

Abstract
The trichothecene deoxynivalenol (DON) binds to eukaryotic ribosomes and triggers p38-driven proinflammatory gene expression in the macrophage—a response that is dependent on both double-stranded RNA-activated protein kinase (PKR) and hematopoietic cell kinase (Hck). Here we elucidated critical linkages that exist among the ribosome and these kinases during the course of DON-induced ribotoxic stress in mononuclear phagocytes. Similar to PKR inhibitors, Hck inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyramidine (PP2) suppressed p38 activation and p38-driven interleukin 8 (IL-8) expression in the U937 human monocyte cell line. U937 cells stably transfected with a PKR antisense vector (U9K-A1) displayed marked reduction of DON-induced p38 activation and IL-8 expression as compared to cells transfected with empty vector (U9K-C2), with both responses being completely ablated by PP2. Western analysis of sucrose density gradient fractions revealed that PKR and Hck interacted with the 40S ribosomal subunit in U9K-C2 but not U9K-A1 cells. Subsequent transfection and immunoprecipitation studies with HeLa cells indicated that Hck interacted with ribosomal protein S3. Consistent with U937 cells, DON induced p38 association with the ribosome and phosphorylation in peritoneal macrophages from wild-type but not PKR-deficient mice. DON-induced phosphorylation of ribosome-associated Hck in RAW 264.7 murine macrophages was also suppressed by 2-aminopurine (2-AP). Both 2-AP and PP2 inhibited DON-induced phosphorylation of p38 as well as two kinases, apoptosis signal-regulating kinase 1 and mitogen-activated protein kinase 3/6, known to be upstream of p38. Taken together, PKR and Hck were critical for DON-induced ribosomal recruitment of p38, its subsequent phosphorylation, and, ultimately, p38-driven proinflammatory cytokine expression.
Keywords: deoxynivalenol (DON), translation, ribosome, protein kinase
Trichothecene mycotoxins and other translational inhibitors activate mitogen-activated protein kinase (MAPK) signaling pathways that are essential for cell survival and apoptosis via a novel mechanism termed the ribotoxic stress response (Iordanov et al., 1997; Shifrin and Anderson, 1999; Yang et al., 2000). Deoxynivalenol (DON), a trichothecene produced during Fusarium graminearum infection of wheat, barley, and corn, commonly contaminates cereal-based foods and is of worldwide concern because of its potential to adversely affect human health (Pestka and Smolinski, 2005). The capacity of DON to aberrantly modulate innate immune function by its action in monocytes and macrophages is of particular interest.
Upon binding to the ribosome in mononuclear phagocytes, DON concurrently inhibits translation and induces activation of p38, extracellular signal–regulated kinase (ERK), and c-Jun N-terminal kinase (JNK) MAPKs (Moon and Pestka, 2002; Zhou et al., 2003). Studies with pharmacological inhibitors have demonstrated that p38 is essential for DON-induced proinflammatory gene expression as well as apoptosis (Islam et al., 2006; Moon and Pestka, 2002; Moon et al., 2003). Two kinases, double-stranded (ds) RNA-activated protein kinase (PKR) (Gray et al., 2008; Zhou et al., 2003) and hematopoietic cell kinase (Hck) (Zhou et al., 2005b), have been reported to be upstream of p38 in the DON-induced ribotoxic stress response.
PKR is a serine/threonine protein kinase, induced by dsRNA, interferon, lipopolysaccharide, tumor necrosis factor α and protein kinase R activating protein, an intracellular activator of PKR. (Balachandran and Barber, 2007; Sadler and Williams, 2007). PKR is present in many cell types and involved in regulation of antiviral responses, protein translation, transcription, and cellular fate. This enzyme has a functional dsRNA-binding domain (DRBD) and a catalytic domain. The DRBD interacts with dsRNA, induces autophosphorylation, and facilitates PKR association with the ribosome. The catalytic domain mediates phosphorylation of substrates including elongation initiation factor 2 (eIF2) α and regulates translation (Kim et al., 2005; Samuel, 1993). The DRBD is believed to exert an autoinhibitory effect on the catalytic domain such that dsRNA binding leads to structural alteration of PKR and its activation (Wu and Kaufman, 1997). The autoinhibitory region of PKR can also be structurally altered by non-dsRNA stimuli (Lemaire et al., 2006; Vattem et al., 2001).
In mononuclear phagocytes, PKR mediates DON-induced MAPK activation as well as downstream effects such as proinflammatory gene expression or apoptosis (Zhou et al., 2003). Induction of MAPK phosphorylation and apoptosis by DON and the translational inhibitor anisomycin are suppressed by the PKR inhibitor 2-aminopurine (2-AP) in RAW 264.7 murine macrophages. Similarly, U937 human monocytes treated with pharmacologic inhibitors of PKR or those lacking functional PKR exhibit depressed induction of p38 phosphorylation and downstream interleukin 8 (IL-8) expression by DON and by the ribosome-inactivating proteins ricin and Shiga toxin (Gray et al., 2008; Islam et al., 2006). Lastly, PKR-deficient U937 cells exhibit suppressed p38 phosphorylation and apoptosis in response to DON or anisomycin (Zhou et al., 2003).
Hck, a member of the Src kinase family, is a nonreceptor protein tyrosine kinase expressed primarily in myeloid cells. Hck functions in cytoskeletal rearrangement, phagocytosis, gene transcription, cell proliferation, and apoptosis (Guiet et al., 2008; Quintrell et al., 1987). Hck contains an SH2 domain, SH2 linker, SH3 domain, and tyrosine kinase domain (Arold et al., 2001; Moarefi et al., 1997; Pellicena et al., 1998). The SH3 domain of Hck plays an important role in protein-protein interaction and interacts with PXXP motifs of several cellular proteins (Gouri and Swarup, 1997). Hck activation is regulated by intracellular autoinhibitory interaction between SH2 and SH3 domains (Yadav and Miller, 2007).
Hck also mediates ribotoxin-induced MAPK activation and related downstream effects in monocytes and macrophages (Zhou et al., 2005a). DON-induced MAPK activation and the downstream proinflammatory cytokine expression are suppressed by the Src inhibitors 4-aminoamino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine (PP1) and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyramidine (PP2) or by Hck short inhibitory RNA in RAW 264.7 macrophages. Induction of p38, JNK, and ERK phosphorylation by the translation inhibitors anisomycin and emetine are similarly inhibited by PP1. Furthermore, Hck inhibition significantly inhibits DON-induced caspase activation and apoptosis.
Recently, we have observed that, in mononuclear phagocytes, DON induces recruitment of p38 and other MAPKs to the ribosome whereupon they are phosphorylated (Bae and Pestka, 2008). Despite the apparent importance of PKR and Hck in mediating p38 activation in mononuclear phagocytes during the ribotoxic stress, intriguing questions remain regarding their relationship to p38 activation or how the ribosome might facilitate interaction among these three kinases. The purpose of this study was to elucidate linkages that exist between the ribosome and PKR, Hck, and p38 following stimulation with DON in human and mouse mononuclear phagocytes.
MATERIALS AND METHODS
Chemicals and reagents.
DON and other chemicals were purchased from Sigma-Aldrich, Inc. (St Louis, MO) unless otherwise noted. Inhibitors were obtained from Calbiochem (La Jolla, CA). Human- and mouse-specific PKR antibodies and mouse-specific Hck antibodies were supplied by Santa Cruz Biotech (Santa Cruz, CA). Human-specific Hck was purchased from BD (Franklin Lakes, NJ). Human- and mouse-specific phospho-PKR antibodies were from EMD Chemicals, Inc. (Gibbstown, NJ).
Cell culture.
All cultures were maintained at 37°C in a humidified 6% CO2 incubator. U937 human monocyte cultures (ATCC, Rockville, MD) were grown in RPMI-1640 supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco BRL, Gaithersburg, MD). U937 cultures stably transfected with a constitutively expressed anti-sense PKR expression plasmid (U9K-A1) or with an empty plasmid (U9K-C2) (Cheung et al., 2005) were kindly supplied by Dr Allan Lau (University of Hong Kong). U9K-A1 and U9K-C2 cells were maintained in supplemented RPMI-1640 containing 0.5 mg/ml geneticin (Gibco BRL).
RAW 264.7 murine macrophage cultures (TIB 77, ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 10% (vol/vol) heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco BRL).
Peritoneal macrophage cultures were elicited by ip injecting wild-type (WT) control (C57Bl/6J) and PKR knockout (KO; Hsu et al., 2004; Scheuner et al., 2003) mice with 1.5 ml of thioglycollate (3%, wt/vol). PKR KO mice were kindly provided by Dr Randal Kaufman (University of Michigan, Ann Arbor). After 72 h, macrophages were obtained by peritoneal lavage with cold Hank's buffer (Invitrogen, Carlsbad, CA) and collected by centrifugation at 450 × g for 5 min. Cells were resuspended in supplemented DMEM and cultured overnight prior to experiment initiation.
Experimental design.
In typical experiments, U937 cells (1 × 105/ml in 225-cm2 cell culture flasks) (Corning, Lowell, MA), peritoneal macrophages, or RAW 264.7 cells (5 × 105/ml in 100-mm cell culture dishes) (Corning) were cultured overnight and then treated with DON for various time intervals prior to analysis. DON concentrations for U937 (500–1000 ng/ml), peritoneal macrophages (500 ng/ml), and RAW 264.7 (250 ng/ml) cells were selected based on maximal MAPK phosphorylation and downstream gene expression responses observed in previous studies (Gray and Pestka 2007; Gray et al., 2008; Islam et al., 2006; Moon and Pestka, 2002; Zhou et al., 2003, 2005b).
For some experiments, cells were treated with inhibitors of PKR (5.0mM 2-AP or 2.5μM C16), Hck (0.25–25μM PP2), or p38 (2.0μM SB203580) either concurrently with or 45 min prior to DON stimulation. A nonfunctional chemical analogue of C16 was used as a negative control to confirm that changes observed with C16 were due to the inhibition of PKR rather than a nonspecific effect. Inhibitor concentrations were selected based on their use in previous studies effects (Gray et al., 2008; Zhou et al., 2003, 2005a). Cytotoxicity did not occur at these concentrations, as confirmed by the 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide viability assay.
Quantitative real-time PCR.
RNaqeous kits (Ambion, Austin, TX) were used to isolate RNA from cell pellets. Briefly, cells were lysed, nucleic acids precipitated with ethanol, and RNA trapped in a glass fiber filter. The RNA was eluted and stored at −80°C. Reverse transcription real-time PCR was performed using One-Step PCR Master Mix and IL-8 (NCBI NM 000584.2) or the ß-2 microglobulin (NCBI B2M-NM 004048.2; housekeeping control) Taqman Gene Expression Assay (NCBI NM 000584.2) (NCBI B2M-NM 004048.2) or the ß-2 microglobulin (housekeeping control) Taqman Gene Expression Assay (Applied Biosystems, Foster City, CA). Reaction conditions and PCR program followed manufacturer's instructions using an ABI 7900HT (384 wells) at the Michigan State University Research Technology and Support Facility. Fold change was determined using a relative quantitation method (Pestka and Smolinski, 2005).
Enzyme-linked immunosorbent assay.
After treatment, cell cultures were centrifuged for 10 min at 300 × g, and the supernatant fraction was collected. OptELISA kits (Pharmingen, San Diego, CA) were used for IL-8 protein measurement according to manufacturer's instructions with two modifications. First, the highest standard utilized was 1600 pg/ml, instead of 400 pg/ml. Second, to economize on reagents, 50 μl of antibody dilutions and samples were used per well instead of 100 μl. All samples were read at 450 nm in a Vmax Kinetic Microplate Reader (Molecular Devices, Menlo Park, CA).
Ribosome isolation.
Cytoplasmic fractions were prepared by sucrose density gradient ultracentrifugation (Galban et al., 2003). Briefly, cells were washed with ice-cold PBS twice and lysed in ice-cold polysome extraction buffer (0.3M NaCl, 15mM MgCl2, 15mM Tris-HCl [pH 7.6], 1% [wt/vol] Triton X-100, 0.1 mg/ml cycloheximide and 1 mg/ml heparin, 0.01% [vol/vol], and phosphatase inhibitors A and B (Santa Cruz Biotech). Cell lysates were centrifuged at 10,000 × g for 15 min to clear the resultant supernatant of nuclei, mitochondria, and debris. Protein was measured using a Bio-Rad DC Protein Assay Kit (Bio-Rad, Hercules, CA). Lysate (1 mg) was layered over 9-ml linear sucrose gradient solution (10–50%) in a 11.5-ml Sorvall centrifuge tube and centrifuged at 35,000 × g for 3 h at 4°C in Sorvall TH-641 rotor. The gradient was fractionated at a rate of 0.5–1.0 ml/min by upward displacement using an ISCO system (Te ledyne ISCO, Lincoln, NE) comprised of a syringe pump, EM-1 UV monitor for continuous measurement of absorbance at 254 nm, and a fraction collector.
Western analysis.
Proteins were separated on 4% (wt/vol) polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). After incubating with blocking buffer (Li-Cor, Lincoln, NE), membranes were incubated with murine antibodies or rabbit antibodies to immobilized proteins of interest overnight at 4°C. After washing, blots were incubated with secondary IRDye 680 goat anti-rabbit and/or IRDye 800CW goat anti-mouse IgG antibodies (Li-Cor) for 1 h at 25°C. Infrared fluorescences from these two antibody conjugates were simultaneously measured using a Li-Cor Odyssey Infrared Imaging System, and relative phosphorylation was determined using Odyssey Analysis Software. Ribosomal fractions were identified with mouse and rabbit antibodies to ribosomal protein (RP) S6 (Cell Signaling Technology) and RPL7 (Bethyl Laboratories, Inc., Montgomery, TX), respectively, followed by IRDye 680 goat anti-species conjugates (Bae and Pestka, 2008).
Plasmids, transfection, and immunoprecipitation.
Plasmids containing DNAs for full-length human RPS3 in-frame with a sequence coding for enhanced green fluorescent protein (EGFP) (pEGFPc1; Clontech, Mountain View, CA) (Kim et al., 2005) or human Hck (Trible et al., 2006) (provided by Dr Thomas Smithgall, University of Pittsburgh) were reconstructed in pTRE-Tight Expression Plasmids. HeLa cells (Clontech) were maintained in DMEM (Sigma) supplemented with 10% (vol/vol) FBS, 4mM L-glutamine, 100 μg/ml G418, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco BRL). Cells were transiently transfected with the plasmids using Lipofectamine (Invitrogen), cultured for an additional 16 h, and then lysed in 1 ml nondenaturing lysis buffer with 1 μg/ml protease inhibitors (pepstatin, aprotinin, leupeptin) (Cell Signaling Technology). After preclearing with mouse IgG and protein-A–coupled Sepharose beads for 10 min at 25°C, cell lysates were immunoprecipitated with anti-green fluorescent protein (GFP) mAb (Abcam, Cambridge, MA) or anti-Hck and then analyzed by Western analysis with Hck and RPS3 antibodies.
Statistics.
Data were analyzed with SigmaStat v 3.1 (Jandel Scientific, San Rafael, CA) using one-way ANOVA with Student-Newman-Keuls method for pairwise comparisons unless otherwise noted; p < 0.05 was considered significant.
RESULTS
DON-Induced IL-8 Expression Is Hck-Dependent in U937 Cells
As reported by Gray et al. (2008), PKR inhibitors, 2-AP and C16, suppressed DON-induced IL-8 mRNA expression and protein expression (Supplementary fig. S1). To determine if Hck also played a role in DON-induced IL-8 mRNA expression, U937 cells were treated with the Src family inhibitor PP2 (2.5μM). DON-induced IL-8 mRNA was significantly inhibited by PP2 pretreatment of (Fig. 1). DON-induced IL-8 protein expression was suppressed in U937 cells cotreated with the p38 inhibitor SB203580 (Supplementary fig. S2). Pretreatment with either PKR (2-AP; Fig. 2A) or Hck (PP2) (Fig. 2B) inhibitors markedly suppressed DON-induced p38 phosphorylation of p38.
FIG. 1.

Hck inhibition suppresses DON-induced IL-8 mRNA expression in U937 cells. Cells were pretreated with PP2 (2.5μM) or dimethyl sulfoxide vehicle (VEH) for 45 min before addition of 0 or 1000 ng/ml DON. IL-8 mRNA expression was measured by real-time PCR after a 6-h DON exposure. Data are mean ± SEM (n = 3). Asterisk indicates significantly different than VEH (p < 0.05). Representative of three independent experiments.
FIG. 2.

PKR and Hck inhibition suppresses DON-induced p38 phosphorylation in U937 cells. Cells were incubated for 45 min with (A) 2-AP (5.0mM) or water vehicle or (B) PP2 (0 or 2.5μM) or with dimethyl sulfoxide vehicle before treating with 0 or 500 ng/ml DON for 15 min. Protein in cell lysate was analyzed by Western blotting for p38 and phospho-p38. Representative of three independent experiments.
PKR inhibitor results were confirmed using U937 cells stably transfected with either an expression plasmid constitutively expressing antisense PKR (U9K-A1) or an empty expression plasmid (U9K-C2). U9K-A1 cells exhibited reduced DON-induced p38 phosphorylation as compared to the U9K-C2 control cells (Fig. 3A). Pretreatment with PP2 (0.25–25μM) ablated DON-induced p38 phosphorylation in U9K-C2 and the residual p38 phosphorylation in U9K-A1 cells. U9K-A1 cells had significantly reduced levels of DON-induced IL-8 protein, as compared to the U9K-C2 control cells (Fig. 3B). DON-induced IL-8 protein expression in U9K-A1 cells was decreased further upon treatment with PP2 (0.25–2.5μM).
FIG. 3.

PKR antisense expression and Hck inhibition suppress DON-induced p38 phosphorylation and IL-8 production in U937 cells. U937 cells expressing control (U9K-C2) or PKR antisense vector (U9K-A1) were pretreated with PP2 (0.25–25μM) or dimethyl sulfoxide vehicle for 45 min before treating with 0 or 500 ng/ml DON. (A) Cells were lysed with SDS after 30 min DON treatment and proteins analyzed by Western blotting for phospho-p38. (B) Culture supernatant was collected after a 6-h DON treatment, and IL-8 protein was assessed by ELISA. Data are mean ± SEM (n = 3). Bars without same letter differ (p < 0.05). Representative of three independent experiments.
PKR and Hck Interact with the 40S Ribosomal Subunit in U937 Cells
DON has previously been shown to induce p38 mobilization to the 40S subunit where it is then phosphorylated (Bae and Pestka, 2008). While PKR is known to associate with the 40S ribosomal subunit (Zhu et al., 1997), the capacity of Hck to interact with specific ribosomal subunits is unknown. To address this question, U937 cells were treated with DON or vehicle for 1 min lysates fractionated on a sucrose density gradient and fractions subjected to Western analysis. Those fractions containing small (40S) and large (60S) ribosomal subunits were confirmed using antibodies against RPS6 and RPL7, respectively (Fig. 4). PKR and Hck were constitutively found in the 40S ribosomal fraction, and both were phosphorylated within 1 min after DON treatment. Comparatively, little or no PKR or Hck was detectable in the 60S ribosomal unit fractions.
FIG. 4.

DON induces phosphorylation of ribosome-associated PKR and Hck in U937 cells. Cultures were treated with DON (0 or 500 ng/ml) for 1 min and subjected to sucrose density gradient fractionation. Fractions were analyzed by Western blotting. Lanes are aligned with density fractions depicted in above blot. Data are representative of three separate experiments.
Functional PKR Is Required for Hck Interaction with the 40S Ribosomal Subunit in U937 Cells
U9K-C2 control and U9K-A1 PKR antisense-containing cells were used to determine whether PKR affected Hck interaction with the ribosome. While Hck was found in lysates of both cell lines, PKR was only detectable in U9K-C2 lysates (Fig. 5A). Following sucrose gradient fractionation, PKR and Hck comigrated with the 40S ribosomal fraction in U9K-C2 control cells (Fig. 5B) but were nearly undetectable in the U9K-A1 40S fraction (Fig. 5C).
FIG. 5.
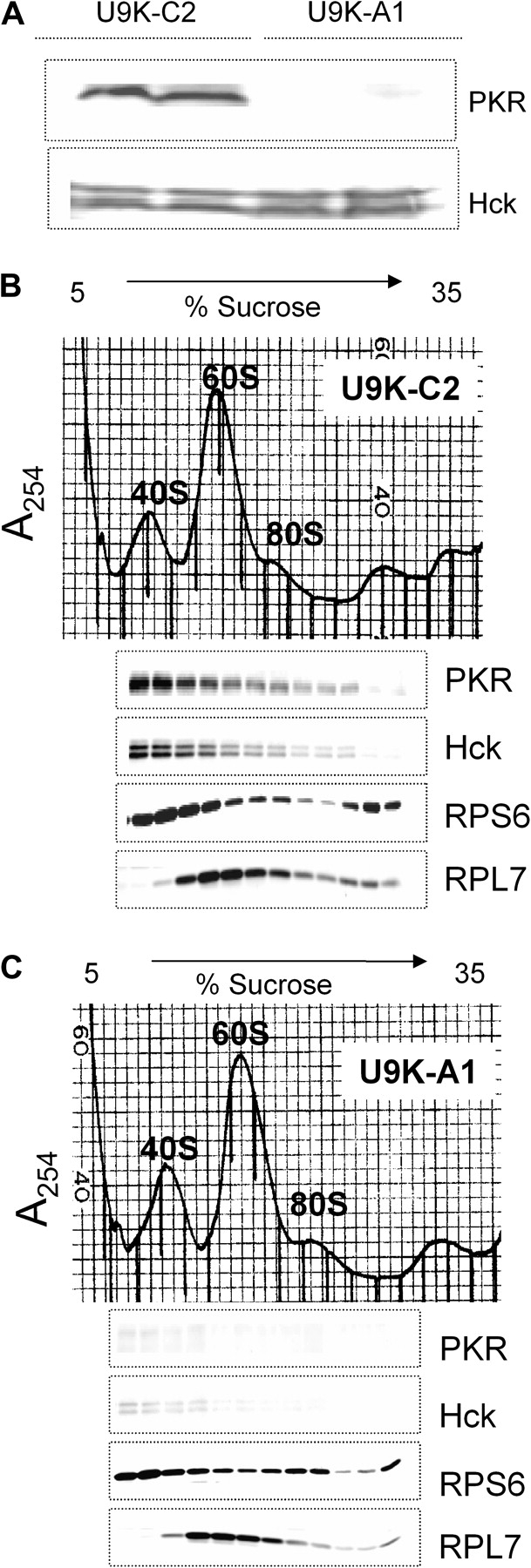
PKR is required for Hck interaction with the ribosome in U937 cells. (A) U9K-C2 control and U9K-A1 PKR-antisense expressing cells were lysed with polysome extraction buffer, and cytoplasmic fractions were collected after centrifugation at 10,000 × g for 15 min. Protein was analyzed by Western blotting with PKR and Hck antibodies. (B) U9K-C2 or (C) U9K-A1 cells were treated with 0 or 500 ng/ml DON for 1 min. Ribosomal fractions were separated by sucrose gradient, and protein was analyzed by Western blotting. Lanes are aligned with density fraction profile shown above. Data are representative of three separate experiments.
Ribosomal Protein S3 Interacts with Hck in Transfected HeLa Cells
The PXXP motif is critical for interaction of proteins with Src family kinases such as Hck. Bioinformatic analysis was used to determine numbers and location of PXXP motifs contained in over 70 known RPs (Fig. S3). Of these, RPS3 was found to contain the largest number (i.e., five) of PXXP motifs—all of which were located in close proximity to one another, suggesting this protein to be a strong candidate for mediating Hck interaction with the 40S subunit. To ascertain whether RPS3 could directly interact with Hck, GFP-labeled RPS3 and Hck were expressed in HeLa cells, which do not express native Hck (Fig. 6). Direct interaction of RPS3 and Hck was suggested upon Western blotting of anti-GFP or anti-Hck immunoprecipitates with anti-RPS3 or anti-Hck.
FIG. 6.

Hck interacts with the RPS3. RPS3-GFP and Hck were overexpressed in HeLa cells. Lysates were immunoprecipitated with anti-GFP or Hck and analyzed by Western blotting with antibodies specific for Hck or RPS3. Results are representative of two separate experiments.
DON-Induced p38 Interaction with the Ribosome and Phosphorylation Suppressed in Macrophages from PKR-Deficient Mice
Peritoneal macrophages from WT and PKR KO mice were treated with DON, and relative p38 phosphorylation was compared in cell lysates from the two cultures (Fig. 7A). Macrophages from PKR KO mice exhibited less DON-induced p38 phosphorylation than did macrophages from WT mice. The p38 association with the pooled ribosomal subunits and monosome fractions was increased in peritoneal macrophages from WT mice following DON treatment, whereas p38 interaction with the pooled ribosome fraction was not present in macrophages from PKR KO mice (Fig. 7B). Similarly, DON-induced p38 phosphorylation increased in the pooled ribosomal fraction of macrophages from WT but not in those from PKR KO mice.
FIG. 7.

DON-induced MAPK interaction with the ribosome is suppressed in peritoneal macrophages from PKR-deficient mice. Peritoneal macrophages from WT and PKR KO mice were cultured with 0 or 500 ng/ml DON for 15 min. (A) Cells were lysed and analyzed by Western blotting. (B) Ribosomal proteins were separated by sucrose density gradient fractionation and pooled ribosomal fractions analyzed by Western blotting. Data are representative of three separate experiments.
PKR Is Also Required for Hck Interaction with Ribosome and Phosphorylation in RAW 264.7 Murine Macrophages
The role of PKR in phosphorylation of ribosome-associated Hck was further confirmed in RAW 264.7 macrophages that were pretreated with either PKR or Hck inhibitor and then stimulated with DON. Following sucrose density gradient separation, the pooled ribosomal fraction was subjected to Western blotting. Phosphorylated Hck was not detectable in pooled ribosomal fractions from RAW 264.7 cells treated with 2-AP or PP2 (Fig. 8). RPL7 detection again confirmed the presence of equal amounts of sample in each lane.
FIG. 8.

PKR inhibition suppresses DON-induced Hck phosphorylation in ribosomal fractions of RAW 264.7 cells. Cells were treated with PKR inhibitor, 2-AP (5mM), or HCK inhibitor, PP2 (2.5μM), for 45 min before adding DON (250 ng/ml) for 5 min. Ribosomes were fractionated on sucrose gradient, and pooled ribosomal fractions analyzed by Western blotting. Data are representative of three separate experiments.
DON-Induced PKR and Hck Phosphorylations Occur with Onset of p38-Ribososme Association in RAW 264.7 Cells
Phosphorylated PKR and Hck were observed in the pooled ribosome fraction after 5 min of DON treatment in RAW 264.7 cells but disappeared within 15 min (Fig. 9); p38 associated with the RS+M fractions at 5, 15, and 30 min after DON exposure times. Equal protein loading in the pooled ribosome fractions was again confirmed using RPL7 antibody.
FIG. 9.

PKR and Hck phosphorylation precedes p38 interaction with the ribosome in RAW 264.7 cells. Cells were treated with 250 ng/ml DON for 0, 5, 15, or 30 min. Ribosomes were fractionated on sucrose gradient, and pooled ribosomal fractions analyzed by Western blotting with antibodies against phospho-PKR, phospho-Hck, p38, or RPL7. Data are representative of three separate experiments.
DON-Induced Phosphorylation of p38 and Its Upstream Kinases Are Dependent on PKR and Hck in RAW 264.7 Cells
Apoptosis signal-regulating kinase 1 (ASK1) and mitogen-activated protein kinase 3/6 (MKK3/6) are kinases known to be upstream of p38. DON was found to induce ASK1 and MKK3/6 phosphorylation in RAW 264.7 cells (Fig. 10A and B). Both the PKR inhibitor 2-AP (Fig. 10A) and the Hck inhibitor, PP2 (Fig. 10B), suppressed DON-induced ASK1 and MKK3/6 phosphorylation. ASK1 and MKK3/6 might thus serve as signaling cascade components that link PKR and/or Hck with p38.
FIG. 10.

PKR and Hck inhibition suppresses DON-induced phosphorylation of ASK1, MKK3/6, and p38 in RAW 264.7 cells. Cells were incubated for 45 min with (A) 2-AP (5mM) or dimethyl sulfoxide vehicle or with (B) PP2 (2.5μM) or dimethyl sulfoxide vehicle prior to treating with 0 or 250 ng/ml DON for 15 min. Cell lysates were analyzed by Western blotting with specific antibodies. Data are representative of three separate experiments.
DISCUSSION
While PKR is known to associate with the ribosome, this is the first report that Hck interacts with the 40S ribosomal subunit and that this occurs in a PKR-dependent fashion. Both of the latter proteins appear to be required for optimal DON-induced p38 activation and p38-driven expression of IL-8 and potentially other proinflammatory genes. Furthermore, upon DON treatment, PKR and Hck were rapidly and transiently phosphorylated while in association with the ribosome, which is consistent with previous findings in whole-cell lysates in RAW 264.7 and U937 cells (Zhou et al., 2003, 2005b). The results support the contention that 40S subunit serves as a scaffold for Hck and PKR. A putative kinase signaling cascade associated with the ribosome scaffold following DON exposure is depicted in Figure 11.
FIG. 11.
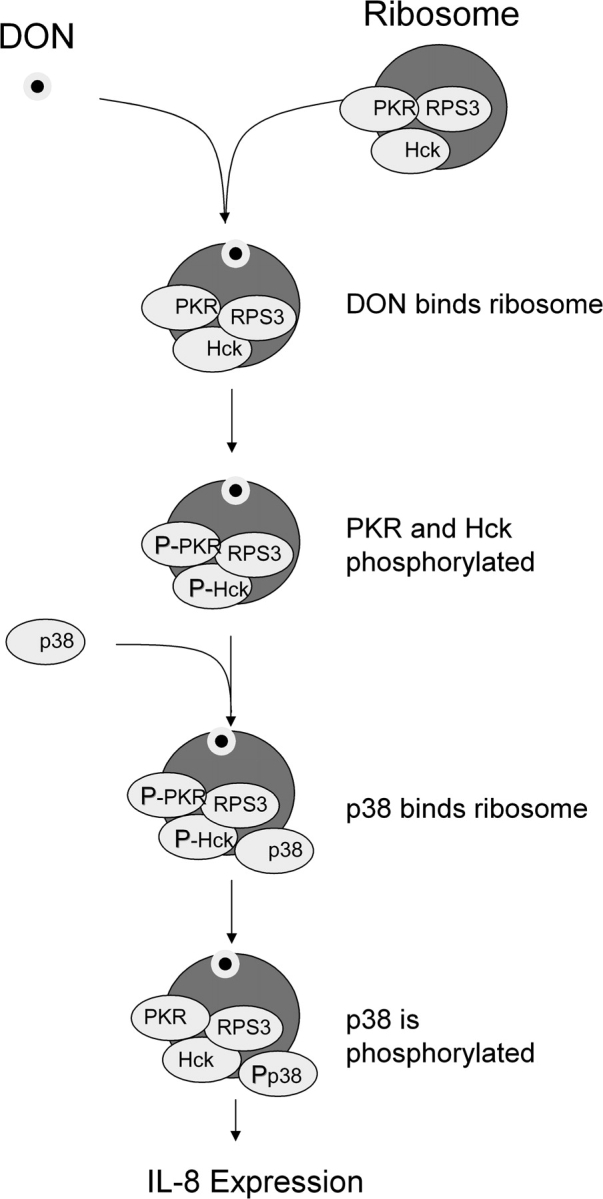
Ribosome functions as scaffold for PKR, Hck, and p38 in DON-induced ribotoxic stress response. DON-induced ribotoxic is proposed to involve: (1) rapid DON uptake and binding to ribosome, (2) activation of ribosomal-associated PKR and Hck, (3) interaction of p38 with the ribosome, (4) p38 phosphorylation, and (5) induction of proinflammatory genes such as IL-8.
The observed colocalization of PKR and Hck to the 40S subunit suggests that these kinases might associate in close proximity. This possibility is supported by the observation that Hck was undetectable in the pooled ribosomal fraction of U9K-A1 cells even though cytoplasmic Hck expression was unaffected by PKR deficiency. Since the PKR inhibitor 2-AP suppressed Hck phosphorylation in the ribosome in RAW 264.7 cells, its activation might be essential for DON-induced Hck phosphorylation.
Several plausible mechanisms might exist to explain how PKR and Hck associate with the ribosome. Zhu et al. (1997) observed that human PKR is primarily localized in the 40S ribosome when the protein is overexpressed in yeast. When PKR is mutated in the DRBD region, it fails to interact with the ribosome, suggesting that PKR interacts with the ribosome via a DRBD. In contrast to PKR, the association of Hck or other Srcs with the ribosome has been heretofore unreported. The SH3 domain of Hck is known to facilitate interaction with several cellular proteins containing PXXP-binding motifs (Gouri and Swarup, 1997). For example, Bruton's tyrosine kinase, open reading frame 3 protein of hepatitis E virus, and Nef protein of HIV interact with SH3 domains of Hck. (Cheng et al., 1994; Collette et al., 2000; Korkaya et al., 2001; Stangler et al., 2007). Prototypical Hck-binding sites contain proline-rich regions with PXXP motifs being particularly critical (Stangler et al., 2007). Upon characterizing potential binding sites of Hck to over 70 RPs, we found that RPS3 was a strong candidate because it contains PXXP motifs, all of which were localized in proline-rich region thus enhancing its potential to bind to the SH3 domain. The capacity of Hck to interact with this candidate protein was strongly suggested following overexpression of these proteins in HeLa cells and immunoprecipitation with specific antibodies. It is particularly notable that RPS3 interacts with eIF2. Since its subunit eIF2α is a well-known PKR substrate which regulates translation initiation (Samuel, 1993), it is reasonable to suggest that PKR and Hck bind to the 40S ribosomal subunit in close proximity.
We have previously demonstrated that p38 initially interacts with the 40S ribosomal subunit in DON-stimulated macrophages (Bae and Pestka, 2008). As observed here, peritoneal macrophages from PKR KO mice exhibit impaired p38 association with ribosome following DON treatment, and overall p38 phosphorylation was reduced in cell lysates from these cells. PKR might thus be critical for p38 ribosomal mobilization and phosphorylation in DON-induced macrophages. These findings correlate with the observations that PKR and Hck are transiently activated in the 40S ribosomal subunit and that both concurrent and subsequent p38 association is detectable in this fraction. Overall, these data are consistent with the possibility that the 40S ribosomal subunit functions as a scaffold for signal transduction during the DON-induced ribotoxic stress response thereby facilitating activation of p38 by PKR and Hck (Fig. 11).
A critical question arising from this work relates to the nature of the signaling cascade that links PKR/Hck and p38. PKR is known to interact with ASK1, a member of the MAPK kinase family, which can activate p38 via MKK3/6. Coimmunoprecipitation studies have demonstrated that PKR associates with ASK1 (Takizawa et al., 2002). Dominant-negative PKR-expressing COS-1 monkey kidney fibroblast cells exhibit suppressed ASK1-induced p38 activation and apoptosis (Matsukawa et al., 2004; Takizawa et al., 2002). In addition to ASK1, Silva et al. (2004) demonstrated that the PKR interacts with MKK6 thereby providing a plausible mechanism for regulating p38 MAPK activation in response to dsRNA stimulation. As shown here, DON-induced ASK1 and MKK3/6 phosphorylation in RAW 264.7 cells was suppressed by the PKR inhibitor, 2-AP. Analogous to the observations for PKR, inhibition of Hck suppressed ASK1 and MKK3/6 phosphorylation. Thus, PKR and Hck might participate in a signaling cascade involving ASK1 and MKK3/6 that mediates p38 activation. Further studies are needed to determine if ASK1 and MKK3/6 also interact with the ribosome and whether they are required for downstream p38 phosphorylation.
It is not yet understood how PKR activation is triggered upon binding of DON to the ribosome. Recently, we observed that both DON and another trichothecene, satratoxin G, associate in noncovalent fashion with both 40S and 60S ribosomal subunits (Bae et al., 2009). The peptidyl transferase ring has been suggested to be a critical binding site for ribotoxins, such as trichothecenes, anisomycin, ricin, and Shiga toxin; and it has been further proposed that these agents promote damage to the peptidyl transferase ring (Foster and Tesh, 2002 and Iordanov et al., 1997). Although DON and the trichothecene T-2 toxin do not directly depurinate or cleave 28S rRNA under cell-free conditions, these toxins induce 18S and 28S rRNA cleavage in RAW 264.7 cells (Li and Pestka, 2008), which might imply an indirect, toxin-facilitated mechanism. Indeed, 28S rRNA cleavage has been mapped using primer extension to the peptidyl transferase region. However, since PKR, Hck, and p38 localize to the 40S ribosomal subunit, a comparable analysis needs to be performed on 18S rRNA. Future investigations will focus on determining how DON binding to the 40S ribosomal subunit induces PKR and Hck activation via the ribosomal interaction.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes for Health (Public Health Service grants ES03553 and DK058833).
Supplementary Material
Acknowledgments
We thank Hag Dong Kim, Hui-Ren Zhou, Hyeonju Cho, Dara Phillips, Chidozie Amuzie, Eleni Beli, and Mary Rosner for technical assistance.
References
- Arold ST, Ulmer TS, Mulhern TD, Werner JM, Ladbury JE, Campbell ID, Noble ME. The role of the Src homology 3-Src homology 2 interface in the regulation of Src kinases. J. Biol. Chem. 2001;276:17199–17205. doi: 10.1074/jbc.M011185200. [DOI] [PubMed] [Google Scholar]
- Bae HK, Pestka JJ. Deoxynivalenol induces p38 interaction with the ribosome in monocytes and macrophages. Toxicol. Sci. 2008;105:59–66. doi: 10.1093/toxsci/kfn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae HK, Shinozuka J, Islam Z, Pestka JJ. Satratoxin G interaction with 40S and 60S ribosomal subunits precedes apoptosis in the macrophage. Toxicol. Appl. Pharmacol. 2009;237:137–145. doi: 10.1016/j.taap.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran S, Barber GN. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol. Biol. 2007;383:277–301. doi: 10.1007/978-1-59745-335-6_18. [DOI] [PubMed] [Google Scholar]
- Cheng G, Ye ZS, Baltimore D. Binding of Bruton's tyrosine kinase to Fyn, Lyn, or Hck through a Src homology 3 domain-mediated interaction. Proc. Natl. Acad. Sci. U.S.A. 1994;91:8152–8155. doi: 10.1073/pnas.91.17.8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung BK, Lee DC, Li JC, Lau YL, Lau AS. A role for double-stranded RNA-activated protein kinase PKR in Mycobacterium-induced cytokine expression. J. Immunol. 2005;175:7218–7225. doi: 10.4049/jimmunol.175.11.7218. [DOI] [PubMed] [Google Scholar]
- Collette Y, Arold S, Picard C, Janvier K, Benichou S, Benarous R, Olive D, Dumas C. HIV-2 and SIV nef proteins target different Src family SH3 domains than does HIV-1 Nef because of a triple amino acid substitution. J. Biol. Chem. 2000;275:4171–4176. doi: 10.1074/jbc.275.6.4171. [DOI] [PubMed] [Google Scholar]
- Foster GH, Tesh VL. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-alpha. J. Leukoc. Biol. 2002;71:107–114. [PubMed] [Google Scholar]
- Galban S, Martindale JL, Mazan-Mamczarz K, Lopez de Silanes I, Fan J, Wang W, Decker J, Gorospe M. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol. Cell Biol. 2003;23:7083–7095. doi: 10.1128/MCB.23.20.7083-7095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouri BS, Swarup G. Interaction of SH3 domain of Hck tyrosine kinase with cellular proteins containing proline-rich regions: evidence for modulation by unique domain. Indian J. Biochem. Biophys. 1997;34:29–39. [PubMed] [Google Scholar]
- Gray JS, Bae HK, Li JC, Lau AS, Pestka JJ. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, Shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 2008;105:322–330. doi: 10.1093/toxsci/kfn128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JS, Pestka JJ. Transcriptional regulation of deoxynivalenol-induced IL-8 expression in human monocytes. Toxicol. Sci. 2007;99:502–511. doi: 10.1093/toxsci/kfm182. [DOI] [PubMed] [Google Scholar]
- Guiet R, Poincloux R, Castandet J, Marois L, Labrousse A, Le C, V, Maridonneau-Parini I. Hematopoietic cell kinase (Hck) isoforms and phagocyte duties—from signaling and actin reorganization to migration and phagocytosis. Eur. J. Cell Biol. 2008;87:527–542. doi: 10.1016/j.ejcb.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Hsu LC, Park JM, Zhang K, Luo JL, Maeda S, Kaufman RJ, Eckmann L, Guiney DG, Karin M. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428:341–345. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, Magun BE. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam Z, Gray JS, Pestka JJ. p38 Mitogen-activated protein kinase mediates IL-8 induction by the ribotoxin deoxynivalenol in human monocytes. Toxicol. Appl. Pharmacol. 2006;213:235–244. doi: 10.1016/j.taap.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Kim HD, Lee JY, Kim J. Erk phosphorylates threonine 42 residue of ribosomal protein S3. Biochem. Biophys. Res. Commun. 2005;333:110–115. doi: 10.1016/j.bbrc.2005.05.079. [DOI] [PubMed] [Google Scholar]
- Korkaya H, Jameel S, Gupta D, Tyagi S, Kumar R, Zafrullah M, Mazumdar M, Lal SK, Xiaofang L, Sehgal D, et al. The ORF3 protein of hepatitis E virus binds to Src homology 3 domains and activates MAPK. J. Biol. Chem. 2001;276:42389–42400. doi: 10.1074/jbc.M101546200. [DOI] [PubMed] [Google Scholar]
- Lemaire PA, Tessmer I, Craig R, Erie DA, Cole JL. Unactivated PKR exists in an open conformation capable of binding nucleotides. Biochemistry. 2006;45:9074–9084. doi: 10.1021/bi060567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Pestka JJ. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008;105:811–818. doi: 10.1093/toxsci/kfn111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J. Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- Moon Y, Pestka JJ. Vomitoxin-induced cyclooxygenase-2 gene expression in macrophages mediated by activation of ERK and p38 but not JNK mitogen-activated protein kinases. Toxicol. Sci. 2002;69:373–382. doi: 10.1093/toxsci/69.2.373. [DOI] [PubMed] [Google Scholar]
- Moon Y, Uzarski R, Pestka JJ. Relationship of trichothecene structure to COX-2 induction in the macrophage: selective action of type B (8-keto) trichothecenes. J. Toxicol. Environ. Health A. 2003;66:1967–1983. doi: 10.1080/713853950. [DOI] [PubMed] [Google Scholar]
- Pellicena P, Stowell KR, Miller WT. Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J. Biol. Chem. 1998;273:15325–15328. doi: 10.1074/jbc.273.25.15325. [DOI] [PubMed] [Google Scholar]
- Pestka JJ, Smolinski AT. Deoxynivalenol: toxicology and potential effects on humans. J. Toxicol. Environ. Health B Crit. Rev. 2005;8:39–69. doi: 10.1080/10937400590889458. [DOI] [PubMed] [Google Scholar]
- Quintrell N, Lebo R, Varmus H, Bishop JM, Pettenati MJ, Le Beau MM, Diaz MO, Rowley JD. Identification of a human gene (HCK) that encodes a protein-tyrosine kinase and is expressed in hemopoietic cells. Mol. Cell Biol. 1987;7:2267–2275. doi: 10.1128/mcb.7.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 2007;316:253–292. doi: 10.1007/978-3-540-71329-6_13. [DOI] [PubMed] [Google Scholar]
- Samuel CE. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 1993;268:7603–7606. [PubMed] [Google Scholar]
- Scheuner D, Gromeier M, Davies MV, Dorner AJ, Song B, Patel RV, Wimmer EJ, McLendon RE, Kaufman RJ. The double-stranded RNA-activated protein kinase mediates viral-induced encephalitis. Virology. 2003;317:263–274. doi: 10.1016/j.virol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Shifrin VI, Anderson P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999;274:13985–13992. doi: 10.1074/jbc.274.20.13985. [DOI] [PubMed] [Google Scholar]
- Silva AM, Whitmore M, Xu Z, Jiang Z, Li X, Williams BR. Protein kinase R (PKR) interacts with and activates mitogen-activated protein kinase kinase 6 (MKK6) in response to double-stranded RNA stimulation. J. Biol. Chem. 2004;279:37670–37676. doi: 10.1074/jbc.M406554200. [DOI] [PubMed] [Google Scholar]
- Stangler T, Tran T, Hoffmann S, Schmidt H, Jonas E, Willbold D. Competitive displacement of full-length HIV-1 Nef from the Hck SH3 domain by a high-affinity artificial peptide. Biol. Chem. 2007;388:611–615. doi: 10.1515/BC.2007.075. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Tatematsu C, Nakanishi Y. Double-stranded RNA-activated protein kinase interacts with apoptosis signal-regulating kinase 1. Implications for apoptosis signaling pathways. Eur. J. Biochem. 2002;269:6126–6132. doi: 10.1046/j.1432-1033.2002.03325.x. [DOI] [PubMed] [Google Scholar]
- Trible RP, Emert-Sedlak L, Smithgall TE. HIV-1 Nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J. Biol. Chem. 2006;281:27029–27038. doi: 10.1074/jbc.M601128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, Staschke KA, Wek RC. Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: Role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2) Eur. J. Biochem. 2001;268:3674–3684. doi: 10.1046/j.1432-1327.2001.02273.x. [DOI] [PubMed] [Google Scholar]
- Wu S, Kaufman RJ. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem. 1997;272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- Yadav SS, Miller WT. Cooperative activation of Src family kinases by SH3 and SH2 ligands. Cancer Lett. 2007;257:116–123. doi: 10.1016/j.canlet.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang GH, Jarvis BB, Chung YJ, Pestka JJ. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol. Appl. Pharmacol. 2000;164:149–160. doi: 10.1006/taap.1999.8888. [DOI] [PubMed] [Google Scholar]
- Zhou HR, Islam Z, Pestka JJ. Induction of competing apoptotic and survival signaling pathways in the macrophage by the ribotoxic trichothecene deoxynivalenol. Toxicol. Sci. 2005a;87:113–122. doi: 10.1093/toxsci/kfi234. [DOI] [PubMed] [Google Scholar]
- Zhou HR, Jia Q, Pestka JJ. Ribotoxic stress response to the trichothecene deoxynivalenol in the macrophage involves the SRC family kinase Hck. Toxicol. Sci. 2005b;85:916–926. doi: 10.1093/toxsci/kfi146. [DOI] [PubMed] [Google Scholar]
- Zhou HR, Lau AS, Pestka JJ. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003;74:335–344. doi: 10.1093/toxsci/kfg148. [DOI] [PubMed] [Google Scholar]
- Zhu S, Romano PR, Wek RC. Ribosome targeting of PKR is mediated by two double-stranded RNA-binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J. Biol. Chem. 1997;272:14434–14441. doi: 10.1074/jbc.272.22.14434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.