, 0.392 mmol) was conducted for 10 h according to general procedure 6 except xylenes was used in place of toluene and the reaction was conducted at 140 °C. The product was formed with 2:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 102 mg (76%) of the title compound was obtained as an orange solid with 2:1 dr, m.p. 118–125 °C. The diastereomers were subsequently separated by careful flash chromatography on silica gel to provide a sample with 18:1 dr. Data are for the major isomer. 1H NMR (400 MHz, CDCl3) δ 7.41−7.29 (m, 5 H), 7.24 (d, J = 8.8 Hz, 2 H), 6.97 (d, J = 8.4 Hz, 2 H), 6.65 (d, J = 6.8 Hz, 2 H), 6.48 (d, J = 9.2 Hz, 2 H), 4.16 (d, J = 13.2 Hz, 1 H), 4.06−4.02 (m, 1 H), 3.71 (s, 3 H), 3.42−3.39 (m, 2 H), 3.20– 3.14 (m, 1 H), 3.03−2.96 (m, 2 H), 2.81 (dt, J = 2.8, 11.2 Hz, 1 H), 2.70−2.66 (m, 1 H), 2.15−2.08 (m, 1 H), 1.28 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 158.1, 152.9, 139.5, 133.8, 133.3, 131.4, 130.5, 129.0, 128.5, 127.2, 120.6, 113.8, 98.0, 63.2, 60.2, 57.8, 55.4, 52.2, 41.8, 30.5, 18.8; IR (film) 2953, 2212, 1602 cm−1. MS (ESI) 412.2370 (412.2389 calcd for C27H29N3O, M + H+).
5.7.28. (±)-(4aS,5R,7aR)-4-(1-benzyl-4-phenyloctahydro-1H-cyclopenta[b]pyrazin-5-yl)benzonitrile (42)
The reaction of (±)-10 (100 mg, 0.342 mmol) with 4-bromobenzonitrile (75 mg, 0.410 mmol) was conducted according to general procedure 6 except with a catalyst loading of 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3, xylenes solvent, and a reaction temperature of 140 °C. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 68 mg (51%) of the title compound was obtained as a white solid, m.p. 145–149 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. 1H NMR (400 MHz, CDCl3) δ 7.40−7.30 (m, 9 H), 7.20−7.16 (m, 2 H), 6.70 (t, J = 7.6 Hz, 1 H), 6.59 (d, J = 8.0 Hz, 2 H), 4.57−4.53 (m, 1 H), 4.25 (d, J = 13.6 Hz, 1 H), 3.82−3.76 (m, 1 H), 2.92 (d, J = 14.0 Hz, 1H), 2.89−2.88 (m, 1 H), 2.85 (t, J = 5.2 Hz, 1 H), 2.78– 2.74 (m, 1 H), 2.41−2.31 (m, 2 H), 2.27−2.13 (m, 2 H), 2.03−1.92 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 149.0, 148.7, 138.8, 131.7, 130.7, 129.4, 129.0, 128.6, 127.3, 119.5, 117.1, 112.7, 109.6, 64.6, 61.7, 59.2, 51.1, 44.9, 43.4, 29.7, 28.7; IR (film) 3060, 2947, 2225, 1597 cm−1. MS (ESI) 394.2272 (394.2283 calcd for C27H27N3, M + H+).
5.7.29. (±)-(4aS,5R,7aR)-4-(1-benzyl-4-phenyloctahydro-1H-cyclopenta[b]pyrazin-5yl)phenyl]phenylmethanone (43)
The reaction of (±)-10 (100 mg, 0.342 mmol) with 4-bromobenzophenone (114 mg, 0.410 mmol) was conducted according to general procedure 6 except with a catalyst loading of 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3, xylenes solvent, and a reaction temperature of 140 °C. The product was formed with >20:1 dr as judged by 1H NMR analysis of a sample taken from the crude reaction mixture. Upon purification, 104 mg (64%) of the title compound was obtained as a white solid, m.p. 147–150 °C. This material was judged to be of >20:1 dr by 1H NMR analysis. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 7.2 Hz, 1 H), 7.85 (d, J = 8.0 Hz, 1 H), 7.81−7.76 (m, 2 H), 7.72 (m, J = 7.6 Hz, 1 H), 7.62−7.43 (m, 5 H), 7.39– 7.28 (m, 5 H), 7.17 (t, J = 7.6 Hz, 1 H), 6.68 (t, J = 7.2 Hz, 1 H), 6.62 (d, J = 8.4 Hz, 2 H), 4.59−4.56 (m, 1 H), 4.27 (d, J = 13.6 Hz, 1 H), 3.86−3.80 (m, 1 H), 2.92 (d, J = 13.6 Hz, 2 H), 2.86 (t, J = 5.2 Hz, 1 H), 2.76 (d, J = 11.6 Hz, 1 H), 2.47−2.32 (m, 2 H), 2.30−2.17 (m, 2 H), 2.03−1.96 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 196.9, 149.0, 148.4, 139.1, 138.2, 135.3, 132.3, 130.9, 130.2, 129.32, 129.27, 129.0, 128.6, 128.3, 127.2, 116.9, 112.8, 64.6, 61.8, 59.2, 51.2, 44.8, 43.5, 29.7, 28.8; IR (film) 3027, 2945, 1652, 1598 cm−1. MS (ESI) 473.2591 (473.2593 calcd for C33H32N2O, M + H+).
5.7.30. (±)-2-(4-methoxybenzyl)-1-(4-methoxyphenyl)-1,2,3,4-tetrahydroquinoxaline (46a)
The reaction of 45a (82 mg, 0.322 mmol) with 4-bromoanisole (41 μL, 0.322 mmol) was conducted for 10 h according to general procedure 6 except using (±)-BINAP (2 mol %) as the ligand. This procedure afforded 60 mg (52%) of the title compound as an orange oil. 1H NMR (300 MHz, CDCl3) δ 7.10 (d, J = 9.0 Hz, 3 H), 6.94−6.92 (m, 2 H), 6.84−6.75 (m, 4 H), 6.71−6.57 (m, 3 H), 3.84−3.73 (m, 8 H), 3.20 (qd, J = 3.5, 11.0 Hz, 2 H), 2.93– 2.89 (m, 1 H), 2.77– 2.73 (m, 1 H); 13C NMR (75 MHz, CDCl3) δ 158.3, 156.1, 142.2, 134.9, 132.0, 131.7, 130.6, 126.5, 119.7, 118.8, 118.5, 115.0, 114.8, 114.0, 61.3, 55.7, 55.5, 41.9, 37.4; IR (film) 3394, 2952, 1507 cm−1. MS (ESI) 361.1914 (361.1916 calcd for C23H24N2O2, M + H+).
5.7.31. (±)-2-(4-Methoxybenzyl)-1-phenyl-1,2,3,4-tetrahydroquinoxaline (46b)
The reaction of 45b (77 mg, 0.344 mmol) with 4-bromoanisole (44 μL, 0.344 mmol) was conducted for 10 h according to general procedure 6 except using (±)-BINAP (2 mol %) as the ligand. This procedure afforded 70 mg (62%) of the title compound as an orange oil. 1H NMR (300 MHz, CDCl3) δ 7.23−7.12 (m, 4 H), 6.94−6.90 (m, 4 H), 6.87−6.76 (m, 3 H), 6.69– 6.60 (m, 2 H), 3.92−3.87 (m, 2 H), 3.80 (s, 3 H), 3.28−3.15 (m, 2 H), 2.95−2.88 (m, 1 H), 2.78−2.71 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 158.3, 149.5, 135.6, 131.7, 130.6, 129.4, 129.3, 122.7, 122.2, 121.2, 120.8, 118.3, 115.2, 114.0, 60.2, 55.4, 42.0, 37.1; IR (film) 3407, 2954, 1589 cm−1. MS (ESI) 331.1795 (331.1810 calcd for C22H22N2O, M + H+).
5.7.32. (±)-2-[(1-Benzyl-1H-indol-5-yl)methyl]-1-phenyl-1,2,3,4-tetrahydroquinoxaline (46c)
The reaction of 45b (77 mg, 0.344 mmol) with 1-benzyl-5-bromo-1H-indole (98.3 mg, 0.344 mmol) was conducted for 10 h according to general procedure 6 except using (±)-BINAP (2 mol %) as the ligand. This procedure afforded 70 mg (62%) of the title compound as an orange solid, m.p. 60−65 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (s, 1 H), 7.30−7.23 (m, 3 H), 7.19 (d, J = 8.5 Hz, 1 H), 7.11−7.08 (m, 3 H), 7.06−7.01 (m, 4 H), 6.95−6.86 (m, 2 H), 6.78−6.72 (m, 2 H), 6.48 (d, J = 3.0 Hz, 1 H), 5.30 (s, 2 H), 4.06−4.00 (m, 1 H), 3.32−3.26 (m, 2 H), 3.11−3.07 (m, 1 H), 2.97−2.93 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 149.2, 137.8, 135.5, 135.4, 130.6, 129.7, 129.3, 129.2, 128.9, 128.6, 127.7, 126.9, 123.7, 122.8, 122.1, 121.6, 120.9, 120.2, 118.2, 115.2, 109.8, 101.5, 60.4, 50.3, 41.8, 38.0; IR (film) 3408, 2916, 1589 cm−1. MS (ESI) 430.2279 (430.2283 calcd for C30H27N3, M + H+).
5.7.33. (±)-4-(2-benzyl-3,4-dihydroquinoxalin-1[2H]-yl)benzonitrile (46d)
The reaction of 45c (150 mg, 0.602 mmol) with bromobenzene (70 μL, 0.662 mmol) was conducted for 10 h according to general procedure 6. Upon purification, 125 mg (64%) of the title compound was obtained as an orange solid, m.p. 55–60 °C. 1H NMR (500 MHz, CDCl3) δ 7.35−7.30 (m, 4 H), 7.27−7.24 (m, 2 H), 7.22−7.20 (m, 2 H), 7.08−7.06 (m, 1 H), 6.94−6.91 (m, 1 H), 6.78−6.75 (m, 2 H), 6.72−6.67 (m, 1 H), 4.07 (s, 1 H), 4.05−4.01 (m, 1 H), 3.36−3.28 (m, 2 H), 2.96−2.91 (m, 1 H), 2.79−2.75 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 152.2, 139.1, 136.5, 133.3, 129.5, 128.8, 126.8, 125.1, 124.0, 122.7, 119.9, 119.3, 118.0, 115.5, 102.3, 58.3, 43.4, 37.3; IR (film) 3400, 2216, 1597 cm−1. MS (ESI) 326.1646 (326.1657 calcd for C22H19N3, M + H+).
5.8. General procedure 7: synthesis of benzopiperazines via Pd-catalyzed tandem N-arylation and carboamination with two different aryl bromides
A flame-dried Schlenk tube equipped with a magnetic stir bar, was purged under a stream of nitrogen and charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), 2-(di-tert-butylphosphino)biphenyl (2 mol %), and sodium tert-butoxide (2.1 equiv). The Schlenk tube was purged with nitrogen and a solution of N-allylphenylenediamine (1 equiv) and bromobenzene (1 equiv) in toluene (0.2 M substrate concentration) was added via syringe. The mixture was heated to 80 °C with stirring until the starting material had been consumed as judged by GC analysis. The mixture was cooled to rt, (±)-BINAP (3 mol %) was added, and the mixture was heated to 85 °C with stirring for 10 min. The second aryl halide (1 equiv) was added via syringe and the mixture was heated to 105 °C with stirring until the second aryl halide had been consumed as judged by GC analysis. The mixture was cooled to room temperature, saturated aqueous ammonium chloride (4 mL) was added, and the layers were separated. The aqueous layer was extracted with ethyl acetate (4 × 5 mL) and the combined organic layers were then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel.
5.8.1. (±)-2-(4-methoxybenzyl)-1-phenyl-1,2,3,4-tetrahydroquinoxaline (46b)
General procedure 7 was used for the coupling of N-allyphenylenediamine (44) (100 mg, 0.675 mmol), bromobenzene (106 mg, 0.675 mmol), and 4-bromoanisole (126 mg, 0.675 mmol). This procedure afforded 112 mg (49 %) of the title compound as an orange oil. Data were identical to those reported above.
5.8.2. (±)-2-[(1-Benzyl-1H-indol-5-yl)methyl]-1-phenyl-1,2,3,4-tetrahydroquinoxaline (46c)
General procedure 7 was used for the coupling of N-allyphenylenediamine (44) (100 mg, 0.675 mmol), bromobenzene (106 mg, 0.675 mmol), and 1-benzyl-5-bromo-1H-indole (193 mg, 0.675 mmol). This procedure afforded 120 mg (41%) of the title compound as an orange solid. Data were identical to those reported above.
5.9. Isolation and characterization of side products 14 and 15
Side products 14 and 15 were isolated by careful chromatography of the crude mixture of products obtained from the Pd2(dba)3/P(o-tol)3-catalyzed reaction of 1a with 4-bromoanisole. These compounds were characterized by 1H NMR and 2-D COSY analysis. Data are as follows:
5.9.1. (S)-4-allyl-6-(4-methoxybenzyl)-2-methyl-1-phenyl-1,2,3,4-tetrahydropyrazine (14)
1H NMR (400 MHz, CDCl3) δ 7.33−7.31 (m, 4 H), 7.20−7.12 (m, 5 H), 6.99−6.97 (m, 2 H), 6.89−6.85 (m, 1 H), 6.80−6.77 (m, 2 H), 6.75−6.73 (m, 2 H), 6.69 (d, J = 8.0 Hz, 1 H), 6.60 (dd, J = 1.0, 8.5 Hz, 2 H), 5.82 (s, 1 H), 4.08−3.98 (m, 2 H), 3.79 (s, 3 H), 3.56−3.52 (m, 1 H), 3.36 (d, J = 10.5, 15.5 Hz, 1 H), 3.14 (d, J = 15 Hz, 1 H), 2.74−2.66 (m, 2 H), 2.61−2.59 (m, 1 H), 2.49 (dd, J = 5.0, 13 Hz, 1 H).
5.9.2. (S)-4-allyl-2,6-dimethyl-1-phenyl-1,2,3,4-tetrahydropyrazine (15)
1H NMR (500 MHz, CDCl3) δ 7.34−7.19 (m, 10 H), 7.13−7.10 (m, 2 H), 6.82 (dt, J = 1.0, 7.0 Hz, 1 H), 6.56 (d, J = 8.0 Hz, 2 H), 5.68 (s, 1 H), 4.04−3.95 (m, 2 H), 3.67−3.64 (m, 1 H), 3.07 (dd, J = 4.0, 13 Hz, 1 H), 2.73−2.68 (m, 2 H), 2.62 (dd, J = 2.5, 10.5 Hz, 1 H), 1.73 (s, 3 H).
5.10. Assignment of stereochemistry
5.10.1. 2-Isopropylpiperazines 20–22
The stereochemistry of 2-isopropylpiperazine 21 was assigned on the basis of 2D NOESY experiments. The key nOe signals are shown below. The stereochemistry of piperazines 20 and 22 were assigned based on analogy to 21.
5.10.2. 2-Benzyloxymethylpiperazines 23 and 24
The stereochemistry of 2-benzyloxymethylpiperazine 24 was assigned on the basis of 2D NOESY experiments. The key nOe signals are shown below. The stereochemistry of piperazine 23 was assigned based on analogy to 24.
5.10.3. 2-Methylpiperazines 26 and 40
The stereochemistry of 2-methylpiperazine 26 was assigned on the basis of 2D NOESY experiments. The key nOe signals are shown below. The stereochemistry of piperazine 40 was assigned based on analogy to 26.
5.10.4. 2-Methylpiperazines 25, 35–37, and 39; 2-benzylpiperazines 13, 16–19, and 30; 2-isobutylpiperazine 29
The stereochemistry of 2-methylpiperazine 25 was assigned on the basis of 2D NOESY experiments. The key nOe signals are shown below. The stereochemistry of 2-methylpiperazines 35–37 and 39, 2-benzylpiperazines 13, 16–19, and 30, and 2-isobutylpiperazine 29 was assigned based on analogy to 25.
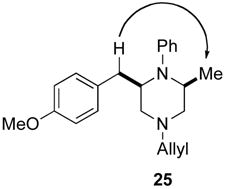
5.10.5. N1-Boc piperazines 27, 28, and 38
The stereochemistry of 2,6-disubstituted-N1-Boc piperazines 27, 27b, 28, 28b, and 38 was assigned following isolation of trans-2,6-disubstituted-N1-Boc piperazine 28b, which was the minor stereoisomer formed in the reaction between 7d and 4-bromobenzonitrile. The stereochemistry of this minor stereoisomer was determined by 2D NOESY experiments. The key nOe signals are shown below. The connectivity of 28 was determined by 2D COSY experiments, and the stereochemistry was assigned as cis. The stereochemistry of 27 and 38 was assigned based on analogy to 28.
5.10.6. 2,3-Disubstituted piperazine 41
The stereochemistry of 2,3-disubstituted piperazine 41 was assigned on the basis of 1D nOe and 2D NOESY experiments. The key nOe signals are shown below.
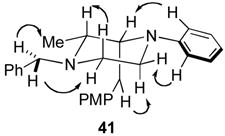
5.10.7. Bicyclic piperazines 42 and 43
The stereochemistry of bicyclic piperazine 42 was assigned on the basis of 2D NOESY experiments. The key nOe signals are shown below. The stereochemistry of 43 was assigned based on analogy to 42.
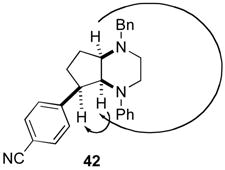
Acknowledgments
The authors thank the NIH-NIGMS (GM 071650) for generous financial support of this work. Additional support was provided by the Camille and Henry Dreyfus Foundation (New Faculty Award, Camille Dreyfus Teacher Scholar Award), Research Corporation (Innovation Award), Eli Lilly, Amgen, GlaxoSmithKline, and 3M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- i.Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- ii.Nilsson JW, Thorstensson F, Kvarnström I, Oprea T, Samuelsson B, Nilsson I. J Comb Chem. 2001;3:546–553. doi: 10.1021/cc010013o. [DOI] [PubMed] [Google Scholar]
- iii.(a) Dinsmore CJ, Beshore DC. Tetrahedron. 2002;58:3297–3312. [Google Scholar]; (b) Jung ME, Rohloff JC. J Org Chem. 1985;50:4909–4913. [Google Scholar]
- iv.For syntheses of nonracemic 2,6-dialkylpiperazines, see: Schanen V, Cherrier MP, de Melo SJ, Quirion JC, Husson HP. Synthesis. 1996:833–837.Mickelson JW, Belonga KL, Jacobsen EJ. J Org Chem. 1995;60:4177–4183.Mickelson JW, Jacobsen EJ. Tetrahedron: Asymmetry. 1995;6:19–22.
- v.For syntheses of racemic 2,6-dialkylpiperazines, see: Berkheij M, van der Sluis L, Sewing C, den Boer DJ, Terpstra JW, Hiemstra H, Bakker WII, van den Hoogenband A, van Maarseveen JH. Tetrahedron Lett. 2005;46:2369–2371.Mouhtaram M, Jung L, Stambach JF. Tetrahedron. 1993;49:1391–1400.
- vi.Michael has recently described a concise, highly diastereoselective asymmetric synthesis of trans-2,6-disubstituted piperazines bearing C-2 methyl groups that is complementary to the transformations described in this paper. See: Cochran BM, Michael FE. Org Lett. 2008;10:329–332. doi: 10.1021/ol702891p.
- Vii.Mercer GJ, Sigman MS. Org Lett. 2003;5:1591–1594. doi: 10.1021/ol034469l.Ferber B, Prestat G, Vogel S, Madec D, Poli G. Synlett. 2006:2133–2135.Viso A, Fernandez de la Pradilla R, Flores A, Garcia A, Tortosa M, Lopez-Rodriguez ML. J Org Chem. 2006;71:1442–1448. doi: 10.1021/jo052077h. and references cited therein.
- viii.A portion of these studies have been previously communicated. See: Nakhla JS, Wolfe JP. Org Lett. 2007;9:3279–3282. doi: 10.1021/ol071241f.
- ix.For related Pd-catalyzed carboamination reactions between aryl or alkenyl halides and alkenes bearing pendant nitrogen nucleophiles that afford pyrrolidines, isoxazolidines, pyrazolidines, indolines, or imidazoldin-2-ones, see: Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.Lira R, Wolfe JP. J Am Chem Soc. 2004;126:13906–13907. doi: 10.1021/ja0460920.Bertrand MB, Wolfe JP. Org Lett. 2006;8:2353–2356. doi: 10.1021/ol0606435.Fritz JA, Wolfe JP. Tetrahedron. 2008;64:6838–6852. doi: 10.1016/j.tet.2008.04.015.Bertrand MB, Neukom JD, Wolfe JP. J Org Chem. 2008;73:8851–8860. doi: 10.1021/jo801631v.Giampietro NC, Wolfe JP. J Am Chem Soc. 2008;130:12907–12911. doi: 10.1021/ja8050487.Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J Org Chem. 2009;74:2533–2540. doi: 10.1021/jo8027399.For reviews, see: Wolfe JP. Eur J Org Chem. 2007:571–582.Wolfe JP. Synlett. 2008:2913–2937.
- x.For examples of Cu-catalyzed intramolecular carboamination of N-(arylsulfonyl)-2-allylanilines and related derivatives, see: Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u.Sherman ES, Chemler SR, Tan TB, Gerlits O. Org Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+.For Pd(II)-catalyzed alkoxycarbonylation of alkenes bearing tethered nitrogen nucleophiles, see: Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J Org Chem. 1997;62:2113–2122. doi: 10.1021/jo961988b.For carboamination reactions between alkenes and N-allylsulfonamides, see: Scarborough CC, Stahl SS. Org Lett. 2006;8:3251–3254. doi: 10.1021/ol061057e.For carboamination of vinylcyclopropanes, see: Larock RC, Yum EK. Synlett. 1990:529–530.For 1,1-carboamination of alkenes, see: Harris GD, Jr, Herr RJ, Weinreb SM. J Org Chem. 1992;57:2528–2530.Harris GD, Jr, Herr RJ, Weinreb SM. J Org Chem. 1993;58:5452–5464.Larock RC, Yang H, Weinreb SM, Herr RJ. J Org Chem. 1994;59:4172–4178.
- xi.Ma D, Zhang Y, Yao J, Wu S, Tao F. J Am Chem Soc. 1998;120:12459–12467. [Google Scholar]
- xii.Use of biphasic conditions led to significant erosion of enantiomeric purity. See: Lu Z, Twieg RJ. Tetrahedron Lett. 2005;46:2997–3001.
- xiii.DEPBT = 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one. Use of other reagents (e.g., DCC/HOBT or CDI) resulted in partial epimerization to afford products with ∼85–90% ee. See: Li H, Jiang X, Ye Yh, Fan C, Romoff T, Goodman M. Org Lett. 1999:91–94. doi: 10.1021/ol990573k.
- xiv.(a) Jiang L, Buchwald SL. In: Metal-Catalyzed Cross-Coupling Reactions. 2nd. de Meijere A, Diederich F, editors. Wiley-VCH; Weinheim, Germany: 2004. pp. 699–760. [Google Scholar]; (b) Hartwig JF. Synlett. 2006:1283–1294. [Google Scholar]; (c) Schlummer B, Scholz U. Adv Synth Catal. 2004;346:1599–1626. [Google Scholar]
- xv.In order to achieve complete consumption of starting materials, a ratio of 4 ligands/Pd atom was employed, as 2:1 ligand/Pd ratios led to only 85–90% conversion.
- xvi.Attempted transformations of N1-acetyl or N1-pivaloyl protected substrates afforded only trace amounts of products.
- xv.Initial studies on carboaminations of amides such as 5a–c suggest these transformations may be feasible, but further optimization is required; low yields (ca. 10–20%) of the desired products were obtained.
- For one-pot syntheses of N-aryl indolines or N-aryl pyrrolidines from primary amines or anilines bearing pendant alkenes, see: (a) reference 9b; Yang Q, Ney JE, Wolfe JP. Org Lett. 2005;7:2575–2578. doi: 10.1021/ol050647u.Ney JE, Hay MB, Yang Q, Wolfe JP. Adv Synth Catal. 2005;347:1614–1620. doi: 10.1002/adsc.200505172.
- xix.Ney JE, Wolfe JP. J Am Chem Soc. 2005;127:8644–8651. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xx.For additional discussion of syn- vs. anti-aminopalladation of alkenes, see: Liu G, Stahl SS. J Am Chem Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u.Minatti A, Muniz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j.
- xxi.Alkene insertion into late M–H bonds is usually much faster than alkene insertion into late M–C bonds. See: Brookhart M, Hauptman E, Lincoln DM. J Am Chem Soc. 1992;114:10394–10401.Siegbahn PEM, Stromberg S, Zetterberg K. Organometallics. 1996;15:5542–5550.
- xxii.The insertion of alkenes into Pd–C bonds in intramolecular Heck reactions occurs through a conformation in which the alkene π–bond and the Pd–C bond are eclipsed. Other possible transition states in which the orientation of the alkene is perpendicular to the M–C bond are significantly higher in energy See: Overman LE, Abelman MM, Kucera DJ, Tran VD, Ricca DJ. Pure Appl Chem. 1992;64:1813–1819.Link JT. Org React. 2002;60:157–534.
- xxiii.The X-ray crystal structure of a known Pd(Ar)(NArR′) complex supports the illustrated trigonal planar geometry of the sp2-hybridized nitrogen atom. See: Yamashita M, Hartwig JF. Am Chem Soc. 2004;126:5344–5345. doi: 10.1021/ja0315107.
- xxiv.Examination of molecular models suggests that low diastereoselectivities would be obtained in reactions that afford cis-2,5-disubstituted pyrrolidines if alkene aminopalladation occurred with the nonbonding nitrogen electrons directed towards the alkene π-system.
-
xxv.An alternative explanation for formation of the cis-2,3-disubstituted piperazine 41 as the major stereoisomer, with low selectivity, would involve cyclization through a chair-like transition state in which the alkene was oriented in a pseudoaxial position (75). However, the alkene pi-system is perpendicular to the Pd–N bond in 75, which is anticipated to lead to high transition state energy. As such, we currently favor the boat-like transition state model for these transformations. For discussion of related transition states in alkene carbopalladation processes, see reference 22.
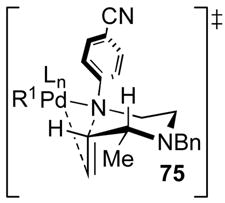
- xxvi.Anderson WK, Lai G. Synthesis. 1995:1287–1290. [Google Scholar]