

Abstract
We have applied cryoreduction/EPR/ENDOR techniques to characterize the active-site structure of the ferrous-oxy complexes of human (hIDO) and Shewanella oneidensis (sIDO) indoleamine 2,3-dioxygenases, Xanthomonas campestris (XcTDO) tryptophan 2,3-dioxygenase, and the H55S variant of XcTDO in the absence and in the presence of the substrate l-Trp and a substrate analogue, l-Me-Trp. The results reveal the presence of multiple conformations of the binary ferrous-oxy species of the IDOs. In more populated conformers, most likely a water molecule is within hydrogen-bonding distance of the bound ligand, which favors protonation of a cryogenerated ferric peroxy species at 77 K. In contrast to the binary complexes, cryoreduction of all of the studied ternary [enzyme-O2-Trp] dioxygenase complexes generates a ferric peroxy heme species with very similar EPR and 1H ENDOR spectra in which protonation of the basic peroxy ligand does not occur at 77 K. Parallel studies with l-Me-Trp, in which the proton of the indole nitrogen is replaced with a methyl group, eliminate the possibility that the indole NH group of the substrate acts as a hydrogen bond donor to the bound O2, and we suggest instead that the ammonium group of the substrate hydrogen-bonds to the dioxygen ligand. The present data show that substrate binding, primarily through this H-bond, causes the bound dioxygen to adopt a new conformation, which presumably is oriented for insertion of O2 into the C2−C3 double bond of the substrate. This substrate interaction further helps control the reactivity of the heme-bound dioxygen by “shielding” it from water.
Introduction
The l-kynurenine pathway is the major route of l-tryptophan (l-Trp) catabolism in mammals and leads to the formation of NAD+. The initial, rate-limiting step in this pathway is the oxidative cleavage of l-Trp to N-formylkynurenine, which is catalyzed by either of two heme-containing enzymes, indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO).(1) The dioxygenase mechanism involves binding of O2 to ferrous heme to give a typical ferrous-oxy heme complex that, in the presence of l-Trp, leads to cleavage of the C2−C3 double bond of the substrate to give product, Scheme 1A, without the requirement for additional reducing equivalents.
Scheme 1. (A) Oxidation of Tryptophan Catalyzed by Heme Dioxygenases and (B) Structures of the Tryptophan Analogues Used in This Work.

In contrast with other heme enzymes (e.g., cytochromes P450, NOS, heme oxygenase) and non-heme dioxygenases (catechol and naphthalene dioxygenases), the reaction mechanism of IDO and TDO is still poorly understood. Spectroscopic studies (reviewed in ref (1)) and sequence alignments with various IDOs2−5 originally implicated the presence of an active-site distal histidine, which was proposed to participate in the initial reaction step by abstracting a proton from the indole nitrogen and by hydrogen bond stabilization of the ferrous-oxy complex (as in the globins). Recent evidence, however, casts doubt on both of these ideas because not all dioxygenases contain an active-site histidine. In fact, functional6,7 and computational(8) data both seem to suggest that an active-site base is not involved in the reaction mechanism, as previously proposed, and this would be consistent with the known chemistry of indoles, which do not react in this fashion.(9) The formation of an initial oxy complex is clearly crucial in the mechanism, but we know little about the factors affecting formation and stability of the ferrous-oxy species. In fact, the ferrous-oxy complexes are variously stable in different dioxygenases, and the reasons for these differences are not clear. This might be linked to the presence or absence of the active-site histidine in IDOs/TDOs, because introduction of a histidine into human IDO destabilizes the otherwise observable ferrous-oxy complex.(10) This instability of the key catalytic ferrous-oxy complex for some dioxygenases is sufficiently great that at ambient temperatures it cannot be observed, and as a result conclusions on the mode of O2 binding have been drawn primarily from structural models of ferrous enzymes11,12 or from spectroscopic work on the related CO-bound enzyme.13−15 Unfortunately, the νO−O mode of heme-bound dioxygen in histidine-coordinated hemoproteins is, in general, not Raman active, which restricts the application of resonance Raman (RR) spectroscopy to these systems. However, a recent RR study found that the νO−O mode was Raman active in the ternary [hIDOII-O2-Trp] complex but not in the binary [hIDOII-O2] complex, and this was attributed to an H-bonding interaction between the heme-bound O2 and the surrounding environment in the ternary complex.(16) Such findings highlight the need for more detailed understanding of the effects of substrate on the interaction of the heme-bound dioxygen with its surroundings.
It has been demonstrated that EPR and ENDOR spectroscopy of cryoreduced ferrous-oxy hemeproteins can provide valuable structural information on the EPR-silent oxy precursors (see, for example, refs (17−20)). In this approach, EPR-silent ferrous-oxy samples are reduced radiolytically at 77 K, and the resulting paramagnetic states are trapped in the conformation of the diamagnetic parent state, thus providing a sensitive EPR probe of the structure of the ferrous-oxy precursors.(21) Here, we have applied these techniques to study the substrate-free ferrous-oxy complexes, and the ternary complexes with substrate or substrate analogues, of several dioxygenases. Our intention was to observe the effect of substrate/analogue on the catalytically competent ferrous-oxy species in the heme dioxygenases. The results reveal important information on the surrounding environment of the heme-bound dioxygen and the interactions present in the ternary complex.
Materials and Methods
Materials
l-Tryptophan (l-Trp), 1-methyl-l-tryptophan (Me-Trp), 5-fluoro-l-tryptophan (5F-Trp), ethylene glycol, and all of the chemicals used for buffers (Sigma-Aldrich) were of the highest analytical grade (more than 97% purity) and were used without further purification. Aqueous solutions were prepared using either purified water from an Elgastat option 2 water purifier or Millipore Q Ultrapure water. All protein samples were concentrated using Centricon concentrators (30 000 molecular weight cutoff).
Protein Expression and Purification
Recombinant human indoleamine 2,3-dioxygenase (hIDO), Shewanella oneidensis IDO (sIDO), Xanthomonas campestris tryptophan 2,3-dioxygenase (XcTDO), and the H55S variant of XcTDO (H55S XcTDO, the histidine 55-to-serine mutant) were expressed in Escherichia coli and purified as described previously.10,12 Protein and heme concentrations were determined by the Bradford and pyridine hemochrome methods.22,23 The concentrations of enzyme solutions were determined spectrophotometrically using absorption coefficients for the ferric form of the enzymes: hIDO, ε406 = 172 mM−1 cm−1;(24) sIDO, ε405 = 92 mM−1 cm−1; XcTDO, ε404 = 180.5 mM−1 cm−1;(12) and H55S XcTDO, ε405= 132 mM−1 cm−1.(12)
Sample Preparation
Ferric hIDO in 50 mM KPi buffer (pH 8.0) in complex with l-Trp or Me-Trp was prepared by addition of l-Trp (40 mM stock solution, 20 μL) or Me-Trp (32 mM stock solution, 20 μL) to ferric hIDO (2 mM, 20 μL), followed by the addition of glycerol (40% stock, 40 μL) to yield a final protein concentration of 0.5 mM. Ferric XcTDO, H55S XcTDO, and sIDO samples were prepared in 50 mM Tris buffer (pH 8.5) and then diluted by addition of an equal volume of ethylene glycol to yield a final protein concentration of 0.5 mM. Ferric l-Trp-bound XcTDO, H55S XcTDO, and sIDO samples were prepared in the same way but in 50 mM Tris buffer (pH 8.5) containing 10 mM l-Trp.
All ferrous heme samples were prepared under anaerobic conditions in a glovebox. Ferrous hIDO samples (50 mM KPi buffer (pH 8.0) were prepared to 2 mM and then diluted by addition of ethylene glycol (final concentration 50%) and dithionite (final concentration 0.55 mM) to yield a final protein concentration of 0.5 mM. Either 10 mM l-Trp or 4 mM Me-Trp was present in the solution. Ferrous sIDO samples were prepared in the same way but in 50 mM Tris buffer (pH 8.5); ferrous XcTDO samples and H55S XcTDO samples were concentrated in the same buffer and then diluted by addition of ethylene glycol that contained dithionite (final concentration 1 mM) and either no substrate, 12 mM l-Trp, 10 mM Me-Trp, or 5F-Trp (final protein concentration 1 mM).
Ferrous-oxy complexes were generated in EPR tubes at −30 °C by bubbling 20 mL of precooled O2 (−30 C) through 200 μL of ferrous protein solution for about 1 min. Deuterated samples were prepared by exchanging the protein of interest into D2O and adjusting the pH to the required value (as above for the non-deuterated equivalent sample, recognizing that pD = pH − 0.4) using NaOD and DCl.(25) Deuterated ethylene glycol and l-Trp/Me-Trp were added, as above, to create the specified samples.
Irradiation of the frozen solutions of the ferrous-oxy dioxygenase complexes at 77 K typically was performed for 20−24 h at a rate of 0.15 Mrad/h (total dose of 3−3.5 Mrad) using a Gammacell 220 60Co unit as described previously.(21) We performed annealing at multiple temperatures over the range of 77−270 K by placing the EPR sample in a bath at the desired temperature (usually n-pentane cooled with liquid N2) for 1 min.
EPR and ENDOR Spectroscopy
X-band continuous wave EPR spectra were recorded on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910 continuous He flow cryostat. Q-band (35 GHz) EPR and ENDOR spectra were recorded on a modified Varian E-110 spectrometer equipped with a helium immersion Dewar at 2 K and operating in dispersion mode using 100 kHz field modulation under “rapid passage” conditions (as described in ref (21)). For ENDOR, in some instances, the radio frequency (rf) bandwidth was broadened to 60 kHz to improve the signal-to-noise ratio.(18) EPR spectra of cryoreduced samples exhibit strong signals from free radicals created by the irradiation; this region is deleted from the spectra, for clarity.
For a single orientation of a paramagnetic center, the first-order ENDOR spectrum of a proton with I = 1/2 in a single paramagnetic center consists of a doublet with frequencies given by
Here, νn is the nuclear Larmor frequency and A is the orientation-dependent hyperfine coupling constant of the coupled nucleus. The doublet is centered at the Larmor frequency and separated by A when νn > A/2, as in case for the 1H spectra presented here. The full hyperfine tensor of the coupled nucleus can be obtained by analyzing a “2D” field-frequency set of orientation-selective ENDOR spectra across the EPR envelope, as described in ref (21).
Results
Ferrous-Oxy (Binary) Complexes
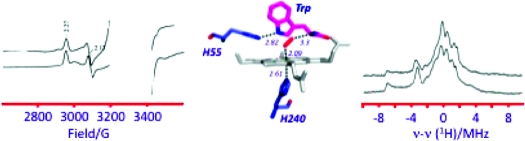
The EPR spectrum of ferrous-oxy hIDO [hIDOII-O2] which was radiolytically reduced at 77 K is presented in Figure 1. The spectrum is a superposition of multiple rhombic S = 1/2 EPR signals with a range of gmax-values, 2.35 ≤ gmax ≤ 2.20. Its shape is independent of the degree of cryoreduction, indicating that it is determined by the presence of multiple conformers of the parent diamagnetic ferrous-oxy complex, and not by the relative probabilities of reduction of the conformers of the oxy heme parents. Two signals stand out as perhaps more intense and thus representing more highly populated parent conformers; they are characterized by g = [2.32, 2.17, 1.95] (signal A) and g = [2.348, 2.26, 1.922] (signal B) (Table 1), values that are typical of a ferric hydroperoxy heme species (Fe(III)-OOH−).19,21,26 A minority of the conformers have signals with gmax ≤ 2.26, characteristic of cryogenerated ferric peroxy heme (Fe(III)-OO2−) intermediates. The assignment of species A and B as ferric hydroperoxy species was corroborated by 1H ENDOR spectroscopy. The trapped A and B species show exchangeable 1H ENDOR spectra with Amax = 12 and 10 MHz, respectively (Figure S1, Supporting Information), values that are characteristic of the hydroperoxy ligand.(18)
Figure 1.

X-band EPR spectra of cryoreduced binary ferrous-oxy hIDO and sIDO complexes and cryoreduced ternary complexes of ferrous-oxy hIDO, XcTDO, and H55S XcTDO with Trp, Me-Trp, and 5F-Trp. Instrument conditions: T = 25 K; microwave power, 2 mW; modulation amplitude, 5 G; microwave frequency, 9.365 GHz.
Table 1. g-Values and Maximal Proton Coupling for Cryoreduced Ferrous-Oxy hIDO, sIDO, XcTDO, H55S XcTDO, and the Oxyglobins.
| complex | species | g1 | g2 | g3 | Amax (MHz) |
|---|---|---|---|---|---|
| [hIDOII-O2] | A | 2.320 | 2.170 | 1.947 | 10 |
| B | 2.35 | 2.26 | 1.922 | 9 | |
| other | 2.3−2.2 | ||||
| [sIDOII-O2] | A | 2.32 | 2.20 | 1.94 | |
| B | 2.30 | 2.18 | 1.95 | ||
| C | 2.27 | 2.16 | ∼1.95 | ||
| [hIDOII-Trp-O2] | 2.27 | 2.17 | 1.946 | 15 | |
| [hIDOII-Me-Trp-O2] | 2.27 | 2.173 | 1.946 | 15 | |
| [XcTDOII-5F-Trp-O2] | 2.27 | 2.17 | 1.95 | 15 | |
| [H55SXcTDOII-Trp-O2] | 2.27 | 2.17 | 1.95 | 15 | |
| [H55SXcTDOII-Me-Trp-O2] | 2.27 | 2.17 | 1.95 | 15 | |
| HbA oxy β-chains | peroxy | 2.25 | 2.14 | 1.95 | 21 |
| hydroperoxy | 2.31 | 2.16 | 1.93 | 12 |
These observations therefore suggest that cryoreduction of the ferrous-oxy moiety of the major conformational sub-states, including A and B, is accompanied by its protonation at 77 K, but this is not the case for some minor ones.
During annealing at temperatures of 116 and 145 K, the heterogeneous population of cryogenerated species relaxes to species A and B (Figure 2); further stepwise annealing, between temperatures of 145 and 190 K, results in the conversion of A to B (Figure 2). The latter completely disappears after annealing at temperatures above 210 K, and this latter process is accompanied by increasing contents of a high-spin form and low-spin ferric state with g = [2.82, 2.27, 1.65], characteristic of substrate-free ferric hIDO (Figure S3). [Note: Low-spin forms of ferric hIDO and tryptophan-bound ferric hIDO display rhombic EPR signals with different g tensors of [2.82, 2.27, 1.65] and [2.52, 2.19, 1.86], respectively (Figure S2).(24) We have found that both low-spin species show exchangeable in D2O 1H ENDOR signals with maximum hyperfine coupling Amax ≈ 10 and 12 MHz, respectively, characteristic of iron(III)-coordinated H2O or OH−. The changes in the g-tensor components caused by the bound Trp are likely due to perturbations of the sixth axial ligand induced by the substrate. This assumption is in agreement with reported results of RR studies on ferric IDO.(15)]
Figure 2.

X-band EPR spectra of cryoreduced oxy ferrous hIDO annealed at indicated temperatures. Instrument conditions: T = 25 K; microwave power, 2 mW; modulation amplitude, 5 G; microwave frequency, 9.365 GHz.
The decay of the ferric hydroperoxy intermediate (B) can occur by protonation of the proximal oxygen and dissociation of H2O2, and/or by protonation of the distal oxygen, with heterolytic or homolytic cleavage of the O−O bond and formation of a ferryl (Compound I/II) intermediate, as happens during annealing of cryoreduced oxyglobins at elevated temperatures.(27)
To distinguish between these processes, we took advantage of the recent report that cryoreduction of EPR-silent Compound II produces low-spin FeIII intermediates having O2− or OH− as the sixth axial ligand.(28) In the case of hIDO, annealing of the cryoreduced ferrous-oxy species at 220 K followed by exposure to additional irradiation at 77 K produces a new EPR signal, g = [2.61, 2.17, 1.84] (Figure S4), close to the values observed for hydroxide-bound ferric hIDO in the presence of l-Trp (g = 2.52, 2.19, 1.86)(24). This species completely disappears after annealing at 200 K for several minutes, converting into a high-spin ferric heme species. We interpret this to mean that, during annealing, the ferric hydroperoxy heme species B of hIDO undergoes protonation at the distal oxygen and converts into Compound II, and this yields the ferric state during a second cryoreduction, as in the case of myoglobin.(27) [Note: The formation of ferric hIDO during annealing of the cryoreduced ferrous-oxy hIDO at temperatures above 190 K may be caused in part by reduction of Compound II by cryogenerated radicals.]
Similar results were obtained for cryoreduced ferrous-oxy sIDO. The EPR spectrum of cryoreduced ferrous-oxy sIDO is better resolved than that for ferrous-oxy hIDO, but nonetheless it contains three rhombic EPR signals (A, B, and C, Table 1), again reflecting the presence of distinct conformational sub-states in the oxy precursor (Figures 1, S5). As for hIDO, the EPR signals A and B (Table 1) can be assigned to a ferric hydroperoxy species, while signal C is from a ferric peroxo intermediate. As can be seen in Figure 1 and Table 1, the g-tensor components for these species and their relative populations differ from those for cryoreduced ferrous-oxy hIDO, unsurprisingly indicating structural differences among conformational sub-states in the ferrous-oxy forms of the two dioxygenases. During progressive annealing at 145 K, species C completely decays; the cryogenerated ferric hydroperoxy intermediates A and B decay at temperatures of 185−195 K. The decay of species A−C is accompanied by the appearance of a new transient EPR signal with g = [2.43, 2.21, 1.904] that is characteristic of a low-spin hexa-coordinated ferric heme species, which was not observed in the annealing pattern for cryoreduced ferrous-oxy hIDO (Figure S5). The nature of the sixth axial ligand in this species remains unclear.
Analogous studies with XcTDO and H55S XcTDO could not be carried out because24,29 their ferrous-oxy complexes are unstable(7) and could not be freeze-trapped for cryoreduction.
Ternary [hIDO-O2-X] Complexes
X = l-Trp
In contrast to the ferrous-oxy case above, the EPR spectrum of cryoreduced [hIDOII-O2-Trp] displays just a single new EPR signal, with g = [2.270, 2.170, 1.946] (Figure 1, Table 1), a g-tensor characteristic of a ferric peroxy heme intermediate.19,21,26 This assignment is corroborated by 1H ENDOR data. This intermediate displays a strongly coupled exchangeable proton ENDOR signal, Amax ≈ 15 MHz (Figures 3, S6), the value commonly observed for ferric peroxy heme species.(19) This result for the substrate-bound dioxygenases contrasts with those for heme monooxygenases, whose ternary oxy complexes directly form the ferric hydroperoxy state, in which the proton of the hydroperoxy ligand has a smaller hyperfine coupling, Amax ≤ 12 MHz.18,21
Figure 3.

1H ENDOR spectra for cryoreduced oxy hIDO (species B) and ternary complexes of oxy IDO, XcTDO, and oxy H55S XcTDO with Trp, Me-Trp, and 5F-Trp. Instrument conditions: T = 2 K; microwave frequency, 34.95 GHz; modulation amplitude, 2 G; rf power, 5 W; rf sweep rate, 0.5 MHz/s; rf broadening, 60 kHz; 20 scans.
The peroxo-hIDO/Trp intermediate remains unchanged upon annealing at 180 K. It begins to decay only at T > 190 K, when the decay is accompanied by formation of low-spin ferric heme with g = [2.53, 2.19, 1.86], characteristic of tryptophan-bound low-spin ferric hIDO (Figure 4). Taken together, these results clearly show that the presence of the substrate shields the basic peroxy ligand in the cryoreduced ternary ferrous hIDO-oxy-substrate complex from protonation.
Figure 4.

X-band EPR spectra of cryoreduced ternary [hIDOII-Trp-O2] complex annealed at indicated temperatures. Instrument conditions as in Figure 2.
The formation of a peroxo-ferric intermediate upon cryoreduction of the ternary complex and the relaxation of this intermediate only at relatively high temperatures resemble the behavior of cryoreduced oxy-ferrous globins17,21,30 and contrast with that of the heme monooxygenases. Previous cryoreduction studies of cytochrome P450 and heme oxygenase showed that protonation of the basic peroxo ligand at 77 K requires the presence of an H-bonded proton delivery network in the parent ferrous-oxy complex that includes an ordered water molecule in the active site that is hydrogen-bonded to the terminal oxygen of the dioxygen ligand.17,21 This water facilitates proton transfer to the peroxo moiety at 77 K or below.(17) In contrast, ferrous-oxy globins do not have such a bound water, and when cryoreduced at 77 K they exclusively generate the ferric peroxo species which becomes protonated only at T > 170 K, a temperature at which a molecule of water might diffuse into the active site.(17) These comparisons indicate that, in the dioxygenases, the bound l-Trp substrate excludes water from the vicinity of the O2 of the ternary oxy complex, thereby shielding it from prompt protonation after cryoreduction.
X = Me-Trp
EPR/ENDOR spectroscopy presented above has shown that the peroxy moiety in cryoreduced ternary dioxygenase complexes, and hence the heme-bound dioxygen of the diamagnetic parent complex, accepts an H-bond but that H2O is not the H-bond donor. Then what is? The obvious suggestion is the indole NH group, which has been proposed previously15,31 to hydrogen-bond to the bound O2 ligand. To test this, cryoreduction studies were carried out with the ternary complex formed with l-Me-Trp, in which the indole proton is replaced with a CH3 group, which cannot hydrogen-bond.
The cryoreduced ternary [hIDOII-O2-Me-Trp] complex displays a peroxo-ferriheme EPR signal with g-tensor components very close to those for irradiated [hIDOII-O2-Trp] (Figure 1, Table 1). Cryoreduced [hIDOII-O2-Me-Trp] also shows a 1H ENDOR signal from an H-bond to the cryotrapped peroxy moiety very similar to that seen for the [hIDOII-O2-Trp] complex (Figures 3, S7). The similarity of the spectra shows that the indole NH group is not the source of the ENDOR-detected hydrogen-bonding interaction to the peroxy ligand, and thus is not H-bonded to the parent dioxygen.
XcTDO and H55S XcTDO
The spectroscopic properties of cryoreduced ternary complexes of XcTDO and H55S TDO and their annealing patterns appear to be very similar to those for [hIDOII-Trp-O2]. In the presence of substrate, ferrous XcTDO forms a ternary complex with O2, [XcTDOII-O2-Trp], which has been observed spectrophotometrically in the steady state,(12) but its lifetime is too short for it to be trapped under the experimental conditions used in this study. Instead, we have used the more stable ternary complex with 5F-Trp, [XcTDO-O2-5F-Trp].(12) For the H55S XcTDO variant, the oxy complex is sufficiently stable7,12 that we have been able to examine [H55S XcTDOII-O2-Trp] and [H55S XcTDOII-O2-Me-Trp].
The cryoreduced ternary [XcTDOII-O2-5F-Trp] complex shows a rhombic EPR signal and 1H ENDOR pattern that are very close to those for the cryoreduced [hIDOII-O2-Trp] complex (Table 1, Figures 1, 3). The mutation of His55 has only a subtle effect on the EPR and 1H ENDOR spectra of the cryoreduced oxy heme moieties in the [H55S XcTDOII-O2-Trp] complex (Table 1, Figures 1, 3). Decay of the cryoreduced ternary complexes is observed after annealing at temperatures above 210−220 K (Figure S8). Likewise, EPR and 1H ENDOR spectra for the cryoreduced [H55S XcTDOII-O2-Me-Trp] complex and its annealing pattern are very similar to those for the [H55S XcTDOII-O2-Trp] complex (Figures 1 and 3, and Table 1), which again suggests that, in the ternary [enzyme-O2-substrate] complex, the indole NH group is not involved in H-bonding to the ferric peroxy group.
Unlike the cryoreduced binary [hIDOII-O2] complex, irradiation of the cryoreduced ternary [H55S XcTDOII-O2-Trp/Me-Trp] annealed at 230 K for 2 min does not result in appearance of the EPR signal characteristic of cryoreduced Compound II. Furthermore, HPLC analysis of cryogenerated [H55S XcTDOII-O2-Trp/Me-Trp]− reveals that no monooxygenated products are formed. Accordingly, the data suggest that proton-assisted conversion of the ferric heme-peroxy intermediate into Compound I that could react with bound substrate does not occur during the annealing subsequent to cryoreduction. Conversion of the ferric peroxy intermediate into the ferric form occurs likely through dissociation of H2O2 formed via double protonation of the peroxy ligand during annealing at high temperatures, as occurs to an extent during annealing of cryogenerated ferric peroxy Mb.27,30
14N ENDOR
For more detailed characterization of the peroxo ferriheme moiety in cryoreduced ternary complexes showing a single EPR signal, we collected 14N ENDOR spectra from the heme pyrrole nitrogens coordinated to iron(III). Low-spin ferriheme species as a rule display a ν+14N ENDOR branch at the gmax turning point, consisting of two quadrupole-split doublets associated with two nonequivalent pairs of pyrrole nitrogens.26,32,33 The cryoreduced ternary complexes of hIDO and XcTDO show a more complex 14N ENDOR pattern at gmax consisting of five or more peaks (Figure S9), which we assign to the presence of two or more conformations of oxy heme moieties with distinct iron(III)−pyrrole nitrogen interactions. In addition, despite high similarities between the EPR and 1H ENDOR spectra for all studied ternary complexes, the 14N ENDOR spectrum for cryoreduced [hIDOII-O2-Trp] differs noticeably from those of the XcTDO complexes. These observations indicate (i) the multiconformational nature of the ternary complexes and (ii) the existence of some structural differences between the oxy heme moieties in the ternary complexes of hIDO and XcTDO.
Discussion
The goal of this paper was to explore the structures of catalytically competent ferrous-oxy complexes of the heme dioxygenases with bound substrate through use of cryoreduction/EPR/ENDOR spectroscopy. To provide a reference point, we also examined the complexes without substrate.
Binary Ferrous-Oxy Complexes
The spectroscopic data presented here reveal that the ferrous-oxy complexes of hIDO and sIDO in the absence of substrate exist in multiple conformational substates with differing geometries of the oxy heme moiety, and with differences between the conformational distributions in the different enzymes. In the majority of these conformations, the ferric peroxy heme formed by cryoreduction acquires a proton at 77 K to form the ferric hydroperoxy heme species. As shown above, this indicates that an H2O is located within hydrogen-bonding distance of the heme-bound dioxygen of the oxyferroheme parent state. However, in a minority of ferrous-oxy hIDO conformers, and in the C conformer of ferrous-oxy sIDO, a cryogenerated ferric peroxy heme moiety becomes protonated after annealing at a relatively low temperature (below 145 K). We propose that, in these conformers, there is a water in the vicinity of bound dioxygen, but the relative position of this water with respect to the bound dioxygen is unfavorable for proton transfer to the cryogenerated peroxy-ferric heme moiety without structural relaxation. Only small rearrangements induced by the annealing are then required to make the proton transfer favorable.
Overall these results show that the majority of active sites of the ferrous-oxy dioxygenases studied here contain a water that is within hydrogen-bonding distance of the distal oxygen of the bound dioxygen and that favors proton transfer to cryogenerated ferric peroxy moiety at 77 K.
Ternary Ferrous-Oxy Complexes of Dioxygenases
In contrast to the binary complexes, cryoreduction of the ternary [enzyme-O2-Trp] complexes generates only a ferric peroxy heme intermediate, which does not become protonated until annealing to relatively high temperatures (above 190 K), when water molecules become mobile and can diffuse to the basic iron-bound peroxy ligand, as found in studies of the ferrous-oxy globins and cytochrome P450cam mutants.21,27,30 Moreover, the peroxo-heme center of the cryoreduced complex displays a single EPR signal, different from either of those from the binary complex. As the structure of the cryoreduced enzyme reveals the structure of the parent oxy-ferrous enzyme, this implies that substrate binding causes the bound dioxygen to adopt a new conformation, presumably that appropriate for substrate oxidation. Furthermore, the absence of protonation of the peroxo moiety subsequent to 77 K cryoreduction shows that the distal pocket of the ternary complex does not contain a water with access to the oxy heme. This “protection” from water likely helps to control the reactivity of the heme-bound dioxygen. This proposal is consistent with the model for the ternary ferrous XcTDO-O2-Trp complex based on the crystal structure of the binary complex of ferrous XcTDO with l-Trp.(12) Notably, this shielding of the heme-bound dioxygen from water, as deduced from the observation of the peroxo-heme as the product of cryoreduction, is seen for the ternary complexes of all the dioxygenases studied here. This indicates that the conformation(s) of and water access to the heme Fe(II)-O2 moiety in the active form depend relatively weakly on the amino acid residues making up the distal pocket and are primarily determined by its interaction with the bound substrate.
Neither the replacement of His55 with serine in XcTDO nor methylation of the indole N1 has a detectable effect on the EPR and 1H ENDOR properties of the cryoreduced oxy heme moiety in the ternary complex. This implies that hydrogen-bonding interaction between His55 and the indole NH group of bound substrate seen in the crystal structures of substrate-bound ferrous XcTDO does not cause spectroscopically detectable perturbations in the conformation of the oxy heme moiety.(12)
The absence of differences between the l-Trp and Me-Trp complexes has further implications for the identity of the H-bond donor to the O2 moiety found by 1H ENDOR for all the dioxygenases examined here. Previous studies on the model [hIDOII-CO-Trp] complex using RR spectroscopy suggested the presence of a hydrogen bond between the NH group and the heme-bound CO,13–15 which led to the idea (also suggested from crystallographic work(31)) that the heme-bound dioxygen interacts with and could itself deprotonate the indole NH group. However, 1H ENDOR spectroscopy on the cryoreduced ternary complexes of Me-Trp with ferrous-oxy hIDO and ferrous-oxy H55S XcTDO now reveals that the hydrogen bond in each is unchanged by methylating l-Trp, which eliminates the possibility that the NH group contributes significantly to H-bond stabilization of the bound O2.
It seems unlikely that the hydrogen bond comes from a nearby protein side chain because, with the exception of S167 in hIDO and His55 in XcTDO,12,31 the active site is devoid of polar residues. Furthermore, the replacement of His55 with serine in XcTDO has no effect on the 1H ENDOR pattern. In addition, recent kinetic studies showed that the S167A mutation in hIDO does not affect the formation or stability of the ferrous oxy species,(10) and by implication S167 is not involved in hydrogen bond stabilization of bound dioxygen.
Examination of the structure for substrate-bound ferrous and ferric XcTDO(12) as well as a model created for the ternary complex11,12 instead leads us to suggest that the hydrogen bond to the bound O2 detected by cryoreduction/1H ENDOR for all the ternary complexes with l-Trp and Me-Trp likely can be assigned to a proton of the ammonium group of the substrate, which is situated within hydrogen-bonding distance (Figure 5). This interaction would be analogous to the hydrogen bond between the distal histidine and the O2 ligand in the globins.(28) It would be consistent with our observations12,29 that the substrate analogue indole propionic acid, in which the amine group is missing, does not form product. In principle, it would be possible to test our proposal with indole propionic acid, but unfortunately this was unsuccessful because the ferrous-oxy complex for this substrate analogue is unstable through autoxidation and could not be trapped for the EPR/ENDOR experiments. This proposal would also provide an explanation for results suggesting superoxide character of the heme-bound O2 in the ternary complex,16,34 because charge polarization would be favored by hydrogen-bonding and electrostatic interactions between the positive-charged ammonium group of the substrate and the distal oxygen of bound O2. It also correlates with the reported appearance of a positive electrostatic potential surrounding the heme-bound CO in hIDO when l-Trp is present and the specific effect of the −CH(NH3)+COO− moiety of l-Trp or its analogues on the stretching frequencies of bound CO in rat liver TDO.13,15,35
Figure 5.
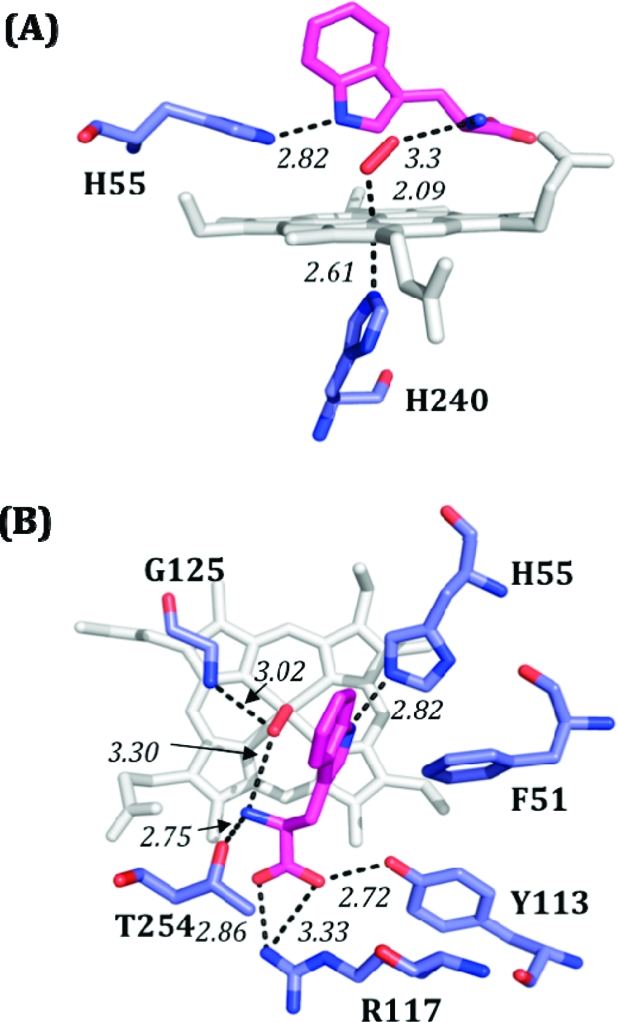
Model of the [XcTDOII-O2-Trp] ternary complex, based on PDB entry 2NW8. Bond distances are shown in italics, l-Trp is shown in magenta, and dioxygen is shown in red. (A) Side view, with heme shown in gray. (B) Overhead view, with heme shown in white. The distance between the N of the Trp ammonium group and the distal O of O2 is ∼2.9 Å.
The EPR and 1H ENDOR results primarily report on the conformation of and H-bonding to the bound dioxygen of the parent oxy heme moiety. These results, and thus this parent conformation, are essentially similar for the ternary complexes of XcTDO and hIDO, even though there are significant differences in their activities and stabilities. In contrast, 14N ENDOR measures the properties of the heme itself and is more sensitive to heme distortions and the conformation of the proximal ligand. The heme pyrrole nitrogens of the cryoreduced [IDO-O2-Trp] and [XcTDO-O2-Trp] ternary complexes in fact show distinct 14N ENDOR spectra (Figure S9), indicative of some differences between the heme conformations in those oxy heme centers. In addition, those spectra indicate that the heme macrocyle of the ternary complexes exhibits multiple conformations, in agreement with recently reported results of RR studies on the [hIDOII-O2-Trp] ternary complexes.(16)
Conclusion
The experimental data presented here show that binding of tryptophan to oxy-ferrous dioxygenases causes the heme-bound dioxygen to adopt a new conformational state, presumably optimized with respect to insertion in the indole C2−C3 bond. The most important finding is that this conformation of the heme-bound dioxygen is primarily stabilized by a hydrogen bond with the terminal ammonium group of tryptophan, and not with a water or with the NH of the Trp indole ring. In particular this is indicated by the findings that both the H55S mutation in XcTDO and methylation of the indole N1 have no detectable effect on the spectroscopic properties of the cryoreduced oxy heme moiety or its annealing behavior. Our observations thus argue against proton abstraction from the NH group as the initial catalytic step. This picture has been emerging recently, as both experimental6,7 and computational8,16 studies on various dioxygenase reactions have suggested that deprotonation of the indole NH does not occur.
These experiments have further revealed that the bound substrate shields the heme-bound dioxygen from access of water and thus itself contributes to catalytic control of the reactivity of the oxy ferroheme. This observation supports those proposed mechanisms for heme dioxygenases that do not require external protons, a base, or additional electrons. In this respect, the heme dioxygenases differ fundamentally from heme monoxygenases for which both an additional electron and proton play essential roles in reductive activation of dioxygen. Indeed, the present results suggest that the bound substrate impedes reductive activation of O2 by dioxygenases.
Acknowledgments
This work was supported by grants from the NIH (B.M.H., HL 13531), The Wellcome Trust (project grant 083636 to E.L.R. and to S.K.C./C.G.M.), BBSRC/EaStChem (studentship to S.J.T.), BBSRC (studentship to N.D.P.), and EPSRC (studentship to N.C.). We are grateful to Dr. Jaswir Basran (University of Leicester) for assistance with protein preparation and Prof. H. Halpern (University of Chicago) for providing access to Gammacell 60Co source.
Supporting Information Available
Supporting Figures S1−S9 and complete ref (12). This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Chem. Rev. 1996, 96, 2841–2887. [DOI] [PubMed] [Google Scholar]
- Kawamichi H.; Suzuki T. J. Protein Chem. 1998, 17, 651–656. [DOI] [PubMed] [Google Scholar]
- Suzuki T. J. Protein Chem. 1994, 13, 9–13. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Kawamichi H.; Imai K. J. Protein Chem. 1998, 17, 817–826. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Takagi T. J. Mol. Biol. 1992, 228, 698–700. [DOI] [PubMed] [Google Scholar]
- Chauhan N.; Thackray S. J.; Rafice S. A.; Eaton G.; Lee M.; Efimov I.; Basran J.; Jenkins P. R.; Mowat C. G.; Chapman S. K.; Raven E. L. J. Am. Chem. Soc. 2009, 131, 4186–4187. [DOI] [PubMed] [Google Scholar]
- Thackray S. J.; Bruckmann C.; Anderson J. L.; Campbell L. P.; Xiao R.; Zhao L.; Mowat C. G.; Forouhar F.; Tong L.; Chapman S. K. Biochemistry 2008, 47, 10677–10684. [DOI] [PubMed] [Google Scholar]
- Chung L. W.; Li X.; Sugimoto H.; Shiro Y.; Morokuma K. J. Am. Chem. Soc. 2008, 130, 12299–12309. [DOI] [PubMed] [Google Scholar]
- Joule J. A.; Mills K.. Heterocyclic chemistry, 4th ed.; Blackwell: Oxford, UK, 2000. [Google Scholar]
- Chauhan N.; Basran J.; Efimov I.; Svistunenko D. A.; Seward H. E.; Moody P. C. E.; Raven E. L. Biochemistry 2008, 47, 4761–4769. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Kang S. A.; Mukherjee T.; Bale S.; Crane B. R.; Begley T. P.; Ealick S. E. Biochemistry 2007, 46, 145–155. [DOI] [PubMed] [Google Scholar]
- Forouhar F.; et al. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batabyal D.; Yeh S.-R. J. Am. Chem. Soc. 2007, 129, 15690–15701. [DOI] [PubMed] [Google Scholar]
- Batabyal D.; Yeh S.-R. J. Am. Chem. Soc. 2009, 131, 3260–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentis A. C.; Thomas S. R.; Takikawa O.; Littlejohn T. K.; Truscott R. J. W.; Armstrong R. S.; Yeh S.-R.; Stocker R. J. Biol. Chem. 2002, 277, 15788–15794. [DOI] [PubMed] [Google Scholar]
- Lewis-Ballester A.; Batabyal D.; Egawa T.; Lu C.; Lin Y.; Marti M. A.; Capece L.; Estrin D. A.; Yeh S. R. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17371–17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov R.; Chemerisov S.; Werst D. E.; Rajh T.; Matsui T.; Ikeda-Saito M.; Hoffman B. M. J. Am. Chem. Soc. 2004, 126, 15960–15961. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Kofman V.; Fujii H.; Yoshida T.; Ikeda-Saito M.; Hoffman B. M. J. Am. Chem. Soc. 2002, 124, 1798–1808. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Kofman V.; Nocek J. M.; Noble R. W.; Hui H.; Hoffman B. M. Biochemistry 2004, 43, 6330–6338. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Macdonald I. D. G.; Makris T. M.; Sligar S. G.; Hoffman B. M. J. Am. Chem. Soc. 1999, 121, 10654–10655. [Google Scholar]
- Davydov R.; Makris T. M.; Kofman V.; Werst D. E.; Sligar S. G.; Hoffman B. M. J. Am. Chem. Soc. 2001, 123, 1403–1415. [DOI] [PubMed] [Google Scholar]
- Berry E. A.; Trumpower B. L. Anal. Biochem. 1987, 161, 1–15. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Papadopoulou N. D.; Mewies M.; McLean K. J.; Seward H. E.; Svistunenko D. A.; Munro A. W.; Raven E. L. Biochemistry 2005, 44, 14318–14328. [DOI] [PubMed] [Google Scholar]
- Glasoe P. K.; Long F. A. J. Phys. Chem. 1960, 64, 188–190. [Google Scholar]
- Davydov R.; Satterlee J. D.; Fujii H.; Sauer-Masarwa A.; Busch D. H.; Hoffman B. M. J. Am. Chem. Soc. 2003, 125, 16340–16346. [DOI] [PubMed] [Google Scholar]
- Garcia-Serres R.; Davydov R. M.; Matsui T.; Ikeda-Saito M.; Hoffman B. M.; Huynh B. H. J. Am. Chem. Soc. 2007, 129, 6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov R.; Osborne R. L.; Kim S. H.; Dawson J. H.; Hoffman B. M. Biochemistry 2008, 47, 5147–5155. [DOI] [PubMed] [Google Scholar]
- Basran J.; Rafice S. A.; Chauhan N.; Efimov I.; Cheesman M. R.; Ghamsari L.; Lloyd Raven E. Biochemistry 2008, 47, 4752–4760. [DOI] [PubMed] [Google Scholar]
- Kappl R.; Hoehn-Berlage M.; Huettermann J.; Bartlett N.; Symons M. C. R. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1985, 827, 327–343. [Google Scholar]
- Sugimoto H.; Oda S.-i.; Otsuki T.; Hino T.; Yoshida T.; Shiro Y. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnenschmidt M.; Bittl R.; Rau H. K.; Haehnel W.; Lubitz W. Chem. Phys. Lett. 2000, 323, 329–339. [Google Scholar]
- Scholes C. P.; Isaacson R. A.; Feher G. Biochim. Biophys. Acta 1972, 263, 448–452. [DOI] [PubMed] [Google Scholar]
- Brady F. O.; Feigelson P. J. Biol. Chem. 1975, 250, 5041–5048. [PubMed] [Google Scholar]
- Uchida K.; Usami M.; Bandow H.; Harada I. Biochim. Biophys. Acta 1992, 1121, 153–159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.