

Abstract
The neuropeptides oxytocin (OXT) and arginine vasopressin (AVP) contribute to the regulation of diverse cognitive and physiological functions including nociception. Indeed, OXT has been reported to be analgesic when administered directly into the brain, the spinal cord, or systemically. Here, we characterized the phenotype of oxytocin receptor (OTR) and vasopressin-1A receptor (V1AR) null mutant mice in a battery of pain assays. Surprisingly, OTR knock-out mice displayed a pain phenotype identical to their wild-type littermates. Moreover, systemic administration of OXT dose-dependently produced analgesia in both wild-type and OTR knock-out mice in three different assays, the radiant-heat paw withdrawal test, the von Frey test of mechanical sensitivity, and the formalin test of inflammatory nociception. In contrast, OXT-induced analgesia was completely absent in V1AR knock-out mice. In wild-type mice, OXT-induced analgesia could be fully prevented by pretreatment with a V1AR but not an OTR antagonist. Receptor binding studies demonstrated that the distribution of OXT and AVP binding sites in mouse lumbar spinal cord resembles the pattern observed in rat. AVP binding sites diffusely label the lumbar spinal cord, whereas OXT binding sites cluster in the substantia gelatinosa of the dorsal horn. In contrast, quantitative real-time reverse transcription (RT)-PCR revealed that V1AR but not OTR mRNA is abundantly expressed in mouse dorsal root ganglia, where it localizes to small- and medium-diameter cells as shown by single-cell RT-PCR. Hence, V1ARs expressed in dorsal root ganglia might represent a previously unrecognized target for the analgesic action of OXT and AVP.
Introduction
Oxytocin (OXT) and arginine vasopressin (AVP) are two closely related nonapeptides synthesized in the paraventricular and supraoptic nuclei of the hypothalamus (Swanson and Kuypers, 1980; Sofroniew, 1983). They are either transported to the posterior pituitary and secreted into the bloodstream, exerting a variety of hormonal effects, or released into the CNS where they act as modulators of neuronal transmission (Leng and Ludwig, 2008; Raggenbass, 2008). Recently, the actions of OXT and AVP in the CNS have received increasing attention because of the documented role of these peptides in the regulation of complex social and sexual behavior in mammals (Donaldson and Young, 2008; Young et al., 2008), including humans (Baumgartner et al., 2008; Petrovic et al., 2008; Walum et al., 2008). Concomitantly, a growing literature has demonstrated the analgesic effects of OXT and AVP in both human and rodent species (Honda and Takano, 2009; Koshimizu and Tsujimoto, 2009). Indeed, OXT was reported to be analgesic in a variety of pain tests when administered into the brain (Ge et al., 2002; Wang et al., 2003; Gao and Yu, 2004), the spinal cord (Yu et al., 2003; Miranda-Cardenas et al., 2006; Condés-Lara et al., 2007), or systemically (Lundeberg et al., 1994; Reeta et al., 2006). In the rat, oxytocin receptor (OTR) binding sites are abundant in laminae I and II of the dorsal horn (Tribollet et al., 1989, 1997; Reiter et al., 1994; Véronneau-Longueville et al., 1999; Liu et al., 2003), and it has been suggested that OXT activates inhibitory spinal interneurons presynaptically to induce analgesia (Robinson et al., 2002; Rojas-Piloni et al., 2007; Breton et al., 2008).
The central and peripheral effects of OXT are thought to be mediated by its binding to a single isoform of the OTR (Gimpl and Fahrenholz, 2001), which activates the phospholipase Cβ (PLCβ) signal transduction pathway (Arnaudeau et al., 1994; Ku et al., 1995; Phaneuf et al., 1995). Centrally, AVP exerts its effects mainly through the vasopressin-1A receptor (V1AR), which is also coupled to the PLCβ pathway (Schöneberg et al., 1998; Thibonnier et al., 1998; Birnbaumer, 2000). Importantly, both OXT and AVP, as well as the OTR and V1AR, display a high degree of sequence homology, and both peptides can therefore activate both receptors (Chini and Manning, 2007).
In the present studies, we used two lines of transgenic knock-out (KO) mice lacking the OTR or the V1AR, respectively, to investigate the role of these receptors in pain. In a series of behavioral and pharmacological experiments, baseline nociceptive sensitivity and OXT-induced analgesia were compared between KO and wild-type (WT) littermates. The finding that OXT-induced analgesia is normal in OTR KO mice but impaired in V1AR KO mice suggested a V1AR-mediated effect of OXT, and this was confirmed by experiments using OTR- and V1AR-selective antagonists. To determine the putative site of action of OXT, we mapped the autoradiographic distribution of OTRs and V1ARs in mouse spinal cord and determined gene expression levels in dorsal root ganglia (DRGs) using real-time quantitative reverse transcription (RT)-PCR as well as a single-cell-based RT-PCR approach.
Materials and Methods
Animals.
All procedures were performed according to national guidelines and approved by a McGill University animal use committee. OTR KO mice and WT controls were obtained from Larry J. Young (Emory University, Atlanta, GA) (Takayanagi et al., 2005) and V1AR KO mice and WT controls were obtained from Jaqueline N. Crawley (National Institute of Mental Health, Bethesda, MD) (Hu et al., 2003). Both lines, bred congenic on a C57BL/6 background for >10 generations, were maintained as heterozygote (HET) × HET breeding colonies, and all offspring was genotyped by custom PCR analysis of DNA isolated from tail tissue. The nociceptive battery (see below) was performed on WT, HET, and KO genotypes; other experiments compared only WT and KO mice.
For V1AR, the wild-type allele was identified by a 987 bp PCR product using the following primers: 5′-CAGGAAATCTTCTGTAACATACTTGGTTTGG, 5′-CAAGATCCGCACAGTGAAGATGACC. The KO allele was identified by a 620 bp PCR product using the following primers: 5′-CAGGAAATCTTCTGTAACATACTTGGTTTGG, 5′-ACCCCTTCCCAGCCTCTGAGCCCAGAAGCGAAGG. Both reactions were run in the same tube under the following conditions: 95°C for 4 min (95°C for 1 min, 60°C for 1 min, 72°C for 1 min) times 40 cycles, 72°C for 5 min.
For OTR, the wild-type allele was identified by a 630 bp PCR product using the following primers: 5′-CTGGGGCTGAGTCTTGGAAG, 5′-CTGGATACTCCAGTTGGGTGC. The KO allele was identified by 410 bp PCR product using the following primers: 5′-CTGGGGCTGAGTCTTGGAAG, 5′-GTTGGGAACAGGGGTGATTA. Both reactions were run in the same tube under the following conditions: 95°C for 5 min (94°C for 30 s, 55°C for 30 s, 72°C for 1 min) times 35 cycles.
Nociceptive assays.
Adult (6–10 weeks of age) mice of both sexes (Mogil and Chanda, 2005) were used for behavioral testing and allowed to habituate to the testing room for at least 1 h. Males and females were tested separately. Moreover, mice were unable to see each other during testing (Langford et al., 2006). For baseline nociceptive phenotyping, each mouse first underwent the whole series of acute thermal and mechanical assays, with 2 d between consecutive tests. We have determined that these acute assays are associated with no detectable carryover or repeated-testing effects (Mogil et al., 2006). After the acute assays, mice were tested in either the formalin, the zymosan, or the abdominal constriction (writhing) test and killed immediately thereafter.
Thermal nociception was assessed using three different assays, the tail withdrawal, hot-plate, and paw withdrawal tests. (1) Tail withdrawal test: Mice were lightly restrained in a cloth/cardboard “pocket” while the distal half of their tail was dipped into a bath maintained at either 47 or 49°C. Latency to reflexively withdraw the tail from the water was measured three consecutive times (separated by 320 s) and averaged. (2) Hot-plate test: Mice were placed on a metal surface maintained at either 50 or 53°C (IITC model PE34MHC) within a 20-cm-high Plexiglas cylinder (15 cm diameter). The latency to either lick or shake the hindpaw was measured as a nocifensive endpoint. (3) Paw withdrawal test: The noxious stimulus was a high-intensity light beam (IITC; setting, 20%; ≈45 W) aimed at the plantar surface of the hindpaw of an inactive mouse (Callahan et al., 2008) placed within Plexiglas cubicles (9 × 5 × 5 cm high) atop a glass floor. For the assessment of baseline thermal sensitivity, the latency to lift the targeted hindpaw (i.e., paw withdrawal latency) was measured six times (separated by 15 min) on each side.
Mechanical nociception was assessed using two different assays: the tail-clip and the von Frey filament test. (4) Tail-clip test: A modification of the tail-pinch test was used (Tagaki et al., 1966). Each mouse was placed in a cloth/cardboard holder, and a binder clip (exerting ≈700 g of force) was applied to the tail ≈1 cm from the base. The mouse was immediately removed from the holder, placed on a table top, and the latency to lick, bite, grab, or bring the nose to within 1 cm of the clip was measured with a stopwatch to the nearest 0.1 s, after which the clip was immediately removed. (5) von Frey test: Mice were placed within Plexiglas cubicles (9 × 5 × 5 cm high) on a metal mesh floor. Mechanical sensitivity was measured by determining the median 50% hindpaw withdrawal threshold with calibrated von Frey nylon monofilaments (0.015–2.0 g) using the up–down method (Chaplan et al., 1994). Reported values represent the average of four distinct threshold evaluations, two on each side.
Chemical nociception was assessed with the abdominal constriction (writhing) and the formalin test. (6) Writhing test: Mice were placed atop a glass floor within 20-cm-high Plexiglas cylinders (15 cm diameter) and habituated for at least 30 min. Mice were briefly removed, injected intraperitoneally with 0.9% acetic acid in a 10 ml/kg volume, and returned immediately to the cylinder. Each mouse was videotaped for the next 20 min by individual video cameras placed underneath the glass floor, and their behavior was later scored by a blinded observer using The Observer (Noldus). The presence or absence of “writhes” (lengthwise stretches of the torso with a concomitant concave arching of the back) was sampled at 20 s intervals and is reported as the percentage of “positive” samples (i.e., samples containing writhing behavior). (7) Formalin test: Mice were placed atop a glass floor within 20-cm-high Plexiglas cylinders (15 cm diameter) and habituated for at least 30 min. Mice were briefly removed, and 25 μl of a 2.5% formaldehyde solution was injected subcutaneously into the plantar surface of the right hindpaw using a 50 μl microsyringe with a 30 gauge needle. After being returned to the cylinder, each mouse was videotaped from below for the next 60 min, and the presence or absence of licking/biting of the right hindpaw was sampled once per minute by a blinded observer (see above). The early/acute phase of the biphasic formalin test was defined conservatively as the first 5 min of observation, and the late/tonic phase as the last 50 min of observation.
Inflammatory hypersensitivity was assessed using zymosan A from Saccharomyces cerevisiae (Meller and Gebhart, 1997). (8) Zymosan thermal hypersensitivity: Paw withdrawal baseline latencies were assessed as described above. The following day, 20 μl of a 2.5 mg/ml zymosan solution was injected into the right hindpaw and two postinjection latency measures on each paw were taken every hour for 6 h.
Pharmacology.
For pharmacological experiments with OXT or AVP, four baseline measures were taken before and 30 min after injection on either the radiant-heat paw withdrawal or the von Frey test. For the paw withdrawal test, a cutoff latency of 30 s was used to prevent the possibility of tissue injury. Reported values represent percentage analgesia produced by OXT or AVP and were calculated as follows: [(postdrug latency/threshold − baseline latency/threshold)/(cutoff latency/threshold − baseline latency/threshold)] × 100. The same protocol was used for hyperosmotic challenge experiments; instead of a peptide injection, mice received a 10 ml/kg (intraperitoneal) injection of hyperosmotic (1 m) or physiological (150 mm) saline. This protocol has been shown to induce an average increase of 15.8 mOsm/kg in wild-type mice (Ciura and Bourque, 2006), which in turn raises serum AVP levels from 2 to 40 pg/ml (Sharif Naeini et al., 2006). Where used, OTR and V1AR antagonists were injected intraperitoneally, intracerebroventricularly, or intrathecally 10 min before intraperitoneal injection of OXT. Intracerebroventricular injections were delivered using a 2.5 μl volume under light isoflurane/oxygen anesthesia according to the method of Laursen and Belknap (1986). Intrathecal injections were delivered using a 5 μl volume based on the method of Hylden and Wilcox (1980).
Compounds.
OXT and AVP were both obtained from Sigma-Aldrich and were dissolved in a 0.9% saline solution and injected intraperitoneally except where otherwise noted. d(CH2)[Tyr(Me)2]AVP (Kruszynski et al., 1980), a selective V1AR antagonist, and desGly-NH2-d(CH2)5[d-Tyr2,Thr4]OVT (Manning et al., 1995), a selective OTR antagonist, were both kindly donated by Dr. Maurice Manning (University of Toledo, Toledo, OH).
Scratching.
Mice were placed atop a glass floor within 20-cm-high Plexiglas cylinders (15 cm diameter) and allowed to habituate for 30 min. Then, they were removed, lightly anesthetized with isoflurane/oxygen, and given an intracerebroventricular injection of OXT or AVP using a volume of 2.5 μl. Mice were immediately returned to their test cylinders and videotaped by individual video cameras from below for the next 30 min. Blinded experimenters using The Observer scored the cumulative duration of vigorous scratching of the flanks with the contralateral hindpaws.
Receptor binding study.
Three male and three female adult mice of each of the OTR KO, the V1AR KO, and the C57BL/6 (WT) genotypes were killed, and their spinal cords and brains were rapidly dissected and frozen in isopentane at −80°C. The lumbar section of the spinal cord (L4–L6) was cut with a freezing microtome in six series of coronal sections, 14 μm thick, and mounted on chrome-alum-gelatin-coated microscope slides. Two brains of each genotype were also cut in coronal sections of equal thickness at the level of the lateral septum and the ventromedial hypothalamus. These sections served as a control for OTR and V1AR binding. All slides were stored at −80°C until the day of the experiment. The binding procedure was performed as previously described (Tribollet et al., 1997). Before incubation, sections were lightly fixed by dipping the slides for 5 min in a solution of 0.2% paraformaldehyde in 0.1 m PBS, pH 7.4, and then rinsed by two 5 min washes in 50 mm Tris-HCl buffer, pH 7.4, supplemented with 0.1% bovine serum albumin. OXT binding sites were labeled with the radioiodinated OTR antagonist d(CH2)5[Tyr (Me)2,Thr4,Tyr9-NH2]OVT (125I-OTA) at 0.02 nm (Elands et al., 1988). AVP binding sites were labeled with the radioiodinated V1AR antagonist HO-Phaa1-d-Tyr(Me)2-Phe3-Gln4-Asn5-Arg6-Pro7-Arg8-NH2 (125I-VPA) at 0.02 nm (Barberis et al., 1995). The specific activity of radioligands was 2200 Ci/mmol. Their binding properties in mouse spinal and brain sections were assessed in preliminary experiments by using selective agonist compounds as competitors (Chini and Manning, 2007). Results indicated that 125I-VPA and 125I-OTA have the same receptor selectivity as in the rat (i.e., label selectively V1AR and OTR, respectively, when used at the proper concentration) (results not shown). Nonspecific binding was determined in the presence of 2 μm OXT or AVP. Binding was performed in a humid chamber, at room temperature, by covering each slide with 400 μl of the incubation medium (50 mm Tris-HCl, 0.025% bacitracin, 5 mm MgCl2, 0.1% bovine serum albumin) containing 125I-VPA or 125I-OTA, either alone or in the presence of unlabeled peptide. Incubation lasted 1 h at room temperature under gentle agitation and was followed by two 5 min washes in ice-cold incubation medium and a quick rinse in distilled water. The slides were air-dried, apposed to Kodak Biomax MR Films for 14 d, and developed in Kodak D19 for 5 min. Both radioligands and films were obtained from PerkinElmer.
Quantitative gene expression analysis.
Five adult male and five adult female C57BL/6 mice (The Jackson Laboratory) were killed, and their DRGs were rapidly transferred into RNAlater (Ambion). Total RNA was isolated from these tissues with the TRIzol method according to the manufacturer's instructions (Invitrogen) and reverse transcribed along with standards of known concentrations (Ambion). Amplification of Oxtr (assay ID, Mm01182684_m1) and Avpr1a (assay ID, Mm00444092_m1) cDNA was performed using an ABI 7000 with TaqMan probes and primers as described in the manufacturer's protocol (Applied Biosystems). Unknown samples were amplified in quadruplicate and standards in triplicate. Relative quantification was made following the standard curve method. Results were averaged and normalized by dividing mean values of the Oxtr or Avpr1a gene by mean values of Gapdh, the endogenous reference gene.
Single-cell gene expression analysis.
Three adult male and three adult female C57BL/6 mice (The Jackson Laboratory) were killed, and their DRGs were rapidly transferred into PIPES buffer supplemented with 10 mm glucose.
DRGs were cleaned of conjunctive tissue with fine forceps and transferred into oxygenated PIPES to which 0.5 mg/ml proteases X and XIV (Sigma-Aldrich) had been added. After 30 min of enzymatic digestion, DRGs were rinsed for 30 min in oxygenated PIPES, and then triturated and plated on Petri dishes. Cell harvesting was performed under a phasic microscope (Olympus IX71) equipped with an electrode manipulator. Glass electrodes with polished ends were filled with 1.5 μl of PIPES containing 10% RNase inhibitor and used to aspire individual cells. For subsequent determination of size, a picture of each cell was taken at 40× magnification. ImageJ software was used to measure cell diameter. Immediately after harvesting, the content of each cell was expelled into an individual 200 μl PCR tube. DNase treatment and first-strand cDNA synthesis were performed using the Superscript III Reverse Transcriptase kit (Invitrogen). Avpr1a and Gapdh transcripts were amplified with nested multiplex PCR in 74 individual cell samples as previously described by Hoyda et al. (2007) using a QIAGEN Multiplex PCR kit (QIAGEN). Water blanks and samples of the dissociation buffer were used as negative controls. Whole DRG samples were positive controls. Primers were designed based on Ensembl database cDNA sequences using Primer3 software (Rozen and Skaletsky, 2000). The following primers were used for first-round amplification: Avpr1a, 5′-CCTACATGCTGGTGGTGATG-3′ (forward) and 5′-TCTTCACTGTGCGGATCTTG-3′ (reverse); Gapdh, 5′-AACTTTGGCATTGTGGAAGG-3′ (forward) and 5′-CCCTGTTGCTGTAGCCGTAT-3′ (reverse). For second-round (nested) amplification, the forward and reverse primer sequences were as follows: Avpr1a, 5′-GGGCTGAGTTTCGTTCTGAG-3′ and 5′-GAAATGCTCTTCACGCTGCT-3′, respectively; Gapdh, 5′-ACCCAGAAGACTGTGGATGG-3′ and 5′-AGGAGACAACCTGGTCCTCA-3′, respectively. Final amplification products had a predicted size of 338 bp (Avpr1a) and 301 bp (Gapdh) and were separated by electrophoresis on a cyanine dye-stained (SYBR Gold; Invitrogen) 2% agarose gel.
Statistical analyses.
Statistical analysis was performed using Prism (GraphPad). Criterion significance level was set at p < 0.05 for all tests, and post hoc analyses were only performed when appropriate. Variance is illustrated in figures as the SEM.
Data from the behavioral phenotyping were analyzed using one-way ANOVA. Behavioral data from pharmacological experiments were analyzed using two-way ANOVA with genotype and dose as between-subject factors. Dunnett's case-comparison post hoc analysis was used for pairwise comparisons between vehicle and different doses of OXT or AVP within each genotype; where only one dose was used, significance was established by Student's t test. FlashCalc 31.5 software (M. Ossipov, University of Arizona, Tucson, AZ) was used to calculate half-maximal effective dose (ED50) values and associated 95% confidence intervals—using the method of Tallarida and Murray (1981)—for the analgesic efficacy of OXT and AVP in the paw withdrawal test. Oxtr and Avpr1a quantitative gene expression were compared using Student's t test (two-tailed).
Results
Nociceptive phenotype of OTR KO and V1AR KO mice
Table 1 shows that nociceptive sensitivity of OTR KO mice is identical with that of their WT and HET littermates in a battery of eight nociceptive assays, including tests for thermal, mechanical, and chemical/inflammatory nociception. No significant main effects of genotype were detected in any assay. In four assays (53°C hot-plate, 49°C tail withdrawal, 15% paw withdrawal, and von Frey tests), significant main effects of sex were observed—in every case with females more sensitive than males, as has been previously reported (Mogil et al., 2006)—but no significant genotype by sex interactions were observed, so data were pooled from both sexes for presentation.
Table 1.
Phenotyping of WT, HET, and OTR KO mice on a battery of nociceptive assays
| Assay | Intensity | Units | OTR |
V1AR |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| na | WTb | HET | KO | p valuec | na | WTb | HET | KO | p valuec | |||
| Thermal | ||||||||||||
| Tail withdrawal | 49°C | s | 16 | 2.7 ± 0.2 | 2.5 ± 0.1 | 2.6 ± 0.1 | 0.74 | 13–18 | 2.1 ± 0.1 | 2.4 ± 0.1 | 2.1 ± 0.1 | 0.08 |
| 47°C | s | 16 | 4.1 ± 0.1 | 4.1 ± 0.1 | 4.1 ± 0.1 | 0.93 | 13–18 | 3.9 ± 0.1 | 3.9 ± 0.1 | 3.7 ± 0.1 | 0.64 | |
| Hot plate | 53°C | s | 16 | 15.1 ± 1.0 | 15.2 ± 1.4 | 14.3 ± 1.0 | 0.82 | 13–18 | 14.7 ± 1.1 | 14.3 ± 1.0 | 17.4 ± 1.0 | 0.08 |
| 50°C | s | 16 | 29.1 ± 2.9 | 27.8 ± 3.2 | 26.1 ± 2.0 | 0.75 | 13–18 | 27.6 ± 1.3 | 28.1 ± 1.7 | 23.3 ± 3.0 | 0.21 | |
| Paw withdrawal | 20% | s | 16 | 9.5 ± 0.6 | 10.5 ± 0.5 | 11.5 ± 0.7 | 0.08 | 14–16 | 7.4 ± 0.3 | 8.0 ± 0.4 | 8.4 ± 0.4 | 0.14 |
| 15% | s | 16 | 15.1 ± 0.8 | 15.9 ± 0.8 | 7.1 ± 1.0 | 0.26 | 14–16 | 12.3 ± 0.8 | 12.2 ± 0.5 | 12.5 ± 0.5 | 0.88 | |
| Mechanical | ||||||||||||
| Tail clip | ≈700 g | s | 20 | 2.1 ± 0.5 | 4.7 ± 1.4 | 2.8 ± 1.1 | 0.14 | 7–8 | 2.2 ± 0.7 | 1.6 ± 0.5 | 2.4 ± 0.3 | 0.52 |
| von Frey | 0.015–2.0 g | g | 20 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.0 ± 0.1 | 0.54 | 6–8 | 1.1 ± 0.1 | 0.9 ± 0.1 | 1.2 ± 0.1 | 0.08 |
| Chemical | ||||||||||||
| Writhing | 0.9% | %d | 14–15 | 23.6 ± 2.4 | 25.2 ± 3.0 | 18.6 ± 3.2 | 0.28 | 10–12 | 27.8 ± 4.4 | 31.7 ± 3.8 | 26.1 ± 4.5 | 0.62 |
| Formalin (early) | 2.5% | % | 16 | 45.0 ± 6.4 | 37.5 ± 6.0 | 33.8 ± 6.5 | 0.45 | 9–17 | 55.6 ± 8.7 | 52.0 ± 4.9 | 55.0 ± 5.6 | 0.90 |
| Formalin (late) | 2.5% | % | 16 | 18.4 ± 2.8 | 21.7 ± 2.3 | 19.4 ± 2.4 | 0.64 | 9–17 | 24.6 ± 2.9 | 30.8 ± 2.8 | 35.2 ± 1.8 | 0.04* |
| Inflammatory | ||||||||||||
| Zymosan | 2.5 μg | %e | 16 | 67.4 ± 1.8 | n.d. | 68.7 ± 2.0 | 0.62 | 12 | 61.1 ± 4.6 | n.d. | 54.3 ± 3.3 | 0.24 |
n.d., Not determined.
aSample size per genotype.
bAll values represent mean ± SEM.
cFrom a one-way ANOVA of genotype.
dPercentage of positive samples featuring writhing or licking/biting behavior.
ePercentage of maximum possible hyperalgesia using areas over the time–response curve (Mogil et al., 2006).
*p < 0.05.
Table 1 further shows that V1AR WT, HET, and KO mice also generally responded similarly to each other. Significant main effects of genotype were obtained only in one assay, the late phase of the formalin test, in which V1AR KOs displayed significantly increased sensitivity compared with WT mice (p < 0.05). In four assays (49 and 47°C tail withdrawal, 15 and 20% paw withdrawal tests), significant main effects of sex were observed, again with females being more sensitive.
The power to detect genotype differences in both mutant lines was comparatively high, since sample sizes used herein are large compared with standard practices [Mogil et al. (2005), their Fig. 5].
Figure 5.
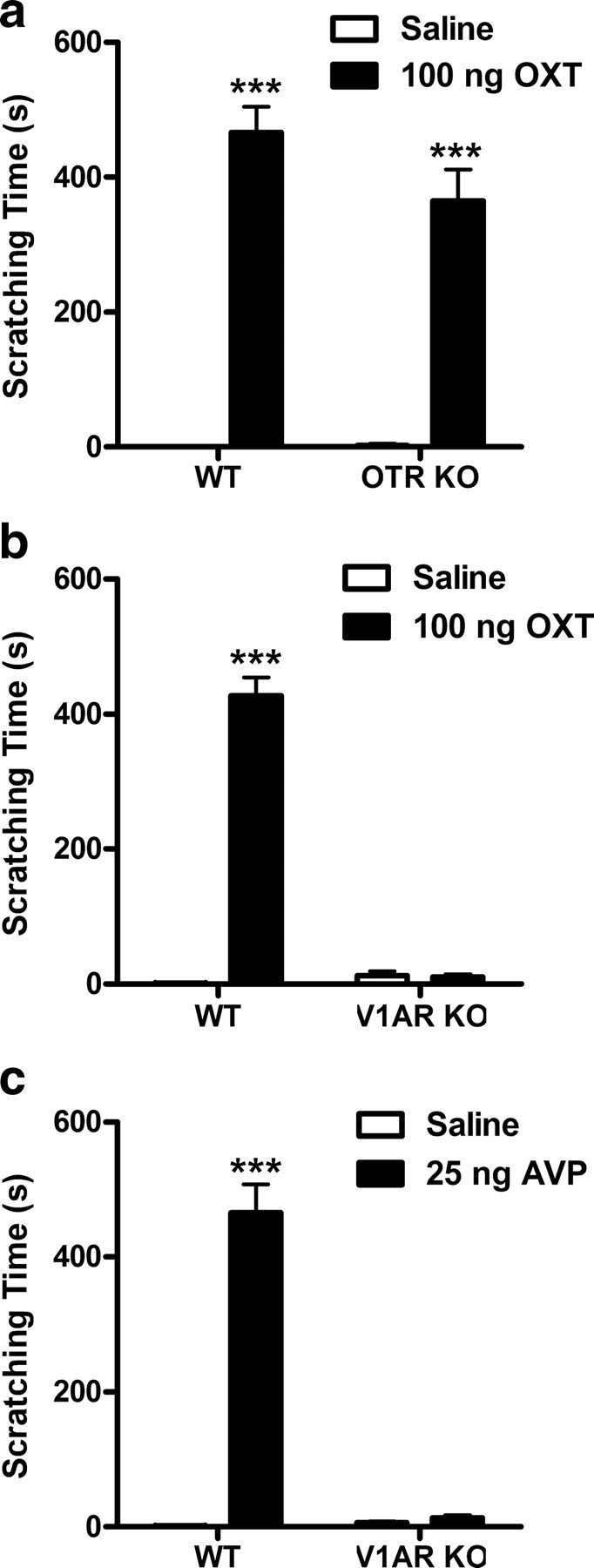
OXT (100 ng) administered intracerebroventricularly produces a robust and long-lasting scratching response in WT and OTR KO (a), but not in V1AR KO mice (b) (n = 7–8/group). AVP (25 ng, i.c.v.) also elicits scratching in WT, but not in V1AR KO mice (c) (n = 7–8/group). ***p < 0.001, compared with saline vehicle. Error bars indicate SEM.
Oxytocin- and vasopressin-induced analgesia in OTR KO and V1AR KO mice
OXT injected systemically dose-dependently increased paw withdrawal latencies in WT and OTR KO mice in the paw withdrawal test of thermal nociception (Fig. 1a). Two-way ANOVA confirmed a main effect of oxytocin dose (F(3,73) = 23.4; p < 0.001). According to Dunnett's case comparison post hoc tests, effective doses of OXT were 2 and 8 mg/kg for both genotypes (p < 0.01). A dose of 8 mg/kg OXT produced ∼60% of maximum possible analgesia (with the arbitrary cutoff latency chosen); a higher dose (16 mg/kg) (data not shown) was no more effective.
Figure 1.
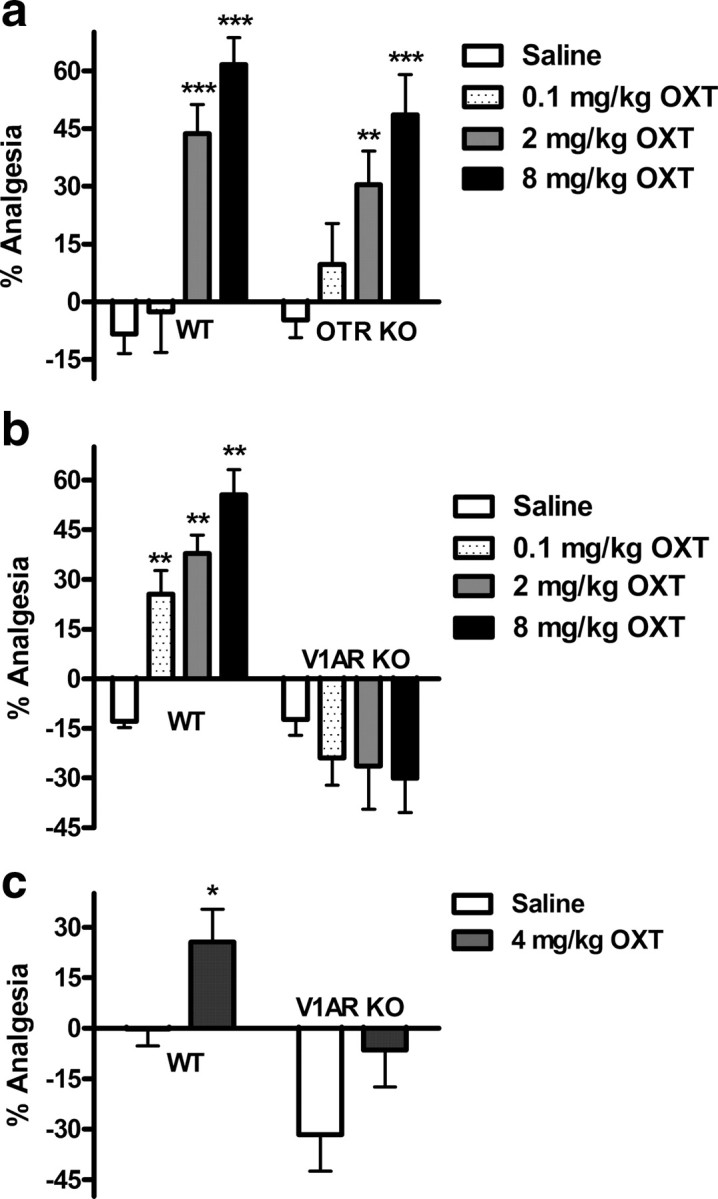
OXT-induced analgesia in the radiant-heat paw withdrawal test (a, b) and von Frey test (c) of mechanical nociception is V1AR dependent. Systemically administered OXT dose-dependently produces analgesia in WT and OTR KO (a), but not in V1AR KO mice (b) (n = 7–16/group). A 4 mg/kg dose of OXT produces analgesia in WT but not V1AR KO mice (c) (n = 8/group). *p < 0.05, **p < 0.01, ***p < 0.001, compared with corresponding saline group. Error bars indicate SEM.
Although still present in V1AR WT mice, OXT-induced analgesia was completely absent in V1AR KO mice (Fig. 1b). Two-way ANOVA revealed a main effect of oxytocin dose (F(3,79) = 3.6; p < 0.05) and of genotype (F(1,79) = 78.1; p < 0.001), as well as a significant dose by genotype interaction (F(3,79) = 10.6; p < 0.001). Follow-up one-way ANOVA and Dunnett's post hoc testing on data from V1AR WT mice revealed that OXT effectively induced analgesia at doses of 0.1, 2, and 8 mg/kg (p < 0.01) compared with saline.
The V1AR-dependent analgesic effect of systemic OXT was confirmed using the von Frey test of mechanical sensitivity. Using a dose of 4 mg/kg, we observed a significant analgesia (i.e., increase in paw withdrawal threshold) in V1AR WT mice compared with saline or zero (one-way Student's t(14) = 2.3, p < 0.05, or t(7) = 2.6, p < 0.05, respectively) that was absent in their KO littermates (Fig. 1c).
Systemic OXT was similarly analgesic in the formalin test of inflammatory nociception. In OTR KO and WT mice, there was a main effect of oxytocin on nocifensive (licking/biting) behavior in both the early (Fig. 2a) (F(1,24) = 5.4; p < 0.05) and late phase (Fig. 2b) (F(1,24) = 23.7; p < 0.01) of the test. In contrast, although still reducing the pain behavior in V1AR WT mice, no OXT-induced analgesia was seen in V1AR KOs in either the early (Fig. 2c) or the late (Fig. 2d) phase of the formalin test. In the early phase, only the main effect of genotype was significant (F(1,24) = 15.4; p < 0.001), although the genotype by oxytocin interaction approached significance (F(1,24) = 2.5; p = 0.13). In the late phase, significant main effects of oxytocin (F(1,24) = 12.9; p < 0.01) and genotype (F(1,24) = 5.5; p < 0.05), as well as a significant interaction (F(1,24) = 4.5; p < 0.05) were observed. In WTs only, OXT significantly reduced licking behavior in the late phase (p < 0.01).
Figure 2.

OXT-induced analgesia in the formalin test is V1AR dependent. A dose of 4 mg/kg OXT reduces licking behavior in WT and OTR KO mice in both the early (a) and late (b) phases (n = 6–8/group). In V1AR KO mice, the analgesic effect of OXT is abolished in both the early (c) and late (d) phases (n = 6–8/group). *p < 0.05, **p < 0.01, compared with corresponding saline group. Error bars indicate SEM.
Systemically administered AVP also induced analgesia in WT, but not in V1AR KO mice (Fig. 3a). A two-way ANOVA revealed a significant main effect of genotype (F(1,49) = 14.2; p < 0.001) and a significant dose by genotype interaction (F(3,49) = 2.9; p < 0.05). AVP was found to be more potent than OXT, since 50% analgesia was obtained with a dose of 0.5 mg/kg already (p < 0.05 compared with saline). Again, higher AVP doses demonstrated no greater efficacy (data not shown). All these conclusions are also supported by estimated ED50 values and potency ratios (Table 2).
Figure 3.

Exogenous and endogenous AVP analgesia is V1AR dependent. AVP injected systemically is analgesic in WT, but not in V1AR KO mice (a) (n = 6–9/group). The graph in b shows the effect of osmotic stress, known to release endogenous AVP. Intraperitoneal injection of 1 m but not 150 mm (physiological) saline produces analgesia in WT but not V1AR KO mice (n = 6–10/group). *p < 0.05, compared with corresponding saline group; **p < 0.01, compared with corresponding 150 mm (physiological saline) group or other genotype. Error bars indicate SEM.
Table 2.
Half-maximal effective doses (ED50 values) and associated 95% confidence intervals (CIs) for systemic OXT- and AVP-induced analgesia in the paw withdrawal test
| Analgesic/genotype | ED50 | 95% CI |
|---|---|---|
| Oxytocin-induced analgesia | ||
| OTR WT | 3.3 | 1.6–7.0 |
| OTR KO | 14.3 | 1.3–158 |
| V1AR WT | 6.4 | 1.8–22.4 |
| V1AR KO | n.c. | n.c. |
| Vasopressin-induced analgesia | ||
| V1AR WT | 0.5 | 0.1–2.0 |
| V1AR KO | n.c. | n.c. |
All values are in milligrams per kilogram. Note that these ED50 estimates are extrapolations because of submaximal analgesia, and their absolute values should be treated with caution. n.c., Not calculable.
Systemic hyperosmotic challenge, a known trigger of endogenous AVP release (Robertson et al., 1976), also elicited analgesia in the paw withdrawal test in WT mice at 1 m [one-way t18 = 3.1, p < 0.01, compared with 150 mm (physiological saline)], but not in V1AR KO mice (Fig. 3b).
Oxytocin and vasopressin-1A receptor antagonists
To further address the hypothesis that the analgesic effects of OXT are actually mediated by the V1AR, OTR- or V1AR-selective antagonists were administered 10 min before injection of 4 mg/kg OXT. When given systemically (Fig. 4a), only the V1AR antagonist could dose-dependently prevent OXT-induced analgesia. One-way ANOVA confirmed a main effect of antagonist treatment (F(3,28) = 17.1; p < 0.001). Likewise, a supraspinally administered V1AR antagonist (Fig. 4b) prevented OXT-induced analgesia completely and dose-dependently (F(5,45) = 5.3; p < 0.001). Interestingly, in this compartment, a higher dose of the OTR antagonist displayed a partial inhibitory effect on OXT-induced analgesia. The same results were obtained using spinal injections of OTR and V1AR antagonists (Fig. 4c). Again, OXT-induced analgesia was fully and dose-dependently prevented by the V1AR antagonist (F(4,40) = 16.9; p < 0.001), whereas the OTR antagonist could only partially inhibit the effect of OXT, even when used at higher doses.
Figure 4.
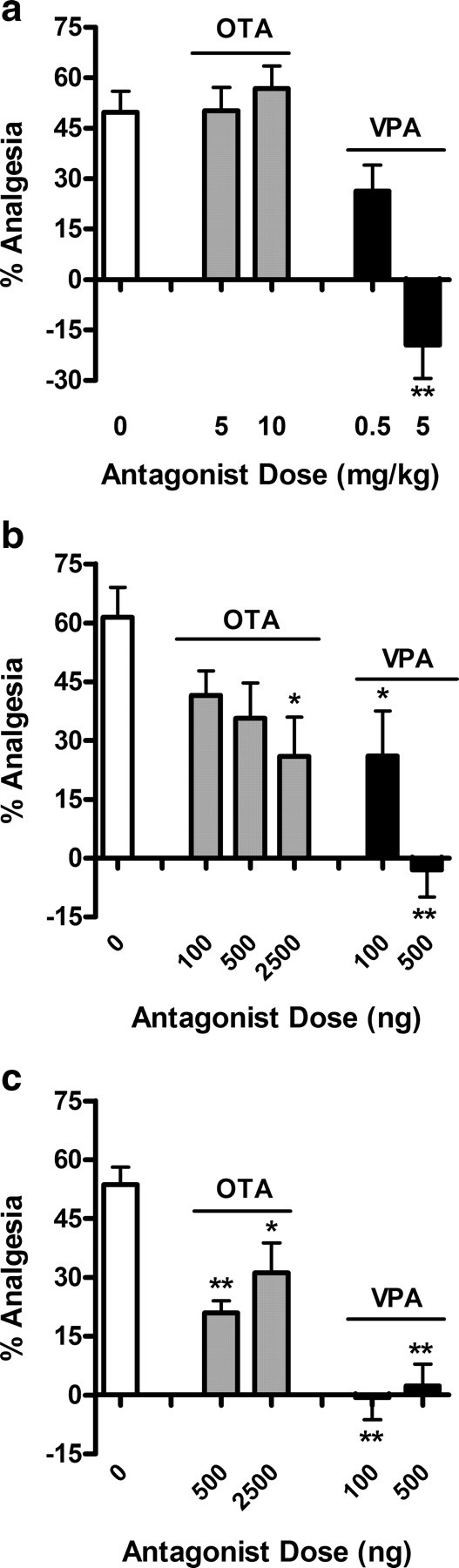
A selective V1AR antagonist (VPA), but not a selective OTR antagonist (OTA), prevents OXT-induced analgesia in the radiant-heat paw withdrawal test (n = 6–8/dose/drug/route). When injected systemically (a), 5 mg/kg VPA fully inhibits, whereas OTA does not affect OXT-induced analgesia. Centrally (intracerebroventricularly) administered (b), a 500 ng dose of VPA is sufficient to block OXT-induced analgesia, whereas up to 2.5 μg of OTA only partly attenuated the effect of OXT. At the spinal level (intrathecal) (c), both antagonists display some efficacy in inhibiting OXT-induced analgesia, but only the VPA is fully effective, at both 100 and 500 ng. *p < 0.05, **p < 0.01, compared with saline group. Error bars indicate SEM.
Oxytocin- and vasopressin-induced scratching
It has been well documented that both OXT and AVP elicit strong scratching and grooming behaviors in rats (Van Wimersma Greidanus et al., 1990) and mice (Meisenberg and Simmons, 1982) when injected into the brain or spinal cord (Thurston et al., 1992). This vigorous response strongly interfered with nociceptive testing in every assay tried, and thus OXT and AVP were administered systemically in analgesia studies. Scratching induced by 100 ng of OXT acutely injected into the third ventricle was present in both WT and OTR KO mice (Fig. 5a) (main effect of oxytocin only: F(1,27) = 210.6, p < 0.001), but absent in V1AR KOs (Fig. 5b). In the latter data set, in addition to significant main effects of oxytocin (F(1,27) = 274.9; p < 0.001) and genotype (F(1,27) = 252.1; p < 0.001), there was also a significant interaction (F(1,27) = 279.6; p < 0.001). Scratching behavior occurred in bouts beginning immediately after injection, sometimes even before the animal had fully recovered from isoflurane anesthesia, and lasted for ∼20 min. Within a 30 min observation period, the cumulative amount of scratching bouts totaled ∼7 min.
Figure 5c shows that, in WT mice only, intracerebroventricular AVP elicited a similar duration of scratching at a dose of 25 ng (p < 0.001 compared with saline). This response was absent in V1AR KO mice. The main effects of vasopressin (F(1,27) = 153.2) and of genotype (F(1,27) = 138.8), as well as the vasopressin by genotype interaction (F(1,27) = 144.0) were all highly significant (all p < 0.001).
Spinal OTR and V1AR distribution
Figures 6 and 7 illustrate the distribution of OXT and AVP receptor binding sites, respectively, in mouse spinal cord at the level of the lumbar enlargement (L4–L6). OXT binding sites are restricted to the superficial layers of the dorsal horn (Fig. 6a). This densely labeled band corresponds to laminae I and II of the dorsal horn, and is absent in sections from OTR KO mice (Fig. 6d). In contrast, Figure 7a shows that AVP binding sites are diffusely present and weakly labeled in all laminae of the spinal cord, with only a slight increase of labeling intensity in the dorsal horn. As expected, AVP binding sites are absent in sections from V1AR KO mice (Fig. 7c). Tissue was obtained from both male and female mice, and no obvious sex difference in spinal distribution of OXT or AVP binding sites was observed (data not shown).
Figure 6.

Autoradiographic localization of specific 125I-OTA binding sites in mouse brain and spinal cord. In WT (a) and V1AR KO mice (c), OTR binding is dense in all superficial laminae of the lumbar spinal cord. OTR binding is absent in OTR KO mice (d). In WT mice, OTR binding is dense in the ventromedial hypothalamus (e, arrow). OTR binding is displaced by coincubation of radioligand with 2 μm cold OXT (b, f). Scale bars: d, 500 μm; f, 1500 μm.
Figure 7.

Autoradiographic localization of specific 125I-VPA binding sites in mouse brain and spinal cord. In WT (a) and OTR KO mice (d), V1AR binding is diffuse in all regions of the lumbar spinal cord. V1AR binding is absent in V1AR KO mice (c). In WT mice, V1AR binding is dense in the lateral septum (e, arrow). V1AR binding is displaced by 2 μm cold AVP (b, f). Scale bars: d, 500 μm; f, 1500 μm.
The specificity of the radioligands used was controlled for by competition with unlabeled ligand (Figs. 6b, 7b), as well as by processing of brain sections of WT animals. Figures 6e and 7e show that 125I-OTA binding sites are enriched in the ventromedial hypothalamus (arrow), whereas 125I-VPA binds to the lateral septum (arrow), both regions where abundant expression of OTR and V1AR, respectively, has been well documented (Barberis and Tribollet, 1996). Binding of radioligands was successfully competed by unlabeled OXT or AVP, respectively (Figs. 6f, 7f).
Oxtr and Avpr1a gene expression in DRG
In the final experiment, Oxtr and Avpr1a gene expression levels were measured in DRG samples of WT mice. Oxtr mRNA was only barely expressed in mouse DRG (0.4 ± 0.2 arbitrary units compared with Gapdh), whereas Avpr1a mRNA expression levels were ≈10-fold higher (4.3 ± 0.1 arbitrary units compared with Gapdh; p < 0.001 by Student's t test). To further characterize Avpr1a expression at the cellular level in the DRG, a single-cell RT-PCR approach was adopted. Figure 8a shows a scatterplot illustrating that, of the total of 74 sampled DRG neurons, 25 (33.8%) expressed Avpr1a. Interestingly, Avpr1a-positive neurons were predominantly of small (<20 μm) or medium (20–30 μm) diameter. In contrast, Avpr1a mRNA was detected in only two (2.8%) large-diameter neurons (>30 μm). The PCR products obtained from nine DRG cells, representative of the large-, medium-, and small-size populations of DRG neurons, are illustrated in Figure 8b, and representative large- and small-diameter cells are pictured in Figure 8, c and d.
Figure 8.
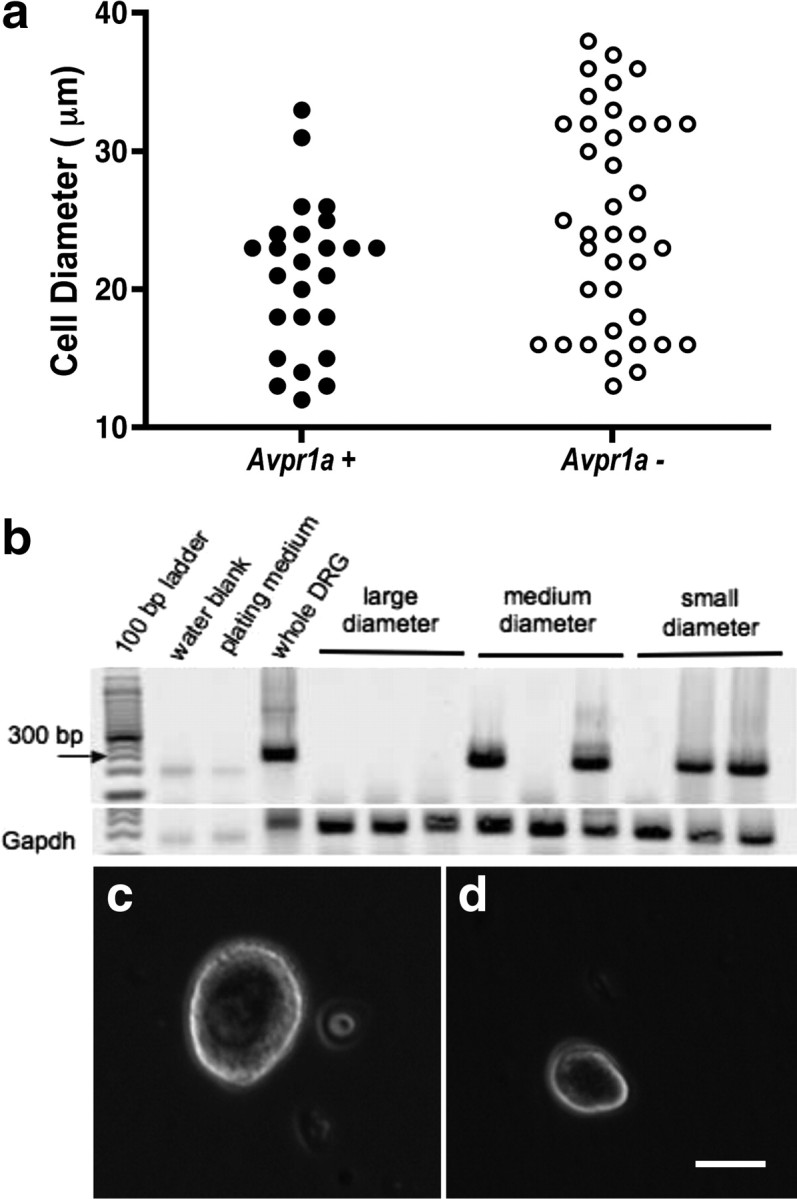
Avpr1a gene expression in individual DRG cells. a, Scatterplot illustrating the proportion of Avpr1a mRNA-expressing DRG neurons of different size. b, Gel showing nine single DRG cells positive or negative for Avpr1a. c, d, Examples of large-diameter (c) and small-diameter (d) dissociated DRG neurons. Scale bar, 15 μm.
Discussion
Using a battery of assays of acute and tonic pain sensitivity, we systematically investigated mutant mice lacking the OTR or the V1AR. Mice of both KO lines displayed nociceptive phenotypes mostly similar to their WT and HET littermates. We note that neuropathic pain states were not assessed here, and thus it remains possible that the OTR and/or the V1AR play as yet undetermined roles in the mediation of mechanical or thermal hypersensitivity in chronic neuropathic pain. The major finding of this study is that systemically administered OXT produced robust, dose-dependent analgesia in OTR KO mice, but not in V1AR KO mice, in three distinct nociceptive assays. Likewise, the analgesic effects of OXT could be fully prevented by a V1AR-selective antagonist, but not by an OTR-selective antagonist. Scratching, a behavior elicited by centrally administered OXT and AVP, was also absent in V1AR KO mice, but intact in OTR KO mice, generalizing our findings beyond nociception itself. Finally, receptor binding and gene transcription experiments demonstrated that, whereas the OTR predominates in pain-relevant regions of the mouse lumbar spinal cord, DRGs mostly express the V1AR. Thus, it appears that OXT acts via the V1AR to modulate pain and scratching behaviors.
Pain sensitivity is normal in OTR KO and V1AR KO mice
A number of studies have reported on the analgesic actions of exogenously administered neurohypophyseal hormones against different pain modalities (Lundeberg et al., 1994; Ge et al., 2002; Yu et al., 2003; Gao and Yu, 2004; Koshimizu and Tsujimoto, 2009), but little is known about the role of endogenous OXT and AVP in pain processing.
The anatomical distribution of OXT and OTRs in rat strongly supports such a role. OTRs are enriched in pain-relevant laminae I and II of the dorsal horn (Reiter et al., 1994; Tribollet et al., 1997; Véronneau-Longueville et al., 1999) and are a likely target of spinal OXTergic projections from the paraventricular nucleus of the hypothalamus (Swanson and Kuypers, 1980; Puder and Papka, 2001; Condés-Lara et al., 2007). A few functional studies have linked paraventricular activity to analgesia, which was mimicked by OXT and prevented by OTR antagonists (Condés-Lara et al., 2006; Miranda-Cardenas et al., 2006; Rojas-Piloni et al., 2007). Here, we used mice deficient in OTRs or V1ARs to investigate the physiological contribution of these neurohypophyseal peptides to pain control and found that the nociceptive phenotype of both KO lines was virtually identical with WT control animals. Although this may indicate that endogenous OXT and AVP do not significantly contribute to (non-neuropathic) pain processing, we cannot exclude the possibility of compensatory mechanisms in the mutant mice.
Systemic oxytocin elicits analgesia through the V1AR
As mentioned above, anatomical data point toward the oxytocinergic system as a strategically located player in spinal pain processing. A number of groups have studied the cellular mechanisms by which spinal OTRs contribute to analgesia (Jo et al., 1998; Condés-Lara et al., 2003; Rojas-Piloni et al., 2007). Recently, it has been suggested that OXT could act on glutamatergic interneurons located in lamina II of the dorsal horn, which in turn activate a larger population of inhibitory GABAergic interneurons to reduce the net activity of spinal nociceptive neurons (Breton et al., 2008). Contrary to what would be expected based on this information, we found that OXT analgesia was fully intact in OTR KO mice, but entirely absent in V1AR KOs. Similarly, when endogenous AVP release was precipitated by a hyperosmotic stressor, OTR KO, but not V1AR KO mice developed analgesia, suggesting that under physiological conditions as well the V1AR is necessary for mediating the analgesic effects of increased circulating neurohypophyseal hormones. Together, these results strongly suggest that, at least systemically, OXT acts via V1ARs, rather than OTRs to produce its analgesic effects. This hypothesis was confirmed by studies using V1AR- and OTR-selective antagonists. It is interesting to note that, when administered systemically, only the V1AR antagonist could prevent OXT-induced analgesia, whereas when injected intracerebroventricularly or intrathecally, the OTR antagonist also displayed a partial inhibitory effect on OXT-induced analgesia. This observation suggests that, centrally, and in particular at the spinal cord level, both receptors contribute to the analgesic properties of OXT, whereas peripherally, the V1AR is solely responsible for OXT-induced analgesia.
In the brain, OXT contributes to the regulation of numerous behaviors and physiological functions (Gimpl and Fahrenholz, 2001; Kiss and Mikkelsen, 2005; Nishimori et al., 2008). We tried to investigate the receptor basis of OXT modulation of anxiety- and depression-like behaviors—using the elevated plus maze and Porsolt forced-swimming tasks, respectively—but we failed to obtain any reliable evidence of dose-dependent OXT effects in WT mice using various routes of administration (data not shown). Interestingly, however, one central effect of OXT, scratching, was also absent in V1AR KOs, while remaining intact in OTR KOs. In light of the growing number of studies on neurohypophyseal hormones as key regulators of pair bonding and affiliative behaviors (Young and Wang, 2004; Storm and Tecott, 2005; Lim and Young, 2006; Caldwell et al., 2008; Donaldson and Young, 2008; Young et al., 2008), our results should be considered before making any definite conclusions as to the receptor type mediating effects of these peptides.
Oxytocin binding sites in superficial layers of the dorsal horn
On the basis of the high sequence homology of both peptides and their receptors, an action of OXT via the V1AR is highly plausible (Chini and Manning, 2007). In contrast, it is difficult to reconcile a V1AR-mediated analgesic effect of OXT with anatomical evidence demonstrating that OTRs are enriched in the superficial layers of the dorsal horn, whereas V1ARs are only weakly expressed in the adult spinal cord (Tribollet et al., 1997). We would note that the vast majority of the anatomical literature is from rat, whereas our pharmacological experiments were performed in mice. Although two studies have systematically compared OTR and V1AR distribution in mouse CNS, one of them focused on forebrain structures (Insel et al., 1991), whereas the other examined spinal distribution only as caudally as the trigeminal nucleus caudalis (Tribollet et al., 2002). One putative explanation for the V1AR dependency of OXT-induced analgesia would be a species difference in receptor distribution. Indeed, whereas OXTergic and AVPergic projections are evolutionary well conserved, marked species differences in brain receptor distribution patterns have been reported (Tribollet et al., 1988, 1992; Dubois-Dauphin et al., 1996; Yoshida et al., 2009).
To investigate whether the spinal distribution of OTRs and V1ARs in mice is distinct from that seen in the rat, an autoradiographic study on lumbar spinal cord sections was performed. Results clearly show that, globally, murine OTR and V1AR distribution is similar to that seen in rat, with OTRs, but not V1ARs, clustering in superficial layers of the dorsal horn. Although a slightly higher density of AVP binding sites in dorsal versus ventral parts of the spinal cord was observed, the intensity of dorsal V1AR labeling remains weak and is not a likely explanation for the above-reported V1AR-dependent OXT analgesia.
Avpr1a mRNA is expressed in dorsal root ganglia
One possible limitation of the present study is that OXT was given intraperitoneally in analgesia experiments, rather than directly into the brain or spinal cord. The rationale for the peripheral route of administration was to circumvent the characteristic and vigorous scratching response elicited by both intracerebroventricular and intrathecal OXT injections (Meisenberg and Simmons, 1982; Van Wimersma Greidanus et al., 1990; Thurston et al., 1992) to maintain the animal in a state allowing for accurate algesiometric testing (Wilson and Mogil, 2001). Since OXT and AVP are low-molecular-weight peptides, it is highly likely that they can cross the blood–brain barrier. Indeed, Jin et al. (2007) have reported that subcutaneously injected OXT can affect centrally mediated behaviors. Alternatively, intraperitoneally injected OXT could act peripherally. In the peripheral nervous system, nerve endings innervating the skin originate in primary sensory neurons located in the DRG. In fact, these cell bodies represent the first locus of nociceptive processing, since they relay incoming pain signals to supraspinally projecting second-order sensory neurons of the dorsal horn (Morris et al., 2004). Our gene expression studies showed that Avpr1a mRNA levels greatly exceed those of Oxtr in the mouse DRG. This is in contrast to the dorsal horn, where OTR expression greatly exceeds V1AR expression. Single-cell gene expression experiments revealed that ∼34% of DRG neurons are positive for Avpr1a mRNA. Based on their size (<30 μm), these neurons probably belong to the population of small- and medium-sized unmyelinated nociceptors. Thus far, the role of V1ARs in the DRG has received minimal attention, but Hu et al. (2004) reported a hyperpolarizing effect of AVP in DRG neurons. Thus, it is tempting to speculate that peripherally administered AVP and OXT both produce analgesia through V1AR-mediated inhibition of DRG neurons. Additional experiments will be needed to explore this hypothesis. In particular, it would be useful to conduct behavioral pain tests after AVP or OXT administration to discrete regions of the CNS via microinjection or viral-mediated delivery.
Conclusions
In addition to demonstrating that analgesia induced by systemic OXT is in fact mediated by the V1AR, we identify V1ARs expressed in DRGs as a potential target for this effect. Moreover, the scratching behavior provoked by intracerebroventricular OXT is also mediated by the V1AR. These findings invite reflection on the receptor substrates and cellular mechanisms of other effects commonly attributed to OXTergic signaling.
Footnotes
This work was supported by the National Institutes of Health (J.S.M.), the Canadian Institutes for Health Research (R.Q.), and the Swiss National Foundation for Scientific Research (E.T.). We express our gratitude to Dr. Maurice Manning for donating V1AR and OTR antagonists.
References
- Arnaudeau S, Leprêtre N, Mironneau J. Oxytocin mobilizes calcium from a unique heparin-sensitive and thapsigargin-sensitive store in single myometrial cells from pregnant rats. Pflugers Arch. 1994;428:51–59. doi: 10.1007/BF00374751. [DOI] [PubMed] [Google Scholar]
- Barberis C, Tribollet E. Vasopressin and oxytocin receptors in the central nervous system. Crit Rev Neurobiol. 1996;10:119–154. doi: 10.1615/critrevneurobiol.v10.i1.60. [DOI] [PubMed] [Google Scholar]
- Barberis C, Balestre MN, Jard S, Tribollet E, Arsenijevic Y, Dreifuss JJ, Bankowski K, Manning M, Chan WY, Schlosser SS, Holsboer F, Elands J. Characterization of a novel, linear radioiodinated vasopressin antagonist: an excellent radioligand for vasopressin V1a receptors. Neuroendocrinology. 1995;62:135–146. doi: 10.1159/000126998. [DOI] [PubMed] [Google Scholar]
- Baumgartner T, Heinrichs M, Vonlanthen A, Fischbacher U, Fehr E. Oxytocin shapes the neural circuitry of trust and trust adaptation in humans. Neuron. 2008;58:639–650. doi: 10.1016/j.neuron.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Birnbaumer M. Vasopressin receptors. Trends Endocrinol Metab. 2000;11:406–410. doi: 10.1016/s1043-2760(00)00304-0. [DOI] [PubMed] [Google Scholar]
- Breton JD, Veinante P, Uhl-Bronner S, Vergnano AM, Freund-Mercier MJ, Schlichter R, Poisbeau P. Oxytocin-induced antinociception in the spinal cord is mediated by a subpopulation of glutamatergic neurons in lamina I-II which amplify GABAergic inhibition. Mol Pain. 2008;4:19. doi: 10.1186/1744-8069-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell HK, Lee HJ, Macbeth AH, Young WS., 3rd Vasopressin: behavioral roles of an “original” neuropeptide. Prog Neurobiol. 2008;84:1–24. doi: 10.1016/j.pneurobio.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BL, Gil AS, Levesque A, Mogil JS. Modulation of mechanical and thermal nociceptive sensitivity in the laboratory mouse by behavioral state. J Pain. 2008;9:174–184. doi: 10.1016/j.jpain.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Chini B, Manning M. Agonist selectivity in the oxytocin/vasopressin receptor family: new insights and challenges. Biochem Soc Trans. 2007;35:737–741. doi: 10.1042/BST0350737. [DOI] [PubMed] [Google Scholar]
- Ciura S, Bourque CW. Transient receptor potential vanilloid 1 is required for intrinsic osmoreception in organum vasculosum lamina terminalis neurons and for normal thirst responses to systemic hyperosmolality. J Neurosci. 2006;26:9069–9075. doi: 10.1523/JNEUROSCI.0877-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condés-Lara M, González NM, Martínez-Lorenzana G, Delgado OL, Freund-Mercier MJ. Actions of oxytocin and interactions with glutamate on spontaneous and evoked dorsal spinal cord neuronal activities. Brain Res. 2003;976:75–81. doi: 10.1016/s0006-8993(03)02690-8. [DOI] [PubMed] [Google Scholar]
- Condés-Lara M, Rojas-Piloni G, Martínez-Lorenzana G, Rodríguez-Jiménez J, López Hidalgo M, Freund-Mercier MJ. Paraventricular hypothalamic influences on spinal nociceptive processing. Brain Res. 2006;1081:126–137. doi: 10.1016/j.brainres.2006.01.050. [DOI] [PubMed] [Google Scholar]
- Condés-Lara M, Martínez-Lorenzana G, Rojas-Piloni G, Rodríguez-Jiménez J. Branched oxytocinergic innervations from the paraventricular hypothalamic nuclei to superficial layers in the spinal cord. Brain Res. 2007;1160:20–29. doi: 10.1016/j.brainres.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Donaldson ZR, Young LJ. Oxytocin, vasopressin, and the neurogenetics of sociality. Science. 2008;322:900–904. doi: 10.1126/science.1158668. [DOI] [PubMed] [Google Scholar]
- Dubois-Dauphin M, Barberis C, de Bilbao F. Vasopressin receptors in the mouse (Mus musculus) brain: sex-related expression in the medial preoptic area and hypothalamus. Brain Res. 1996;743:32–39. doi: 10.1016/s0006-8993(96)01019-0. [DOI] [PubMed] [Google Scholar]
- Elands J, Barberis C, Jard S, Tribollet E, Dreifuss JJ, Bankowski K, Manning M, Sawyer WH. 125I-labelled d(CH2)5[Tyr(Me)2,Thr4,Tyr-NH29]OVT: a selective oxytocin receptor ligand. Eur J Pharmacol. 1988;147:197–207. doi: 10.1016/0014-2999(88)90778-9. [DOI] [PubMed] [Google Scholar]
- Gao L, Yu LC. Involvement of opioid receptors in the oxytocin-induced antinociception in the central nervous system of rats. Regul Pept. 2004;120:53–58. doi: 10.1016/j.regpep.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Ge Y, Lundeberg T, Yu LC. Blockade effect of mu and kappa opioid antagonists on the anti-nociception induced by intra-periaqueductal grey injection of oxytocin in rats. Brain Res. 2002;927:204–207. doi: 10.1016/s0006-8993(01)03346-7. [DOI] [PubMed] [Google Scholar]
- Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- Honda K, Takano Y. New topics in vasopressin receptors and approach to novel drugs: involvement of vasopressin V1a and V1b receptors in nociceptive responses and morphine-induced effects. J Pharmacol Sci. 2009;109:38–43. doi: 10.1254/jphs.08r30fm. [DOI] [PubMed] [Google Scholar]
- Hoyda TD, Fry M, Ahima RS, Ferguson AV. Adiponectin selectively inhibits oxytocin neurons of the paraventricular nucleus of the hypothalamus. J Physiol. 2007;585:805–816. doi: 10.1113/jphysiol.2007.144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HY, Sun ZP, Zhao YM, Si JQ, Zheng Y. Effect of arginine vasopressin on membrane potential of dorsal root ganglion neurons in rats (in Chinese) Sheng Li Xue Bao. 2004;56:107–111. [PubMed] [Google Scholar]
- Hu SB, Zhao ZS, Yhap C, Grinberg A, Huang SP, Westphal H, Gold P. Vasopressin receptor 1a-mediated negative regulation of B cell receptor signaling. J Neuroimmunol. 2003;135:72–81. doi: 10.1016/s0165-5728(02)00442-3. [DOI] [PubMed] [Google Scholar]
- Hylden JLK, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Insel TR, Gelhard R, Shapiro LE. The comparative distribution of forebrain receptors for neurohypophyseal peptides in monogamous and polygamous mice. Neuroscience. 1991;43:623–630. doi: 10.1016/0306-4522(91)90321-e. [DOI] [PubMed] [Google Scholar]
- Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, Islam MS, et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature. 2007;446:41–45. doi: 10.1038/nature05526. [DOI] [PubMed] [Google Scholar]
- Jo YH, Stoeckel ME, Freund-Mercier MJ, Schlichter R. Oxytocin modulates glutamatergic synaptic transmission between cultured neonatal spinal cord dorsal horn neurons. J Neurosci. 1998;18:2377–2386. doi: 10.1523/JNEUROSCI.18-07-02377.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss A, Mikkelsen JD. Oxytocin—anatomy and functional assignments: a minireview. Endocr Regul. 2005;39:97–105. [PubMed] [Google Scholar]
- Koshimizu TA, Tsujimoto G. New topics in vasopressin receptors and approach to novel drugs: vasopressin and pain perception. J Pharmacol Sci. 2009;109:33–37. doi: 10.1254/jphs.08r18fm. [DOI] [PubMed] [Google Scholar]
- Kruszynski M, Lammek B, Manning M, Seto J, Haldar J, Sawyer WH. [1-beta-Mercapto-beta,beta-cyclopentamethylenepropionic acid),2-(O-methyl)tyrosine]argine-vasopressin and [1-beta-mercapto-beta,beta-cyclopentamethylenepropionic acid)]argine-vasopressine, two highly potent antagonists of the vasopressor response to arginine-vasopressin. J Med Chem. 1980;23:364–368. doi: 10.1021/jm00178a003. [DOI] [PubMed] [Google Scholar]
- Ku CY, Qian A, Wen Y, Anwer K, Sanborn BM. Oxytocin stimulates myometrial guanosine triphosphatase and phospholipase-C activities via coupling to G alpha q/11. Endocrinology. 1995;136:1509–1515. doi: 10.1210/endo.136.4.7895660. [DOI] [PubMed] [Google Scholar]
- Langford DJ, Crager SE, Shehzad Z, Smith SB, Sotocinal SG, Levenstadt JS, Chanda ML, Levitin DJ, Mogil JS. Social modulation of pain as evidence for empathy in mice. Science. 2006;312:1967–1970. doi: 10.1126/science.1128322. [DOI] [PubMed] [Google Scholar]
- Laursen SE, Belknap JK. Intracerebroventricular injections in mice. Some methodological refinements. J Pharmacol Methods. 1986;16:355–357. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- Leng G, Ludwig M. Neurotransmitters and peptides: whispered secrets and public announcements. J Physiol. 2008;586:5625–5632. doi: 10.1113/jphysiol.2008.159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MM, Young LJ. Neuropeptidergic regulation of affiliative behavior and social bonding in animals. Horm Behav. 2006;50:506–517. doi: 10.1016/j.yhbeh.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Liu X, Tribollet E, Ogier R, Barberis C, Raggenbass M. Presence of functional vasopressin receptors in spinal ventral horn neurons of young rats: a morphological and electrophysiological study. Eur J Neurosci. 2003;17:1833–1846. doi: 10.1046/j.1460-9568.2003.02625.x. [DOI] [PubMed] [Google Scholar]
- Lundeberg T, Uvnäs-Moberg K, Agren G, Bruzelius G. Anti-nociceptive effects of oxytocin in rats and mice. Neurosci Lett. 1994;170:153–157. doi: 10.1016/0304-3940(94)90262-3. [DOI] [PubMed] [Google Scholar]
- Manning M, Miteva K, Pancheva S, Stoev S, Wo NC, Chan WY. Design and synthesis of highly selective in vitro and in vivo uterine receptor antagonists of oxytocin: comparisons with Atosiban. Int J Pept Protein Res. 1995;46:244–252. doi: 10.1111/j.1399-3011.1995.tb00596.x. [DOI] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain. 1997;1:43–52. doi: 10.1016/s1090-3801(97)90052-5. [DOI] [PubMed] [Google Scholar]
- Meisenberg G, Simmons WH. Behavioral effects of intracerebroventricularly administered neurohypophyseal hormone analogs in mice. Pharmacol Biochem Behav. 1982;16:819–825. doi: 10.1016/0091-3057(82)90242-8. [DOI] [PubMed] [Google Scholar]
- Miranda-Cardenas Y, Rojas-Piloni G, Martínez-Lorenzana G, Rodríguez-Jiménez J, López-Hidalgo M, Freund-Mercier MJ, Condés-Lara M. Oxytocin and electrical stimulation of the paraventricular hypothalamic nucleus produce antinociceptive effects that are reversed by an oxytocin antagonist. Pain. 2006;122:182–189. doi: 10.1016/j.pain.2006.01.029. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Chanda ML. The case for the inclusion of female subjects in basic science studies of pain. Pain. 2005;117:1–5. doi: 10.1016/j.pain.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Ritchie J, Sotocinal SG, Smith SB, Croteau S, Levitin DJ, Naumova AK. Screening for pain phenotypes: analysis of three congenic mouse strains on a battery of nine nociceptive assays. Pain. 2006;126:24–34. doi: 10.1016/j.pain.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Morris R, Cheunsuang O, Stewart A, Maxwell D. Spinal dorsal horn neurone targets for nociceptive primary afferents: do single neurone morphological characteristics suggest how nociceptive information is processed at the spinal level. Brain Res Brain Res Rev. 2004;46:173–190. doi: 10.1016/j.brainresrev.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Nishimori K, Takayanagi Y, Yoshida M, Kasahara Y, Young LJ, Kawamata M. New aspects of oxytocin receptor function revealed by knockout mice: sociosexual behaviour and control of energy balance. Prog Brain Res. 2008;170:79–90. doi: 10.1016/S0079-6123(08)00408-1. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Kalisch R, Singer T, Dolan RJ. Oxytocin attenuates affective evaluations of conditioned faces and amygdala activity. J Neurosci. 2008;28:6607–6615. doi: 10.1523/JNEUROSCI.4572-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phaneuf S, Asboth G, Europe-Finner GN, Watson SP, Lopez Bernal A. Second messenger pathways for oxytocin and prostaglandins in human myometrium. Biochem Soc Trans. 1995;23:21S. doi: 10.1042/bst023021s. [DOI] [PubMed] [Google Scholar]
- Puder BA, Papka RE. Hypothalamic paraventricular axons projecting to the female rat lumbosacral spinal cord contain oxytocin immunoreactivity. J Neurosci Res. 2001;64:53–60. doi: 10.1002/jnr.1053. [DOI] [PubMed] [Google Scholar]
- Raggenbass M. Overview of cellular electrophysiological actions of vasopressin. Eur J Pharmacol. 2008;583:243–254. doi: 10.1016/j.ejphar.2007.11.074. [DOI] [PubMed] [Google Scholar]
- Reeta Kh, Mediratta PK, Rathi N, Jain H, Chugh C, Sharma KK. Role of kappa- and delta-opioid receptors in the antinociceptive effect of oxytocin in formalin-induced pain response in mice. Regul Pept. 2006;135:85–90. doi: 10.1016/j.regpep.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Reiter MK, Kremarik P, Freund-Mercier MJ, Stoeckel ME, Desaulles E, Feltz P. Localization of oxytocin binding sites in the thoracic and upper lumbar spinal cord of the adult and postnatal rat: a histoautoradiographic study. Eur J Neurosci. 1994;6:98–104. doi: 10.1111/j.1460-9568.1994.tb00251.x. [DOI] [PubMed] [Google Scholar]
- Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10:25–37. doi: 10.1038/ki.1976.76. [DOI] [PubMed] [Google Scholar]
- Robinson DA, Wei F, Wang GD, Li P, Kim SJ, Vogt SK, Muglia LJ, Zhuo M. Oxytocin mediates stress-induced analgesia in adult mice. J Physiol. 2002;540:593–606. doi: 10.1113/jphysiol.2001.013492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Piloni G, López-Hidalgo M, Martínez-Lorenzana G, Rodríguez-Jiménez J, Condés-Lara M. GABA-mediated oxytocinergic inhibition in dorsal horn neurons by hypothalamic paraventricular nucleus stimulation. Brain Res. 2007;1137:69–77. doi: 10.1016/j.brainres.2006.12.045. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Schöneberg T, Kostenis E, Liu J, Gudermann T, Wess J. Molecular aspects of vasopressin receptor function. Adv Exp Med Biol. 1998;449:347–358. doi: 10.1007/978-1-4615-4871-3_44. [DOI] [PubMed] [Google Scholar]
- Sharif Naeini R, Witty MF, Séguéla P, Bourque CW. An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci. 2006;9:93–98. doi: 10.1038/nn1614. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Morphology of vasopressin and oxytocin neurones and their central and vascular projections. Prog Brain Res. 1983;60:101–114. doi: 10.1016/S0079-6123(08)64378-2. [DOI] [PubMed] [Google Scholar]
- Storm EE, Tecott LH. Social circuits: peptidergic regulation of mammalian social behavior. Neuron. 2005;47:483–486. doi: 10.1016/j.neuron.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Kuypers HG. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J Comp Neurol. 1980;194:555–570. doi: 10.1002/cne.901940306. [DOI] [PubMed] [Google Scholar]
- Takayanagi Y, Yoshida M, Bielsky IF, Ross HE, Kawamata M, Onaka T, Yanagisawa T, Kimura T, Matzuk MM, Young LJ, Nishimori K. Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc Natl Acad Sci U S A. 2005;102:16096–16101. doi: 10.1073/pnas.0505312102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida RJ, Murray RB. Manual of pharmacologic calculations with computer programs. New York: Springer; 1981. [Google Scholar]
- Thibonnier M, Berti-Mattera LN, Dulin N, Conarty DM, Mattera R. Signal transduction pathways of the human V1-vascular, V2-renal, V3-pituitary vasopressin and oxytocin receptors. Prog Brain Res. 1998;119:147–161. doi: 10.1016/s0079-6123(08)61568-x. [DOI] [PubMed] [Google Scholar]
- Thurston CL, Campbell IG, Culhane ES, Carstens E, Watkins LR. Characterization of intrathecal vasopressin-induced antinociception, scratching behavior, and motor suppression. Peptides. 1992;13:17–25. doi: 10.1016/0196-9781(92)90135-p. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Barberis C, Jard S, Dubois-Dauphin M, Dreifuss JJ. Localization and pharmacological characterization of high affinity binding sites for vasopressin and oxytocin in the rat brain by light microscopic autoradiography. Brain Res. 1988;442:105–118. doi: 10.1016/0006-8993(88)91437-0. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Charpak S, Schmidt A, Dubois-Dauphin M, Dreifuss JJ. Appearance and transient expression of oxytocin receptors in fetal, infant, and peripubertal rat brain studied by autoradiography and electrophysiology. J Neurosci. 1989;9:1764–1773. doi: 10.1523/JNEUROSCI.09-05-01764.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribollet E, Dubois-Dauphin M, Dreifuss JJ, Barberis C, Jard S. Oxytocin receptors in the central nervous system. Distribution, development, and species differences. Ann N Y Acad Sci. 1992;652:29–38. doi: 10.1111/j.1749-6632.1992.tb34343.x. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Barberis C, Arsenijevic Y. Distribution of vasopressin and oxytocin receptors in the rat spinal cord: sex-related differences and effect of castration in pudendal motor nuclei. Neuroscience. 1997;78:499–509. doi: 10.1016/s0306-4522(96)00591-x. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Ueta Y, Heitz F, Marguerat A, Koizumi K, Yamashita H. Up-regulation of vasopressin and angiotensin II receptors in the thalamus and brainstem of inbred polydipsic mice. Neuroendocrinology. 2002;75:113–123. doi: 10.1159/000048227. [DOI] [PubMed] [Google Scholar]
- Van Wimersma Greidanus TB, Kroodsma JM, Pot ML, Stevens M, Maigret C. Neurohypophyseal hormones and excessive grooming behaviour. Eur J Pharmacol. 1990;187:1–8. doi: 10.1016/0014-2999(90)90334-3. [DOI] [PubMed] [Google Scholar]
- Véronneau-Longueville F, Rampin O, Freund-Mercier MJ, Tang Y, Calas A, Marson L, McKenna KE, Stoeckel ME, Benoit G, Giuliano F. Oxytocinergic innervation of autonomic nuclei controlling penile erection in the rat. Neuroscience. 1999;93:1437–1447. doi: 10.1016/s0306-4522(99)00262-6. [DOI] [PubMed] [Google Scholar]
- Walum H, Westberg L, Henningsson S, Neiderhiser JM, Reiss D, Igl W, Ganiban JM, Spotts EL, Pedersen NL, Eriksson E, Lichtenstein P. Genetic variation in the vasopressin receptor 1a gene (AVPR1A) associates with pair-bonding behavior in humans. Proc Natl Acad Sci U S A. 2008;105:14153–14156. doi: 10.1073/pnas.0803081105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Lundeberg T, Yu LC. Antinociceptive role of oxytocin in the nucleus raphe magnus of rats, an involvement of mu-opioid receptor. Regul Pept. 2003;115:153–159. doi: 10.1016/s0167-0115(03)00152-6. [DOI] [PubMed] [Google Scholar]
- Wilson SG, Mogil JS. Measuring pain in the (knockout) mouse: big challenges in a small mammal. Behav Brain Res. 2001;125:65–73. doi: 10.1016/s0166-4328(01)00281-9. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Takayanagi Y, Inoue K, Kimura T, Young LJ, Onaka T, Nishimori K. Evidence that oxytocin exerts anxiolytic effects via oxytocin receptor expressed in serotonergic neurons in mice. J Neurosci. 2009;29:2259–2271. doi: 10.1523/JNEUROSCI.5593-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KA, Liu Y, Wang Z. The neurobiology of social attachment: a comparative approach to behavioral, neuroanatomical, and neurochemical studies. Comp Biochem Physiol C Toxicol Pharmacol. 2008;148:401–410. doi: 10.1016/j.cbpc.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LJ, Wang Z. The neurobiology of pair bonding. Nat Neurosci. 2004;7:1048–1054. doi: 10.1038/nn1327. [DOI] [PubMed] [Google Scholar]
- Yu SQ, Lundeberg T, Yu LC. Involvement of oxytocin in spinal antinociception in rats with inflammation. Brain Res. 2003;983:13–22. doi: 10.1016/s0006-8993(03)03019-1. [DOI] [PubMed] [Google Scholar]