

Abstract
An intracellularly expressed defective human immunodeficiency virus type-1 (HIV-1) protease (PR) monomer could function as a dominant-negative inhibitor of the enzyme that requires dimerization for activity. Based on in silico studies, two mutant PRs harboring hydrophilic mutations, capable of forming favorable inter- and intra-subunit interactions, were selected: PRRE containing Asp25Arg and Gly49Glu mutations, and PRRER containing an additional Ile50Arg mutation. The mutants were expressed and tested by PR assays, nuclear magnetic resonance (NMR) and cell culture experiments. The mutant PRs showed dose-dependent inhibition of the wild-type PR in a fluorescent microtiter plate PR assay. Furthermore, both mutants were retained by hexahistidine-tagged wild-type HIV-1 PR immobilized on nickel-chelate affinity resin. For the first time, heterodimerization between wild-type and dominant-negative mutant PRs were also demonstrated by NMR spectroscopy. 1H–15N Heteronuclear Single Quantum Coherence NMR spectra showed that although PRRE has a high tendency to aggregate, PRRER exists mainly as a folded monomer at 25–35 µM concentration, but in the presence of wild-type PR in a ratio of 1:1, heterodimerization occurs with both mutants. While the recombinant virus containing the PRRE sequence showed only very low level of expression, expression of the viral proteins of the virus with the PRRER sequence was comparable with that of the wild-type. In cell culture experiments, infectivity of viral particles containing PRRER protein was reduced by 82%, at mutant to wild-type infective DNA ratio of 2:1.
Keywords: dimerization, human immunodeficiency virus-1 protease, macromolecular inhibitor, NMR, trans-dominant effect
Introduction
The human immunodeficiency virus type 1 (HIV-1) protease (PR) is a target for chemotherapy, because of its critical functions in the life cycle of retroviruses (Oroszlan and Luftig, 1990; Tözsér and Oroszlan, 2003). The PR is a homodimeric aspartic PR with an extended binding site capable of recognizing at least seven residues of the substrate (Oroszlan and Luftig, 1990; Louis et al., 2000). The initial targeting of the PR focused on small molecule inhibitors, especially on substrate-based peptidomimetic inhibitors: all HIV-1 PR inhibitors currently used in therapy belong to this class (Wlodawer, 2002; Randolph and DeGoey, 2004). However, resistance against these drugs develops rapidly, and emerging virus strains are often cross-resistant to other PR inhibitors (Swanstrom and Eron, 2000; Barbaro et al., 2005). Alternative strategies have also been suggested, like the development of non-peptidomimetic compounds targeting the active site (Yehia et al., 2004) or functionally important sites other than the active site, i.e. the dimerization interface (Schramm et al., 1991; Schramm et al., 1999; Caflisch et al., 2000; Hwang and Chmielewski, 2004; Hwang and Chmielewski, 2005) or the flap region (Rezacova et al., 2002; Sperka et al., 2005). Furthermore, the PR may also be targeted by macromolecular inhibitors that could provide higher interaction surface and hence a lower chance for the development of resistance (Babe et al., 1995; McPhee et al., 1996; Rozzelle et al., 2000; Todd et al., 2000a). Designing defective PR monomers and expressing them in the infected cells has been demonstrated to be an effective method (Babe et al., 1995; Junker et al., 1996; McPhee et al., 1996; Rozzelle et al., 2000; Todd et al., 2000a; 2000b). Similar strategy worked for Gag (Trono et al., 1989; Shimano et al., 1999), Tat (Pearson et al., 1990; Modesti et al., 1991; Fraisier et al., 1998), Rev (Bevec et al., 1992; Liu et al., 1994), Env (Buchschacher et al., 1992; Steffy and Wong-Staal, 1993; Buchschacher et al., 1995; Chen et al., 1996) and Vpr (Sawaya et al., 2000) proteins of HIV-1. Small molecule PR inhibitors contribute substantially to the side effects of highly active anti-retroviral therapy (HAART): insulin resistance, dyslipidemia, lipodystrophy, atherosclerosis (Tözsér, 2001; Rudich et al., 2005). Because of the different chemical nature of these macromolecular inhibitors, they are not expected to exert the abovementioned side effects.
Studies on trans-dominant negative HIV-1 PR inhibitors were pioneered by Craik and co-workers (Babe et al., 1995; Junker et al., 1996; McPhee et al., 1996; Rozzelle et al., 2000; Todd et al., 2000a; 2000b). They showed that a mutant form of the PR carrying Asp25Lys, Gly49Trp and Ile50Trp mutations (PRKWW) was the most potent inhibitor for the wild-type (wt) PR in vitro and in cell culture experiments. The substitution of the catalytic aspartate by a positively charged Lys may form a favorable ion-pair with the negatively charged catalytic Asp in the heterodimeric PR, while bulky Trp residues may interact with the hydrophobic S1 and S2' subsites and partially fill the substrate-binding site. These residues are not only expected to promote stronger interactions for wt:PRKWW heterodimer when compared with those for wt:wt homodimer in the absence of substrate or inhibitor, but are also expected to prevent the formation of PRKWW:PRKWW homodimeric form, because of steric and electrostatic repulsions. Proteins having multiple substitutions are preferred over single mutants because of lower risk of in vivo selection to the wild-type revertant form during a gene therapy application.
Here, we present a new strategy for the design of macromolecular inhibitors of HIV-1 PR. The hydrophobic residues introduced to fill up the substrate-binding site of the PR in a substrate-like manner may cause problems during in vitro and in vivo applications. The increased hydrophobicity of the surface of the defective monomer may decrease the solubility of the protein and may increase protein aggregation both in vitro and in vivo, as well as depletion of chaperone proteins. Therefore, hydrophilic residues were preferred in our mutagenesis study of the PR to create defective monomers with improved physicochemical characteristics. Here, we present the design and characterization of mutant forms of HIV-1 PR containing Asp25Arg and Gly49Glu mutations (PRRE), and Asp25Arg, Gly49Glu and Ile50Arg mutations (PRRER).
Materials and methods
Molecular modeling
Mutated residues were introduced into a high-resolution crystal structure of HIV-1 PR (Mahalingam et al., 2002) or into the previously minimized structures (see below) with Sybyl program (Tripos Inc., St Louis, MO, USA). All 20 natural amino acids were changed in the selected position of monomer B. Five hundred Powell iteration steps were applied for the mutated structures using AMBER all atoms force field (Weiner et al., 1986) with 8 Å cutoff. The distance-dependent dielectric constant was set to four. Intermonomer interaction energy was calculated for each mutated structure as the difference of the total energy of the dimer and the sum of the total energies of each monomer. Based on favorable inter- and intramolecular energies, mutants were selected and subjected to the next round of in silico mutagenesis at a different position. The structures were examined on a Silicon Graphics Fuel computer graphics system.
Site-directed mutagenesis of recombinant HIV-1 protease
The construction of the mature HIV-1 PR containing five stabilizing mutations (Q7K, L33I, L63I, C67A and C95A) was described previously (Louis et al., 1999; Mahalingam et al., 1999). The kinetic parameters of this enzyme, designated as wild-type HIV-1 PR, are indistinguishable from that of the native PR (Louis et al., 1999; Mahalingam et al., 1999). Substitution mutations were carried out using the PR template, QuikChange mutagenesis protocol (Stratagene, La Jolla, CA, USA) and appropriate primers (synthesized by Integrated DNA Technologies, Coralville, IA, USA). Mutations were verified by DNA sequencing performed with ABI Prism dye terminator cycle sequencing kit and a model 3100-Avant Genetic Analyzer (both from Applied Biosystems, Foster City, CA, USA).
Protein expression and purification
Freshly prepared Escherichia coli BL21 (DE3) culture of 500 ml bearing one of the plasmid constructs was grown at 37°C upto an absorbance of 0.7–1.0 at 600 nm, in Luria-Bertani medium containing 100 µg/ml ampicillin, and induced for expression with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3.5 h. Cells were harvested by centrifugation at 2000 g for 20 min at 4°C. After removal of the supernatant, the cell pellet was suspended in 30 ml of lysis buffer [50 mM Tris, 1 mM ethylene diaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 0.5% Triton X-100, pH 8.2)], and lysed by sonication on ice (Branson Sonifier 450, Branson Ultrasonic Corporation, Danbury, CT, USA). The lysate was centrifuged at 9000 g for 20 min at 4°C, suspended in lysis buffer containing 3 M urea and sonicated again. This washing step was repeated three times to obtain purified inclusion bodies. The final pellet was dissolved in denaturation buffer (50 mM Tris, 5 mM EDTA, 5 mM DTT, 8 M urea, pH 8.0) and filtered through 0.22 µm pore size filter (Millipore, Billerica, MA, USA). Proteins were purified by high-performance liquid chromatography (HPLC) on a POROS 20R2 high-performance perfusion chromatography column (PerSeptive Biosystems Inc., Framingham, MA, USA) using acetonitrile gradient (0–100%) in water, in the presence of 0.05% trifluoroacetic acid. Purity of selected fractions was assessed by sodium dodecyl sulphate–polyacryl amide gel electrophoresis (SDS–PAGE) using 16% polyacrylamide gels. Solvents of the collected fractions containing the PR or a mutant PR were removed by SpeedVac Concentrator SVC 100H (Savant Instruments Inc., Farmingdale, NY, USA). Dried proteins were dissolved in 6 M guanidine-HCl. Protein concentrations were determined by the Bradford spectrophotometric method (Bradford, 1976) using bovine serum albumin (Sigma–Aldrich, St Louis, MO, USA) as a standard, and stock solutions with equal protein concentrations were prepared for each protein solution.
Fluorescent microtiter plate protease assay
PR assays were performed as described previously (Bagossi et al., 2004). Briefly, 10 µl of denaturated protein solution (0.91–9.1 µM) containing the active and inactive proteins in various ratios in 6 M guanidine-HCl solution was added into microtiter plate wells and mixed with 150 µl of buffer A (250 mM phosphate buffer, pH 5.6 containing 5% glycerol, 1 mM EDTA, 5 mM DTT, 500 mM NaCl) and 20 µl 1% poly(ethylene glycol) (PEG), dissolved in buffer A. The mixture was preincubated at 37°C for 15 min and the reaction was initiated by the addition of 20 µl of 9.55 µM fluorescent peptide substrate dissolved in buffer A. The RE(Edans)-SQNY↓PIVRK-(Dabcyl)R fluorescent peptide substrate (FSP-407; arrow indicates the cleavage site of HIV-1 PR) was synthesized by Dr Ivo Blaha (Ferring Leciva, Prague, Czech Republic). The increase in fluorescence was detected at 460 nm, using 355 nm excitation wavelength in a Wallac 1420 Victor2 fluorimeter-luminometer (Wallac Oy, Turku, Finland) at 37°C. Primary data analysis including inner filter correction was performed with the program KiDet (Bagossi et al., 2004). A truncated PR incapable of dimerization (PR5–95), bearing deletions of terminal residues 1–4 and 96–99 (Ishima et al., 2001; Ishima et al., 2007), was used as a control protein. Activity percentage relative to the control protein was calculated and plotted against the ratio of inactive:active proteins using the SigmaPlot program (Systat Software, Inc., Point Richmond, CA, USA).
Detection of heterodimer formation by SDS–PAGE
Mutants and N-terminal hexahistidine-tagged wild-type PR [0.5 nmol, X28-PR (Wondrak and Louis, 1996)] in 6 M guanidine-HCl solution were mixed in 1:1 molar ratio or were applied alone in the case of control samples. They were diluted 20-fold in buffer D (50 mM sodium-acetate, pH 5.0 containing 100 mM NaCl, 5% glycerol, 1% Triton X-100) and were incubated at room temperature for 15 min. ProBond Ni-NTA agarose (Invitrogen Corp., Carlsbad, CA, USA) equilibrated in buffer D was added to the protein solution. The suspension was incubated on ice for 30 min. The resin was separated by centrifugation and washed three times with buffer D. SDS–PAGE sample buffer was added to the resin, and heated at 95°C for 10 min. The suspension was centrifuged and proteins in the supernatant were analyzed by SDS–PAGE using Coomassie Brilliant Blue staining.
Nuclear magnetic resonance spectroscopy
Uniformly 15N-labeled PRRE and PRRER proteins for NMR experiments were prepared by growing E. coli BL21 (DE3) bearing the appropriate plasmid in minimal media containing 15N-NH4Cl as the sole nitrogen source. Proteins, including unlabeled PR prepared in Luria-Bertani medium, were purified from inclusion bodies using an established protocol as described previously, involving size-exclusion chromatography under denaturing conditions followed by reverse-phase HPLC (Louis et al., 2003). Peak fractions (∼0.5 mg/ml) were stored in aliquots at −70°C. Proteins were dialyzed extensively against 30–50 mM formic acid or 7 mM HCl and concentrated to ∼2 mg/ml using Millipore YM-10 centriprep and centricon concentrators (Millipore, Beford, MA, USA) and stored at 4°C. Four samples (320 µl each), namely 15N-PRRE, 15N-PRRER and a 1:1 mixture of 15N-PRRE:unlabeled PR and 15N-PRRER:unlabeled PR, were prepared for acquiring 1H–15N Heteronuclear Single Quantum Coherence (HSQC) spectra. Proteins were prepared according to the quench protocol of protein folding (Ishima et al., 2007) to a final concentration of ∼35 µM (in monomer concentration) by mixing one volume of enzyme in 30–50 mM formic acid (or 7 mM HCl) with 1.3–2.3 volumes of 5 mM sodium acetate, pH 6.0, and then with 3–4 volumes of 100 mM sodium acetate, pH 5.8. In the case of mixed samples, the two proteins were mixed in a 1:1 ratio prior to folding. Spectra were recorded using a Bruker 600 MHz spectrometer equipped with a low temperature probe at 20°C.
Modification of plasmids of HIV-1 vector system
The original HIV-1 vector system (a kind gift from Dr Didier Trono, Department of Genetics and Microbiology, University of Geneva Medical School, Geneva, Switzerland) (Dull et al., 1998; Trono, 2007) was modified to introduce a more efficient promoter and to contain sequences of mutant PRs. Human cytomegalovirus (CMV) immediate-early promoter sequence was amplified by polymerase chain reaction (PCR) and was inserted between the BamHI and MluI restriction sites in the plasmid pWOX-GFP, upstream to the green fluorescent protein (GFP) initiation codon. The PR coding fragment from the pMDLg/pRRE was cloned between PstI and StyI restriction sites of pT7Blue-3 plasmid (Novagen, Madison, WI, USA). Site-directed mutagenesis was performed using the QuikChange protocol and the sequences were verified by DNA sequencing performed with ABI Prism Dye Terminator cycle sequencing kit and a model 3100-Avant Genetic Analyzer (both from Applied Biosystems). Mutated fragments were cloned back into the pMDLg/pRRE plasmid. N- and C-terminal truncations to mimic PR5–95 coding region were created by overlapping PCR(Ho et al.,1989). The oligonucleotide sequences are available at http://biochemistry.med.unideb.hu/SBBG/OligoDB. The desired sequences were verified by DNA sequencing.
Production of viral particles
Viral particles were produced by transient transfection into 293T cells, maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), using polyethylenimine (Sigma). Cells were grown to approximately 70% confluence and a total of 35.5 µg plasmid was used for the transfection of cells in T75 flasks: 10 µg pWOX-CMV-GFP (transfer vector plasmid), 6.5 µg pMDLg/pRRE (packaging plasmid containing wild-type PR sequence), 2.5 µg pRSV.rev (Rev coding plasmid), 3.5 µg pMD.G (VSV-G envelope protein-coding plasmid) and 13 µg pMDLg/pRRE-M (plasmid containing a mutant PR sequence) or salmon sperm DNA (Sigma), in DMEM containing 1% FBS. The medium was replaced after 5–7 h and the conditioned media containing virus particles were collected after another 24, 48 and 72 h, clarified by centrifugation, filtered through 0.45 µm polyvinylidene fluoride (PVDF) filter (Millipore, Billerica, MA, USA) and concentrated by ultracentrifugation (100 000 g, 2 h). The viral pellet was reconstituted in phosphate-buffered saline (PBS) solution, and stored at −70°C. To determine the total amount of capsid protein (p24) by enzyme-linked immunosorbent assay (Beckman Coulter, Fullerton, CA, USA), the uncleaved Gag was digested with PR after the virus lysis and the completeness of cleavage was verified by western blotting using an anti-CA antibody (a kind gift from Dr Robert Gorelick, Frederick, MD, USA).
Infectivity assay
For infection, 293T cells were seeded in 24-well plates in 300 µl DMEM supplemented with FBS, glutamine and antibiotics (all from Invitrogen). After overnight incubation, cells were infected with viral particles containing equal amount (24 ng) of capsid protein in a volume of 120 µl DMEM. After two days, the medium was completed with 120 µl DMEM containing double the amount of FBS, glutamine and antibiotics. On the seventh day, cells were scraped, washed with PBS and 10 000 cells were counted by flow cytometer (FACS Calibur, BD Biosciences, Franklin Lakes, NJ, USA) to determine the number of GFP-positive cells.
Results and discussion
In this work, we present a new strategy to design trans-dominant macromolecular inhibitors of HIV-1 PR by creating a hydrophilic surface at the active-site and flap interface. Our preliminary work with PRKWW and other mutants showed that substitutions of native residues to hydrophobic ones cause serious difficulties in the purification of the proteins, as a consequence of low solubility and increased tendency to aggregate. To fill in the substrate-binding pockets and to prevent binding of the natural substrates of the PR, hydrophobic residues are the first choice of logic, as the PR prefers hydrophobic P1 and P1' residues in all types of cleavage sites, and hydrophobic P2 and P2' residues in certain cleavage sites. However, increased hydrophobicity may cause increased tendency for aggregation or protein misfolding, a likely reason why attempts to crystallize constructs containing hydrophobic residues failed (Rozzelle et al., 2000) and tethered heterodimers showed the undesirable property of protein loss because of aggregation above ∼1 mg/ml concentration. These features might also be disadvantageous in vivo, when relatively high concentration of the mutant protein must be present to achieve the desired effect. The hydrophobic surface of the defective monomer may also excessively bind to chaperone proteins, which may cause unexpected adverse effects.
In silico mutagenesis of HIV-1 protease
Maximal interaction energy between wild-type PR and the inhibitory mutant is expected when the surface of the mutant monomer is similar to that of the substrate-bound wild-type monomer. However, it is very difficult to mimic properly the surface of a two-chain complex (PR subunit plus peptide substrate) with a single-chain mutant monomer, therefore we attempted only to prevent binding of substrate by building a physical barrier into the substrate-binding pocket. The polarity of this barrier was not as important as in the former strategy (mimicry of substrate-bound enzyme surface), therefore we had the freedom to choose charged or hydrophilic residues to improve the stability and the solubility of the mutant monomer.
Molecular mechanics calculations were run to calculate the intermonomer interaction energy of heterodimeric PR (Table I) and the total energy of mutated monomer (Table II) with substitutions of all 20 natural amino acids at position 25. Substitution of the catalytic residue (Asp25) must be the first one that should be considered for the creation of an inactive monomer. Not surprisingly, positively charged residues (Lys, Arg) gave the best interaction energy values; however, only Trp showed better monomer energy than the original Asp. The Asp25Lys and the Asp25Arg mutations fitted well into our working hypothesis, therefore both were selected for further in silico mutagenesis study.
Table I.
Calculated intermonomer interaction energy difference for defective heterodimeric HIV-1 proteases
| Residue | Interaction energy difference from the starting structure (kcal/mol) | |||
|---|---|---|---|---|
| Starting structure PR | PRD25K | PRD25R | PRRE | |
| Mutated position 25 | 49 | 49 | 50 | |
| Gly | 2.64 | 0.00 | 0.00 | 3.22 |
| Ala | 2.01 | 2.89 | 1.64 | 1.56 |
| Cys | −0.69 | 1.89 | 0.61 | −0.11 |
| Ser | 0.15 | 0.91 | 0.38 | 0.21 |
| Thr | −1.82 | 0.44 | 0.16 | −0.44 |
| Val | 2.06 | 0.79 | 0.29 | −0.15 |
| Ile | 1.15 | 5.34 | 4.32 | 0.00 |
| Leu | 2.41 | −0.01 | 0.67 | −0.60 |
| Met | 0.01 | −1.09 | −0.16 | −2.20 |
| Pro | 1.17 | 0.39 | 1.45 | 1.53 |
| His | −5.09 | 0.41 | 0.17 | −2.29 |
| Phe | −1.25 | −2.12 | −0.68 | −1.49 |
| Tyr | −2.66 | −2.07 | −0.34 | −2.88 |
| Trp | −4.58 | −0.31 | −1.32 | −0.72 |
| Asp | 0.00 | −0.65 | −0.11 | 1.62 |
| Asn | −0.29 | 1.12 | 0.02 | −0.69 |
| Glu | −1.03 | 1.63 | 0.12 | 3.36 |
| Gln | 0.21 | −0.69 | −0.36 | −2.06 |
| Lys | −11.94 | −0.76 | −1.68 | −12.84 |
| Arg | −8.97 | 0.08 | −5.42 | −21.86 |
The best two values in the given position are shown in bold.
Table II.
Calculated total energy difference of the mutated monomer of HIV-1 proteases
| Residue | Total energy of monomer (kcal/mol) | |||
|---|---|---|---|---|
| Starting structure PR | PRD25K | PRD25R | PRRE | |
| Mutated position 25 | 49 | 49 | 50 | |
| Gly | 6.32 | 0.00 | 0.00 | −2.00 |
| Ala | 5.09 | 2.93 | 2.05 | −2.98 |
| Cys | 4.02 | 3.55 | 1.62 | −2.81 |
| Ser | 5.10 | 1.73 | 1.43 | −3.37 |
| Thr | 5.33 | 4.79 | 3.89 | −2.39 |
| Val | 7.25 | 6.70 | 55.33 | −1.06 |
| Ile | 13.61 | 8.35 | 7.38 | 0.00 |
| Leu | 11.52 | 6.71 | 4.59 | 1.27 |
| Met | 4.82 | 4.11 | 3.81 | 0.22 |
| Pro | 33.18 | 40.51 | 24.32 | 8.37 |
| His | 24.53 | 18.26 | 16.86 | 15.73 |
| Phe | 5.60 | 5.33 | 1.40 | 2.36 |
| Tyr | 3.00 | 3.91 | 0.72 | 0.22 |
| Trp | −2.48 | 7.44 | 1.81 | 1.76 |
| Asp | 0.00 | −0.45 | −11.13 | −2.20 |
| Asn | 0.18 | −0.73 | −4.45 | −3.61 |
| Glu | 3.86 | −2.92 | −9.08 | −2.85 |
| Gln | 4.88 | 0.56 | 1.96 | −3.29 |
| Lys | 9.76 | 11.71 | 15.02 | 3.61 |
| Arg | 5.66 | 10.39 | 7.52 | −0.10 |
The best two values in the given position are shown in bold.
The next considered position was 49, which is located near the tip of the flap at the top of the substrate-binding site. Gly49 could be found in the wt monomer and it could be changed even to the largest residue (Trp) without any detrimental effect to the desired inhibition (Babe et al., 1995; Junker et al., 1996; McPhee et al., 1996; Rozzelle et al., 2000; Todd et al., 2000a; 2000b). Our calculations also showed that large hydrophobic residues (Phe, Tyr) and positively charged residues (Lys, Arg) gave good intermolecular interaction energy values (Table I). The former residues were neglected because their hydrophobicities did not fit into our design strategy. The positively charged residues may promote good ion-pairs with the catalytic aspartate of the wt monomer (Table I); however, they might repel the positively charged residue introduced at position 25 in the same monomer (large positive values in the row of Lys and Arg of Table II). Negatively charged residues (Asp, Glu) were found to strengthen the intramonomer interaction with Lys25 or Arg25 (Table II), and these mutations do not affect substantially the inter-subunit interactions (Table I). Structural visualization showed (Fig. 1A) that Arg25 combined with Glu49 might give especially good interaction, since the partially positive-charged nitrogen atoms of Arg25 side chain might properly fit into the space between the side chains of Asp25 of wt monomer and Glu49 of the mutated monomer to form an ion-pair-stabilized triad. The Arg25–Glu49 double mutant (called PRRE) was selected for further rounds of computational mutagenesis and for in vitro experiments.
Fig. 1.
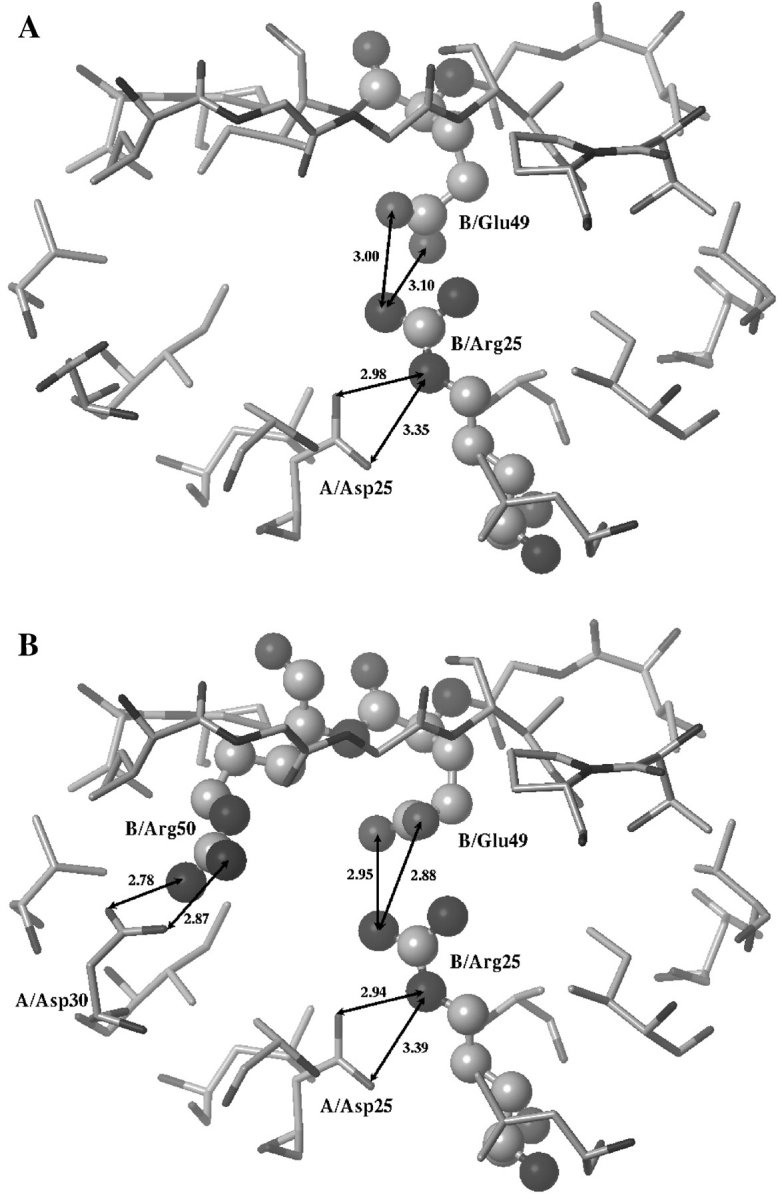
Molecular models of defective heterodimeric proteases (PR) containing a wild-type monomer A and PRRE (A) or PRRER (B) as monomer B. Mutated residues in monomer B are shown as ball and stick representations. Arrows indicate good hydrogen-bond distances with numbers in angstrom. Important residues are labeled.
The next candidate residue for mutagenesis was Ile50, following the pioneering work of Craik and co-workers (Babe et al., 1995; Junker et al., 1996; McPhee et al., 1996; Rozzelle et al., 2000; Todd et al., 2000a; 2000b). Ile50 is located in the tip of the flap, and forms a hydrophobic cover on the top of a substrate or an inhibitor. Our calculations showed that Arg and Lys in this position could result in favorable intermonomer interaction energy (Table I). The long and flexible chain of Arg/Lys may interact with the catalytic aspartate or with Asp30 resulting in good ionic interactions (Fig. 1B). Monomer having Arg50 had more favorable total energy than that of monomer bearing Lys50 (Table II), therefore the Arg25-Glu49-Arg50 triple mutant (called PRRER) was also selected for in vitro experiments. Further amino acid positions including Leu23, Ala28, Asp30, Val32, Ile47, Thr80, Val82, Ile84 and Ala95 were also tested in silico, but no substantial improvement could be achieved by substitution of these residues (data not shown).
To generate a monomer having an enhanced hydrophobic active-site/flap interface, a modified version of the PRKWW mutant was constructed by changing Asp25 to hydrophobic Trp. Trp25 had the best monomer energy in our calculation (Table II) and may interact well with Trp49 and Trp50 through aromatic–aromatic stacking. In this mutant (PRWWW), all mutated residues were changed to hydrophobic ones to fulfill the requirements of a hydrophobic strategy.
In vitro inhibition experiments
The selected mutations were introduced into the PR sequence. Proteins were expressed in E. coli and purified from inclusion bodies. The activity of PR in the presence of variable amounts of mutant proteins was measured using a microtiter plate-based fluorescent method (Bagossi et al., 2004) and activity relative to the same amount of dimerization-defective PR5–95 mutant (Ishima et al., 2001; 2007) was calculated (Fig. 2). PR5–95, which contains deletions of the N- and C-terminal residues, was shown to adopt a monomer fold, but is inactive because of its inability to form dimer (Ishima et al., 2001; 2007). HIV-1 PR is very sensitive to additives: they may activate/stabilize (polyethylene glycol, bovine serum albumin), as well as inactivate (glycerol, dimethyl sulfoxide) the enzyme (Jordan et al., 1992), therefore the appropriate control is crucial in all experimental systems where the activity of the enzyme is measured. The PRWWW mutant showed a weak dose-dependent inhibition, while the PRRE and the PRRER mutants showed similar inhibition as the previously described PRKWW mutant (Babe et al., 1995; Junker et al., 1996; McPhee et al., 1996; Rozzelle et al., 2000; Todd et al., 2000a; 2000b). It should be noted, that in our assay, the complete inhibition of PR could not be reached even at the highest tested (9:1) ratio of mutant to wild-type PR, unlike the case of previously published assay (Rozzelle et al., 2000), where all tested mutants including PRKWW were able to completely inhibit the activity of wild-type PR at ratios higher than 3.5:1. We assume that these differences are because of different assay conditions (buffers, substrates, controls).
Fig. 2.

Dose-dependent inhibition of protease (PR) activity by mutant proteins. PR5–95 was used as control protein. PR5–95 mixed with PR defines 100% activity at the given inactive:active proteins ratio.
To test whether the inhibition was caused by the specific interaction between PR and mutant monomers, we showed that heterodimeric PRs were formed in the 1:1 mixture of His6-tagged X28-PR and PRRER (Fig. 3) or PRRE (data not shown) mutant proteins, as it was shown previously for PRKWW mutant by Craik and co-workers (Rozzelle et al., 2000).
Fig. 3.

Sodium dodecyl sulphate–polyacryl amide gel electrophoresis stained with Coomassie Brilliant Blue showing formation of the heterodimer complex between hexahistidine-tagged wild-type X28-PR and PRRER mutant on the surface of Ni-NTA agarose. 1: eluted fraction from batch of nickel resin and His6-X28-PR; 2: eluted fraction from batch of nickel resin, His6-X28-PR and PRRER at 1:1 protein ratio; 3: eluted fraction from batch of nickel resin and PRRER; M: molecular weight marker, molecular weights are indicated by numbers in kilodalton.
Protein fold information derived from nuclear magnetic resonance studies
In previous studies we have assigned NMR chemical shifts that are characteristic for the mature HIV-1 PR dimer and the monomer (Ishima et al., 2001; 2003). In recent studies, we also defined a simple protocol for PR folding at a low protein concentration of ∼28 µM (in monomer concentration) to facilitate acquiring 1H–15N HSQC spectra using a cryoprobe (Ishima et al., 2007). The same methodology was used to probe monomer folding/dimerization of PRRE and PRRER proteins and to assess if they have the potential to form heterodimers with wild-type PR. The HSQC spectrum of 15N-labeled PRRER in the absence of wt PR is shown in Fig. 4A. The spectrum is typical of a folded monomer as described before (Ishima et al., 2003). In addition, signals observed in the narrow region of 1H 8–8.5 ppm indicate the presence of unfolded fraction of the protein. In contrast, in the presence of equal proportion of unlabeled wt PR (Fig. 4B), some signals characteristic of the PR dimer become apparent (shown in solid boxes). Although additional NMR experiments are required to assign signals of all of the shifted peaks of PRRER in the presence of wt PR, this observation is clearly indicative of heterodimer formation of PRRER with wt PR as only the labeled PRRER will be visible in the spectrum. It is also apparent from the spectrum that the presence of wt PR also significantly facilitates the folding of PRRER as shown by the decrease in the amount of unfolded PRRER (compare Fig. 4A and B).
Fig. 4.

1H–15N Heteronuclear Single Quantum Coherence spectra of mutant proteases acquired either alone (A, C) or in the presence of unlabeled protease (B, D) at a ratio of 1:1 in 50 mM sodium acetate buffer, pH 5.8 at 20°C. Proteins were prepared according to the quench protocol of protein folding (Ishima et al., 2007) to a final concentration of 25–35 µM (in monomer concentration). Peaks unique to the dimer are shown in solid boxes.
Similar NMR experiments were carried out using 15N-labeled PRRE. Differently from the PRRER spectrum, the 1H–15N HSQC spectrum of PRRE in the absence of PR (Fig. 4C) exhibited very weak signals and signals corresponding to a folded protein were not evident. The loss in signal intensities with only side chain peaks observed at 1H 6.9, 7.2 and 7.6 ppm, suggests that the majority of the protein undergoes aggregation even at a low protein concentration of ∼28 µM. Remarkably, in the presence of wt PR (Fig. 4D), PRRE is fully folded, exhibiting peaks similar to those of PRRER, indicating that the PRRE most likely forms a dimer when folded in the presence of wt PR. In addition, no visible peaks corresponding to unfolded protein are evident. From the spectra of PRRER and PRRE (Fig. 4A and C), we propose that the enhanced level of aggregation or lack of folding of PRRE is because of a negative charge effect created by the flap residue mutation Gly49Glu. Interestingly, introduction of a positive charge by the Ile50Arg mutation seems to restore the stability of the fold or diminish aggregation.
The existence of the folded mutant monomer, such as PRRER is beneficial over the unfolded PRRE monomer because (i) the energy gain of the dimerization process is not diminished by the energy of folding and (ii) the unfolded form may be degraded faster in cellular environment. The lack of homodimer formation is also advantageous, because a higher effective concentration of mutant monomer can be maintained. Consistent with the lack of homodimer formation, DMP323, a potent symmetric cyclic urea inhibitor, does not bind and induce dimerization of the mutant proteins (data not shown), suggestive of a steric barrier build into the substrate-binding site.
Test of infectivity inhibition
To test the infectivity of viral particles containing wt and mutant PRs, we used a third-generation HIV-1 vector system (Dull et al., 1998; Trono, 2007). It consists of four separate plasmids (Fig. 5) to maximize the number of recombination events that would be necessary to generate a replication competent virus, and hence to minimize the safety risk. pWOX-GFP is the vector used to introduce the foreign gene (here GFP). We modified the internal promoter of the original construct by the insertion of CMV promoter upstream to the GFP gene. The resultant pWOX-CMV-GFP vector produced substantially higher fluorescent signal than that of the original one in our assay system (data not shown). pMDLg/pRRE is the packaging construct coding for Gag and GagPol polyproteins in the transfected cells under the control of CMV promoter, in the presence of Rev protein, which is encoded in a separate construct of pRSV.rev. The last construct, pMD.G, encodes G protein of vesicular stomatitis virus, a heterologous envelope to pseudotype viral particles.
Fig. 5.

Schematic drawing of plasmid constructs used. Asterisks show the places of genetic manipulations of the original vector system (Dull et al., 1998; Trono, 2007). Abbreviations: U3, R, U5: elements of retroviral long-terminal repeat; SD: splice donor site; SA: splice acceptor site; Ψ: packaging sequence; RRE: Rev-responsive element; cPPT: central polypurine tract; dPPT: distal polypurine tract; EF1a: elongation factor 1 alpha; TATA: TATA-box; GFP: gene of Green Fluorescent Protein; WPRE: Woodchuck hepatitis virus post-transcriptional regulatory element; CMV: human cytomegalovirus immediate-early promoter; RSV: promoter of Rous Sarcoma Virus; polyA: polyadenylation site.
pMDLg/pRRE plasmid was modified by site-directed mutagenesis to harbor the desired mutations. 293T cells were transfected by equal amounts of plasmids of the various constructs and the composition of viral particles were analyzed by western blot using anti-CA antibody (Fig. 6). The Gag (and presumably GagPol) expression was very low in constructs harboring PRRE and PR5–95 mutants. Wild-type PR completely processed Gag and GagPol polyproteins; however, there was no sign for polyprotein processing in viral particles containing PRRER and PRKWW protein. These mutants were incapable of forming active PRs in the immature virus.
Fig. 6.

Western blot analyses of viral particles containing wild-type or mutant proteases (1: wt PR, 2: PR5–95, 3: PRKWW, 4: PRRE, 5: PRRER) using anti-CA antibody.
293T cells were also transfected by plasmid mixtures encoding the mutant and wt PRs in a DNA ratio of 2:1. Concentrations of virus stocks were set to be equal based on p24 assay. 293T cells were infected by viral particles and the quantity of the infected cells was measured by cell sorting based on the fluorescent signal of GFP-containing cells. We used salmon sperm DNA for control experiments, in which 97% of the cells were infected (Fig. 7), which showed the effectiveness of our infection protocol. Infectivity of viral particles containing PRKWW or PRRER proteins were drastically reduced (Fig. 7), having infectivity values about 8 and 15%. In this assay we saw much higher inhibition than at the 2:1 protein ratio of the in vitro experiments (Fig. 2), which had inhibition of infection values of about 80% in vivo. Similar observations were reported by Junker and co-workers (Junker et al., 1996). PRKWW is still a more effective inhibitor than PRRER in cell culture experiment (Fig. 7B) even if not in vitro at higher protein ratios (Fig. 2).
Fig. 7.
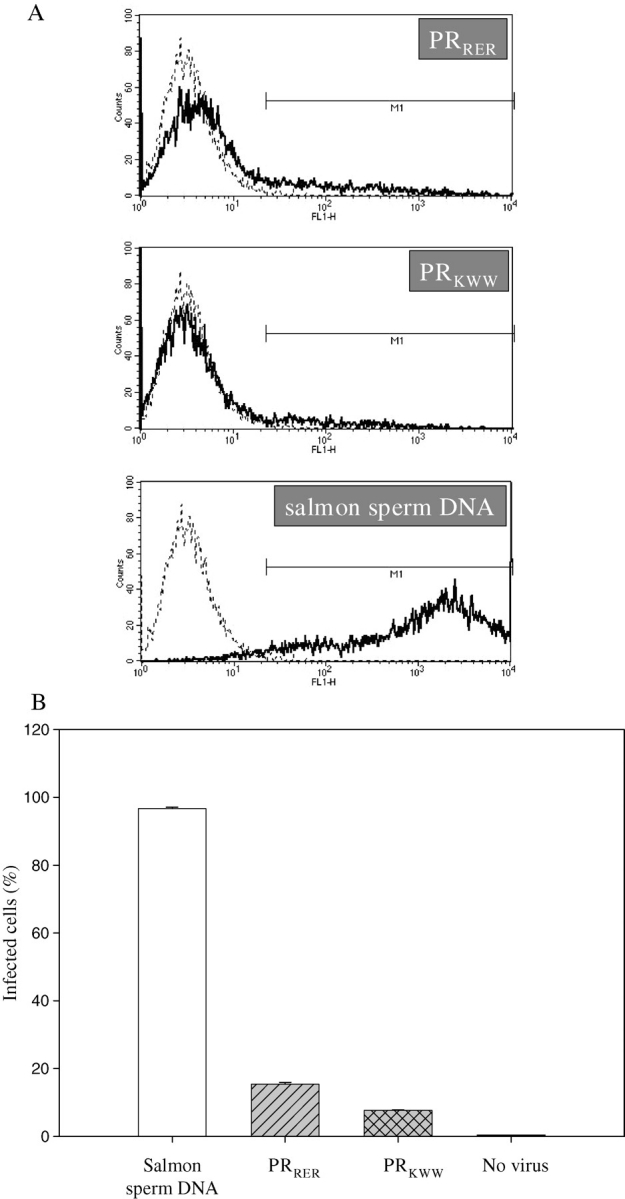
Fluorescence-activated cell sorting (FACS) analyses of 293T cells infected by viral particles containing wild-type protease (produced by cells infected with DNA mix at 2:1 ratio of salmon sperm DNA and wt plasmid) or mixture wt and mutant proteins (produced by cells infected with DNA mix at 2:1 ratio of mutant:wt plasmid). (A) Histograms of representative experiments. Population of non-infected cells is shown with dashed line. (B) Bar chart of summary of parallel experiments of FACS analyses. Error bars are small and therefore not visible in all cases.
In summary, we designed and tested mutant HIV-1 PRs containing hydrophilic/charged residues in the substrate-binding site. These mutants showed trans-dominant inhibitory effects in vitro and one mutant was effective also in cell culture experiments, and they represent a novel hydrophilic strategy for generation of macromolecular inhibitors against HIV-1 PR. Heterodimerization could be detected, for the first time, between a wild-type retroviral PR and trans-dominant negative mutants by NMR spectroscopy using our novel hydrophilic constructs and our facile protein folding protocol (Ishima et al., 2007). The Asp25Arg and the Gly49Glu mutations introduced into analogous positions of other retroviral PRs may exert a similar inhibitory effect on the corresponding wild-type PR. These residues target the catalytic aspartate, thus it may be considered a general strategy for all retroviral PRs.
Funding
Hungarian Science and Research Fund (OTKA F34479, K68288); Intramural Research program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Acknowledgements
We thank Dr Didier Trono (Department of Genetics and Microbiology, University of Geneva Medical School, Geneva, Switzerland) for the HIV vector system, Tamás Sperka and Krisztina Matúz for help in protein purification and Szilvia Petö, Katalin Szabó and Annie Aniana for excellent technical assistance.
Footnotes
Edited by Anthony Wilkinson
References
- Babe L.M., Rose J., Craik C.S. Proc. Natl Acad. Sci. USA. 1995;92:10069–10073. doi: 10.1073/pnas.92.22.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagossi P., Kadas J., Miklossy G., Boross P., Weber I.T., Tozser J. J. Virol. Methods. 2004;119:87–93. doi: 10.1016/j.jviromet.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Barbaro G., Scozzafava A., Mastrolorenzo A., Supuran C.T. Curr. Pharm. Des. 2005;11:1805–1843. doi: 10.2174/1381612053764869. [DOI] [PubMed] [Google Scholar]
- Bevec D., Dobrovnik M., Hauber J., Bohnlein E. Proc. Natl Acad. Sci. USA. 1992;89:9870–9874. doi: 10.1073/pnas.89.20.9870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buchschacher G.L., Jr, Freed E.O., Panganiban A.T. Hum. Gene Ther. 1992;3:391–397. doi: 10.1089/hum.1992.3.4-391. [DOI] [PubMed] [Google Scholar]
- Buchschacher G.L., Jr, Freed E.O., Panganiban A.T. J. Virol. 1995;69:1344–1348. doi: 10.1128/jvi.69.2.1344-1348.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caflisch A., Schramm H.J., Karplus M. J. Comput. Aided Mol. Des. 2000;14:161–179. doi: 10.1023/a:1008146201260. [DOI] [PubMed] [Google Scholar]
- Chen S.S., Ferrante A.A., Terwilliger E.F. Virology. 1996;226:260–268. doi: 10.1006/viro.1996.0654. [DOI] [PubMed] [Google Scholar]
- Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraisier C., Abraham D.A., van Oijen M., Cunliffe V., Irvine A., Craig R., Dzierzak E.A. Gene Ther. 1998;5:946–954. doi: 10.1038/sj.gt.3300676. [DOI] [PubMed] [Google Scholar]
- Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hwang Y.S., Chmielewski J. Bioorg. Med. Chem. Lett. 2004;14:4297–4300. doi: 10.1016/j.bmcl.2004.05.081. [DOI] [PubMed] [Google Scholar]
- Hwang Y.S., Chmielewski J. J. Med. Chem. 2005;48:2239–2242. doi: 10.1021/jm049581j. [DOI] [PubMed] [Google Scholar]
- Ishima R., Ghirlando R., Tozser J., Gronenborn A.M., Torchia D.A., Louis J.M. J. Biol. Chem. 2001;276:49110–49116. doi: 10.1074/jbc.M108136200. [DOI] [PubMed] [Google Scholar]
- Ishima R., Torchia D.A., Lynch S.M., Gronenborn A.M., Louis J.M. J. Biol. Chem. 2003;278:43311–43319. doi: 10.1074/jbc.M307549200. [DOI] [PubMed] [Google Scholar]
- Ishima R., Torchia D.A., Louis J.M. J. Biol. Chem. 2007;282:17190–17199. doi: 10.1074/jbc.M701304200. [DOI] [PubMed] [Google Scholar]
- Jordan S.P., Zugay J., Darke P.L., Kuo L.C. J. Biol. Chem. 1992;267:20028–20032. [PubMed] [Google Scholar]
- Junker U., Escaich S., Plavec I., Baker J., McPhee F., Rose J.R., Craik C.S., Bohnlein E. J. Virol. 1996;70:7765–7772. doi: 10.1128/jvi.70.11.7765-7772.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Woffendin C., Yang Z.Y., Nabel G.J. Gene Ther. 1994;1:32–37. [PubMed] [Google Scholar]
- Louis J.M., Wondrak E.M., Kimmel A.R., Wingfield P.T., Nashed N.T. J. Biol. Chem. 1999;274:23437–23442. doi: 10.1074/jbc.274.33.23437. [DOI] [PubMed] [Google Scholar]
- Louis J.M., Weber I.T., Tözsér J., Clore G.M., Gronenborn A.M. Adv. Pharmacol. 2000;49:111–146. doi: 10.1016/s1054-3589(00)49025-3. [DOI] [PubMed] [Google Scholar]
- Louis J.M., Ishima R., Nesheiwat I., Pannell L.K., Lynch S.M., Torchia D.A., Gronenborn A.M. J. Biol. Chem. 2003;278:6085–6092. doi: 10.1074/jbc.M209726200. [DOI] [PubMed] [Google Scholar]
- Mahalingam B., Louis J.M., Reed C.C., Adomat J.M., Krouse J., Wang Y.F., Harrison R.W., Weber I.T. Eur. J. Biochem. 1999;263:238–245. doi: 10.1046/j.1432-1327.1999.00514.x. [DOI] [PubMed] [Google Scholar]
- Mahalingam B., Boross P., Wang Y.F., Louis J.M., Fischer C.C., Tözsér J., Harrison R.W., Weber I.T. Proteins. 2002;48:107–116. doi: 10.1002/prot.10140. [DOI] [PubMed] [Google Scholar]
- McPhee F., Good A.C., Kuntz I.D., Craik C.S. Proc. Natl Acad. Sci. USA. 1996;93:11477–11481. doi: 10.1073/pnas.93.21.11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesti N., Garcia J., Debouck C., Peterlin M., Gaynor R. New Biol. 1991;3:759–768. [PubMed] [Google Scholar]
- Oroszlan S., Luftig R.B. Curr. Top. Microbiol. Immunol. 1990;157:153–185. doi: 10.1007/978-3-642-75218-6_6. [DOI] [PubMed] [Google Scholar]
- Pearson L., Garcia J., Wu F., Modesti N., Nelson J., Gaynor R. Proc. Natl Acad. Sci. USA. 1990;87:5079–5083. doi: 10.1073/pnas.87.13.5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph J.T., DeGoey D.A. Curr. Top. Med. Chem. 2004;4:1079–1095. doi: 10.2174/1568026043388330. [DOI] [PubMed] [Google Scholar]
- Rezacova P., Brynda J., Fabry M., Horejsi M., Stouracova R., Lescar J., Chitarra V., Riottot M.M., Sedlacek J., Bentley G.A. J. Mol. Recog. 2002;15:272–276. doi: 10.1002/jmr.587. [DOI] [PubMed] [Google Scholar]
- Rozzelle J.E., Dauber D.S., Todd S., Kelley R., Craik C.S. J. Biol. Chem. 2000;275:7080–7086. doi: 10.1074/jbc.275.10.7080. [DOI] [PubMed] [Google Scholar]
- Rudich A., Ben-Romano R., Etzion S., Bashan N. Acta Physiol. Scand. 2005;183:75–88. doi: 10.1111/j.1365-201X.2004.01383.x. [DOI] [PubMed] [Google Scholar]
- Sawaya B.E., Khalili K., Gordon J., Srinivasan A., Richardson M., Rappaport J., Amini S. J. Virol. 2000;74:4877–4881. doi: 10.1128/jvi.74.10.4877-4881.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm H.J., Nakashima H., Schramm W., Wakayama H., Yamamoto N. Biochem. Biophys. Res. Commun. 1991;179:847–851. doi: 10.1016/0006-291x(91)91895-j. [DOI] [PubMed] [Google Scholar]
- Schramm H.J., de Rosny E., Reboud-Ravaux M., Buttner J., Dick A., Schramm W. Biol. Chem. 1999;380:593–596. doi: 10.1515/BC.1999.076. [DOI] [PubMed] [Google Scholar]
- Shimano R., Inubushi R., Oshima Y., Adachi A. Virus Genes. 1999;18:197–201. doi: 10.1023/a:1008054111697. [DOI] [PubMed] [Google Scholar]
- Sperka T., Pitlik J., Bagossi P., Tozser J. Bioorg. Med. Chem. Lett. 2005;15:3086–3090. doi: 10.1016/j.bmcl.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Steffy K.R., Wong-Staal F. J. Virol. 1993;67:1854–1859. doi: 10.1128/jvi.67.4.1854-1859.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanstrom R., Eron J. Pharmacol. Ther. 2000;86:145–170. doi: 10.1016/s0163-7258(00)00037-1. [DOI] [PubMed] [Google Scholar]
- Todd S., Anderson C., Jolly D.J., Craik C.S. Biochim. Biophys. Acta. 2000;a 1477:168–188. doi: 10.1016/s0167-4838(99)00272-1. [DOI] [PubMed] [Google Scholar]
- Todd S., Laboissiere M.C., Craik C.S. Anal. Biochem. 2000;b 277:247–253. doi: 10.1006/abio.1999.4388. [DOI] [PubMed] [Google Scholar]
- Tözsér J. Ann. N. Y. Acad. Sci. 2001;946:145–159. doi: 10.1111/j.1749-6632.2001.tb03909.x. [DOI] [PubMed] [Google Scholar]
- Tözsér J., Oroszlan S. Curr. Pharm. Des. 2003;9:1803–1815. doi: 10.2174/1381612033454478. [DOI] [PubMed] [Google Scholar]
- Trono D. 2007 LVG-Tronolab, Lentivectors, http://tronolab.epfl.ch/page58115.html . [Google Scholar]
- Trono D., Feinberg M.B., Baltimore D. Cell. 1989;59:113–120. doi: 10.1016/0092-8674(89)90874-x. [DOI] [PubMed] [Google Scholar]
- Weiner S.J., Kollman P.A., Nguyen D.T., Case D.A. J. Comp. Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- Wlodawer A. Annu. Rev. Med. 2002;53:595–614. doi: 10.1146/annurev.med.53.052901.131947. [DOI] [PubMed] [Google Scholar]
- Wondrak E.M., Louis J.M. Biochemistry. 1996;35:12957–12962. doi: 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- Yehia N.A., Antuch W., Beck B., Hess S., Schauer-Vukasinovic V., Almstetter M., Furer P., Herdtweck E., Domling A. Bioorg. Med. Chem. Lett. 2004;14:3121–3125. doi: 10.1016/j.bmcl.2004.04.026. [DOI] [PubMed] [Google Scholar]