

Abstract
Esophageal squamous cell carcinoma (ESCC) is an exceptionally drug resistant tumor with a five-year survival rate <5%. From an initial drug screen we identified bortezomib as having robust activity in ESCC lines. Mechanistically, bortezomib induced a G2/M phase cell cycle arrest and p53-independent apoptosis associated with caspase cleavage and Noxa induction. Bortezomib also demonstrated excellent activity in organotypic culture and in vivo models of ESCC. Biochemically, bortezomib treatment activated the p38 and JNK stress-activated MAP kinase pathways and induced phospho-H2AX activity. Although H2AX is known to co-operate with JNK to induce apoptosis following UV-irradiation, knockdown of H2AX did not abrogate bortezomib-induced apoptosis. Instead, blockade of p38 MAP kinase signaling, using either siRNA or a pharmacological inhibitor, reversed bortezomib-induced apoptosis and the upregulation of Noxa.
Radiation therapy is known to activate the p38 MAP kinase pathway, and is a mainstay of ESCC treatment strategies. In a final series of studies we showed that the co-administration of bortezomib with irradiation led to enhanced p38 MAP kinase activity and a significant reduction in colony formation. We therefore suggest that p38 MAP kinase pathway activation is an excellent potential therapeutic strategy in ESCC. It is further suggested that bortezomib could be added to existing ESCC therapeutic regimens.
Keywords: Squamous cell carcinoma, therapy, proteasome, p38 MAPK, bortezomib
Introduction
Esophageal cancers are amongst the most aggressive tumors known with over 75% of newly diagnosed patients dying within the first year. Five-year survival rates are also dismal (5–10%), with over 50% of patients harboring distant metastases at the time of presentation (1). One source for optimism is the current revolution in molecularly targeted cancer therapy. Recently, there has been a great deal of progress in the identification of key “driver” oncogenic mutations and signaling pathway activities that can be targeted by small molecule pharmacological inhibitors. In particular, striking results have been observed with Imatinib (Gleevec®) in chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST) and gefitinib (Iressa ®) and erlotinib (Tarceva ®) in non-small cell lung carcinoma (2–5). To date, such “targeted” strategies have been not been explored extensively in ESCC. As tumors often posses multiple genetic and cell signaling lesions, the inhibition of one signaling pathway is often therapeutically ineffective (6). Rather, more success can be envisioned with agents targeted against multiple cellular pathways. One such multi-pathway inhibitor is the novel proteasome inhibitor bortezomib (Velcade ®). Bortezomib is a dipeptidyl boronic acid that inhibits the 26S proteasome, a large multi-subunit protein complex that degrades poly-ubiquitinated target proteins, such as the cyclins, apoptosis regulators and p53 (7, 8). Although the proteasome performs many important housekeeping functions in normal cells, the increased metabolic activity and rapid cycling seen in transformed cells makes proteasome function critical for tumor cell survival (7). The mechanisms of bortezomib induced cell death remain poorly defined and appear to be cell-type specific or context dependent. In melanoma and head and neck carcinoma cell lines, increased Noxa expression appears to be critical for apoptosis induction (9, 10). There is also evidence that the generation of cellular stress leading to reactive oxygen species (ROS) is a critical feature of bortezomib’s anti-cancer activity (11). In the current study we demonstrate that bortezomib is strongly apoptotic in ESCC lines, and blocks the growth and survival of these cells in 3D organotypic cultures and animal xenograft models, thereby providing complementary in vitro and in vivo findings. We further demonstrate that bortezomib exerts its pro-apoptotic effects through a novel mechanism involving the activation of the p38 MAP kinase pathway.
Materials and Methods
Cell Lines
Esophageal cancer cells, TE cell lines (TE1, −3, −8, −10, −11, −12), and FEF3 (fetal esophageal fibroblasts) are available commercially and through the National Institutes of Health/National Institute of Diabetes Digestive and Kidney Diseases Center for Molecular Studies in the Digestive and Liver Diseases’ Cell Culture Core Facility (University of Pennsylvania, PA) and were cultured as described previously (12). Human microvascular endothelial cells (HMVEC) are available commercially through Cascade Biologics, Inc. (Portland, OR) and were cultured as described previously (13).
Antibodies and Reagents
H2AX, phospho-H2AX, p53, p21 and Noxa antibodies were purchased from Calbiochem (San Diego, CA). p38 MAP kinase, phospho-p38 MAP kinase, caspase-3 and PARP antibodies were purchased from Cell Signaling (Danvers, MA). Mouse monoclonal anti-human CD31 antibody was purchased from Dako North America, Inc. (Carpinteria, CA). Texas-Red conjugated anti-phalloidin antibody was purchased from Molecular Probes (Eugene, OR). Texas Red-conjugated anti-mouse secondary antibody and Fluorescein-conjugated anti-rabbit secondary antibody were from Vector Laboratories (Burlingame, CA). TUNEL, In Situ Cell Death Detection Kit, was used from Boehringer Mannhein/Roche (Indianapolis, IN). Ki67 staining was done using Ki67 antibody from Abcam (Cambridge, MA). For in vitro assays, stock solutions (10 mM) of the JNK inhibitor VIII (Calbiochem), proteasome inhibitor-1 (PI1, Calbiochem), MG-132 (Calbiochem) and SB 230580 (Calbiochem) were prepared in dimethylsulfoxide (DMSO) and were stored at −20°C. Stock solutions of 1 μM Bortezomib (Millenium Pharmaceuticals) were prepared in normal saline and were stored at −20°C. Stock solutions of 10 μM z-VAD-FMK (Sigma) were prepared in DMSO and were stored at −20°C.
In vitro 3D Network Formation Assay and Fluorescence Imaging
Reconstruction of vessel-like structure in 3D collagen gels and subsequent fluorescent staining of networks/cords in whole-mount gels were performed as described previously (13).
Western Blotting Analysis
Experiments were performed as described previously (6).
Irradiation experiments
Genotoxic stress was induced by exposing the U2OS cells to ionizing radiation at a dose of 3 Gy for a three minute period (IR; J.L. Shepherd Mark 1 Model 30, 137 Cesium Irradiator; J.L. Shepherd and Associates). TE12 cells were plated in four groups (control, control irradiated, bortezomib alone, bortezomib + radiation) at 30 or 60 cells per well in 96 well plates (n=6). Bortezomib treated plates received 10 nM of drug for 4 hrs, prior to irradiation. The radiation plates received 2 Gy of radiation. Plates were examined for the presence of colonies after two weeks, and the number of colonies per well scored.
Transfection
To achieve transient suppression of gene expression of H2AX and p38 MAP kinase, Dharmacon SMARTpool siRNAs were used as described previously (12).
Organotypic Cell Culture
Reconstructs of human ESCC were grown as described in (12, 14).
Immunofluorescence microscopy
TE12 cells were seeded onto glass coverslips in six-well plates and incubated overnight. Cells were then fixed and analyzed as described previously (12)
Immunofluorescence detection of Ki67 and TUNEL on the esophageal reconstructs were performed as described in (15).
Generation of Reactive Oxygen Species (ROS)
TE12 cells were treated with 500 μM H2O2 for 1 hr, 10 nM bortezomib for 24hrs and normal saline 24hrs. The cells were trypsinized and incubated with 100 nM CM- H2DCFDA from Invitrogen (Carlsbad, CA) before being washed and resuspended in phosphate-buffered saline (PBS). Cellular ROS was measured by flow cytometry. Data shown are representative of three independent experiments.
Adherent cell Proliferation assay
Cells were plated into a 96-well plate at a density of 2.5 × 104 cells/ml and left to grow overnight. Inhibition of proliferation was analyzed by the MTT assay as described previously (6).
Cell cycle analysis
Cell cycle analysis was performed after treatment with bortezomib 10 nM (0, 8, 24 and 48hrs) as described in (6).
Three-dimensional spheroid growth
Esophageal carcinoma spheroids were prepared using the liquid overlay method as described in (6).
In vivo experiments
The study protocol was approved by the Wistar Institute Animal Care and Use Committee (IACUC). Each group consisted of eight NOD/SCID mice. Sixteen mice were injected s.c. with TE11 cells (2 × 106) into the lower back. When animals developed nodules of about 5 mm in diameter, the study drug administration was initiated (day1). The NOD/SCID mice were assigned randomly to two experimental groups of eight animals each: (a) 200 μl normal saline, (b) 1mg/kg bortezomib (in 200 μl normal saline) by intraperitoneal (i.p.) injection twice a week. The dose chosen in the present study were based on preliminary-dose finding experiments. During the experiment, tumor volumes were assessed twice weekly by caliper measurements. At treatment day 16, 6 hrs after the final drug application, all animals were euthanized. Data shows the mean of the treated and untreated groups +/− s.e. mean from one experiment.
Results
Bortezomib inhibits the proliferation of human esophageal squamous cell carcinoma cell lines through G2/M cell cycle arrest and induction of apoptosis
As targeted therapeutic approaches have not been attempted in ESCC in an extensive fashion, we initiated our studies by screening a panel of ESCC lines against inhibitors of MEK (U0126), PI3K (LY294002), glycogen synthase kinase (GSK) 3-β (DW1/2) (16), the proteasome (MG-132, PI1 and bortezomib), Hedgehog (cyclopamine) and cyclooxygenase-2 (NS-398) (Supplemental Table 1). In our initial screen compounds were tested both in a two-dimensional cell culture assay (MTT) and in a three-dimensional collagen implanted spheroid assay (6). Two-dimensional and three-dimensional models were used as the pharmacological profiles of drugs in two-dimensional cell culture are not predictive of response in three-dimensional cell culture or in vivo (6). Of the compounds tested, only the proteasome inhibitors MG-132, PI1 and Bortezomib continued to show good anti-cancer activity under both model systems (Figure 1 and supplemental figure 1).
Figure 1. Bortezomib inhibits the proliferation of human esophageal squamous cell carcinoma cell lines through G2/M cell cycle arrest and induction of apoptosis.

A: In two-dimensional adherent cell culture, bortezomib reduces growth of the ESCC cell lines in a concentration-dependent fashion. Adherent TE1, TE3, TE8, TE10, TE11 and TE12 cells were treated with increasing concentrations of Bortezomib (1 nM – 10 μM) for 72 hrs before being treated with MTT. The resulting changes in absorbance were read in a plate reader at 490nm and expressed as a percentage of control absorbance. Data show the mean of three independent experiments ± SE mean.
B: Inhibition of cell growth by bortezomib is associated with G2/M cell cycle arrest. Adherent TE12 cells were treated with saline or 10 nM bortezomib for 8, 24 or 48 hrs. Cells treated with bortezomib were found to enter G2/M phase cell cycle arrest with increasing periods of time. The cell cycle profile was obtained using 15,000 cells. Data show represents mean of three independent experiments ± SE mean.
C: Bortezomib blocks growth of TE cells in three-dimensional culture. TE12 cells were grown under non-adherent conditions for 72 hrs until spheroids had formed. Spheroids were then harvested and embedded into a collagen matrix before being treated with normal saline or bortezomib (10 nM–10μM). After 72hrs, spheroids were treated with calcein-AM (viable cells, green), and ethidium bromide (dead, cells, red). Data shown is representative of three independent experiments. Magnification 4X, scale bar: 100 μM.
D: Western blot showing induction of apoptosis in cells treated with bortezomib. TE12 (ESCC) and 1205Lu (melanoma) cells were treated with 10 nM bortezomib for increasing periods of time (0–48 hrs) followed by protein extraction and probing for expression of cleaved caspase-3/PARP, p53, p21 and Noxa. β-actin is shown as a loading control.
Treatment of six human ESCC lines (TE1, TE3, TE8, TE10, TE11, TE12) with increasing concentrations of bortezomib (1 nM – 10 μM) led to a concentration-dependent decrease in cell growth (Figure 1A). Cell cycle analysis on the TE12 cell line showed that treatment with bortezomib (10 nM) led to a time-dependent (0, 8, 24, 48 hrs) increase in the G2/M, S and sub-G1 phases of the cell cycle (Figure 1B). The increase in the sub-G1 population following bortezomib treatment was indicative of the cells undergoing apoptosis. Similar pro-apoptotic effects were also noted in the TE1, TE3 and TE10 cell lines (supplemental figure 1A). Bortezomib was also highly effective in our three-dimensional spheroid model and reduced both the cell viability (as witnessed by loss of green staining and increased red fluorescence) and invasion of the TE12 line in a concentration-dependent manner (Figure 1C). Other proteasome inhibitors (PI1 and MG-132) were also found to have similar effects upon the invasion and survival of two other ESCC lines (TE1 and TE10) in our spheroid model (supplemental figure 1B and not shown).
Treatment of TE12 cells with bortezomib (10 nM) induced the rapid (<12 hrs) cleavage of caspase-3 and PARP (Figure 1D). Similar caspase-3 and PARP cleavage was also seen in TE1 cells treated with the proteasome inhibitor MG-132 (supplemental figure 1C). As the TE12 cell line has mutated p53 (17), we did not observe any increase in the expression of p53 or its downstream target p21 following bortezomib administration. However, both p53 and p21 expression was increased in 1205Lu cells, a melanoma cell line that retains functional p53 (16). The pro-apoptotic protein Noxa was shown to be rapidly upregulated (<8 hrs) in both the TE12 ESCC line and the 1205Lu melanoma line (Figure 1D). There is some suggestion that increased Noxa expression is a consequence of reactive oxygen species (ROS) generation. To investigate this we treated the TE12 cell line with bortezomib and measured the increase in fluorescence of the cell permeable ROS probe CM-H2DCFDA (supplemental figure 2). It was found that although the positive control (hydrogen peroxide) induced a rightward shift in the curve, there was no equivalent increase in fluorescence intensity seen following bortezomib treatment, indicating that this was not inducing apoptosis through ROS generation (supplemental figure 2).
Bortezomib induces apoptosis in a 3D organotypic model of ESCC and inhibits angiogenesis
We next tested bortezomib in two more elaborate ESCC models that allowed us to assess its effects on both the stromal fibroblasts and endothelial cells. In the first model, ESCC were layered on top of a tissue-like matrix consisting of esophageal fibroblasts and collagen. Here, we found that treatment with bortezomib (500 nM or 1 μM) led to a concentration-dependent increase in the level of apoptosis as seen by the enhanced TUNEL staining (Figure 2A,B). Interestingly, the effects of the bortezomib were relatively tumor-cell specific, and there was little positive TUNEL staining seen in the underlying fibroblast layer.
Figure 2. Bortezomib induces apoptosis in a 3D organotypic model of ESCC and inhibits vascular network formation.

A: 3D organotypic cultures were formed in a transwell system, using a collagen and fibroblast matrix on the bottom and ESCC (TE12 cells) seeded on top. After 14 days, the cultures were treated with bortezomib (0, 0.5 μM and 1 μM). After 72hrs the reconstructs were harvested and paraffin-embedded. TUNEL staining was carried out to quantify apoptotic cells after treatment. TUNEL-green, DAPI-blue. Magnification 10X, scale bar: 100 μM.
B: Graph showing quantification of apoptotic TE12 cells in treated reconstructs (as shown in A). Total area of positive TUNEL staining (μM × 103) was assessed using ProImage 6.0 software. Data shown is representative of three independent experiments. *P<0.05
C: Vascular networks were created using human endothelial cells (HMVEC) that were cultured and overlaid with collagen I, followed by a second overlay of collagen containing TE12 cells and esophageal fibroblasts (FEF3). After solidification of the collagen matrix, cells were treated with 0 (normal saline) or 0.5 μM of bortezomib for 72hrs. 3D-angiogenesis models were harvested and immunofluorescence staining was done, showing GFP-tagged FEF3 esophageal fibroblasts-green, HMVECs (CD31-red) and total nuclei (DAPI-blue). All representative images are shown as a 3-color merge, with the monochrome images of CD31 staining. Bortezomib treated 3D-angiogenesis model showed decreased vascular network formation in comparison to the control. Magnification 20X, scale bar: 100 μM.
Many targeted therapies also have unintended beneficial effects upon angiogenesis. To investigate this, we used a model where the interaction of ESCCs and human esophageal fibroblasts co-operate to induce vascular network formation of human microvascular endothelial cells (HMVECs). In control cultures, the interaction of ESCC and fibroblasts induced the HMVECs to lift off the bottom of the plate and grow upwards into the acellular collagen layer, where they formed organized vascular networks, as demonstrated by the increased CD31 staining (Figure 2C). Treatment of the cultures with bortezomib (0.5 μM) completely inhibited the organized vascular network formation, and only a sparse layer of unorganized HMVECs were observed (Figure 2C).
Bortezomib induces regression of established human ESCC xenografts through inhibition of proliferation and apoptosis induction
Next, we grew TE11 cells as tumor xenografts in NOD/SCID mice. After tumor establishment (5 × 5 mm), mice were dosed twice weekly with 1 mg/kg bortezomib by i.p. injection. After 14 days it was found that bortezomib treatment had significantly suppressed tumor growth (tumor growth; vehicle treated: 4.2 ± 0.2-fold. tumor growth; bortezomib treated: 0.6 ± 0.1-fold) and led to significant (P<0.05) tumor regression (Figure 3A–B). To assess the mechanism of action of bortezomib in the xenografts, sections were taken from control and treated tumors and stained for either proliferation (Ki67) or apoptosis induction (TUNEL) (Figure 3C). It was noted that 14-day bortezomib treatment led to a significant (P<0.05) reduction in Ki67 positivity, whilst concurrently increasing TUNEL staining (Figure 3D).
Figure 3. Bortezomib induces regression of established human ESCC xenografts through inhibition of proliferation and apoptosis induction.

TE11 cells were grown as tumor xenografts in NOD/SCID mice. After tumor establishment, mice were dosed twice weekly with 0 (normal saline) or 1 mg/kg bortezomib i.p. (8 mice per group) for 16 days. A: photographs of representative tumors for each group at day 16. B: growth curves normalized to the start volumes. bortezomib treatment led to significant (* P<0.05) levels of tumor regression. C: Immunofluorescent staining of tumors from each group. TUNEL and Ki67 staining was done in paraffin-embedded sections (TUNEL-red, Ki67-green, DAPI-blue). Magnification 10X, scale bar: 50 μM. D: Quantification of immunofluorescence for treated and untreated tumors. Graph shows the percentage of positive cells in each group. * P<0.04.
Treatment of ESCC lines with Bortezomib induces phospho-H2AX foci downstream of caspase-3 activation
It is known that during the DNA damage response, activated H2AX localizes to discrete nuclear foci at the site of DNA double strand breaks. Treatment of the TE12 cells with bortezomib (10 nM) caused a massive upregulation of phospho-H2AX in the nucleus, with high power magnification revealing the discrete focal expression of the H2AX (Figure 4A: Inset). As recent studies have shown that H2AX activation may play an important role in apoptosis induction (18) we transfected TE12 cells with an siRNA against H2AX, which led to near-total protein knockdown after 4 days of treatment (Figure 4B). Any possible role of H2AX in bortezomib-induced apoptosis induction was discounted by the fact that protein knockdown did not reduce either caspase-3 cleavage or DNA laddering (Figure 4C and data not shown). Next, we investigated whether the increase in phospho-H2AX occurred as a downstream consequence of DNA-strand breaks following caspase cleavage. In these studies, the TE12 cells were pre-treated using the pan-caspase inhibitor z-VAD-FMK (1 μM) prior to bortezomib treatment. Here, the inhibition of Bortezomib-induced caspase activation completely blocked the phosphorylation of H2AX, indicating that this occurred following caspase cleavage (Figure 4D).
Figure 4. Knockdown of H2AX expression does not inhibit Bortezomib-induced apoptosis.

A: Immunofluorescence showing γ-H2AX foci formation after bortezomib treatment. TE12 cells were grown on glass slides and treated with normal saline or 10nM Bortezomib for 12hrs. Slides were harvested, fixed, and permeabilized, and before being stained for phospho-H2AX (γ-H2AX-red, DAPI-blue), magnification 20 X, scale bar: 50 μM. Inset: Magnification 60 X, scale bar: 50 μM.
B: Western blot showing the downregulation of H2AX after day 4 of siRNA transfection. Proteins were extracted and resolved followed by probing for H2AX, ATM and β-actin.
C: H2AX knock-down TE12 cells and control cells were treated with 10 nM bortezomib for 24 and 48hrs. Cells were harvested, lysed and probed for caspase-3 cleavage. No difference in caspase-3 cleavage was observed in H2AX knockdown cells compared to control cells.
D: TE12 cells were treated with z-VAD-FMK (1 μM), bortezomib or a combination of both for 48hrs. Cells lysates were then harvested, resolved by Western blotting and probed for cleaved-PARP (c-PARP), cleaved caspase-3 (c-Caspase-3), total caspase-3 (Caspase-3), total PARP (PARP) and phospho-H2AX (p-H2AX). Equal protein loading was confirmed by stripping of the blot and reprobing for actin expression.
p38 MAP kinase activity is critical to Bortezomib-induced apoptosis and interaction with radiation treatment
Previous work has demonstrated that p38 MAP kinase is critical for radiation-induced G2/M arrest (19). As bortezomib induces a G2/M cell cycle arrest in the ESCC lines (Figure 1B), the potential role of p38 MAP kinase in the activity of bortezomib was addressed. Treatment of the cells with Bortezomib (10 nM) induced the phosphorylation (<4 hr) of p38 MAP kinase (Figure 5A). A critical role for p38 MAP kinase in bortezomib-induced apoptotic response was shown by the ability of the specific p38 MAP kinase inhibitor SB 230580 (10 μM) to block caspase-3 cleavage (Figure 5B). Pre-treatment of the cells with SB 230580 also blocked the bortezomib-induced activation of both Noxa and phospho-H2AX (Figure 5B), demonstrating that p38 MAP kinase is a critical pathway necessary for the anti-cancer activity of bortezomib in ESCC lines. We further demonstrated that knockdown of p38 MAPKalpha protein expression using an siRNA was similarly able to block bortezomib-induced caspase cleavage (Figure 5C). Next, it was shown that bortezomib also activated another stress-activated MAP kinase - the JNK pathway (Supplemental Figure 3). However, unlike p38 MAP kinase, there was little evidence for this pathway being directly involved in bortezomib-induced apoptosis, as inhibition of the pathway, using the small molecule inhibitor JNK inhibitor VIII (1 μM), did not attenuate the level of bortezomib induced caspase-3 cleavage.
Figure 5. p38 MAP kinase activity is critical to Bortezomib-induced apoptosis and interaction with radiation treatment.
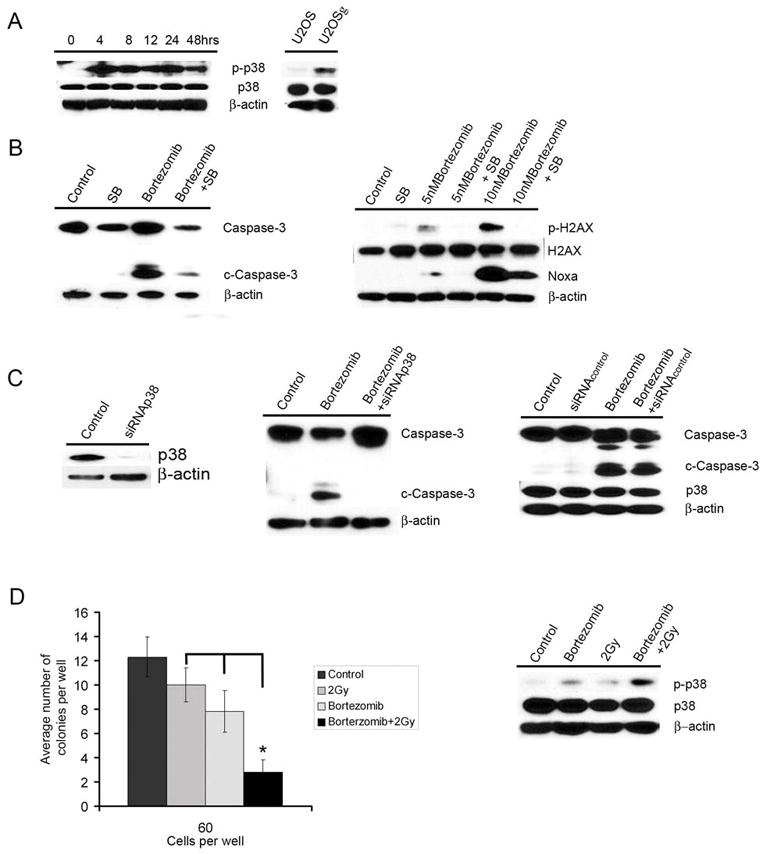
A: Bortezomib treatment led to the rapid upregulation of phospho-p38 MAP kinase (p-p38) activity in TE12 cells. Irradiation of the human osteosarcoma line U2OS also led to the rapid upregulation of p38 activity. Equal protein expression was confirmed by stripping and probing the blot for actin expression.
B: Inhibition of p38 MAP kinase reverses bortezomib-induced apoptosis. TE12 cells were pre-treated with 10 μM SB 230580 for 24hrs before being treated with bortezomib (10 nM) for 48hrs. Cells were harvested, lysed and probed for caspase-3 cleavage, total caspase-3, phospho-H2AX, total H2AX and Noxa expression. Equal protein loading was confirmed by stripping the blot and probing for actin expression.
C: Knockdown of p38 MAP kinase inhibits Bortezomib induced caspase-3 cleavage. TE12 cells were transiently transfected using SMARTpool siRNA p38 MAP kinase (siRNAp38). Control cells were transfected with a scrambled siRNA sequence (siRNAcontrol) After 4 days, cells were treated with bortezomib (10 nM, 48 hrs). Protein was then extracted from the cells, resolved and probed for expression of caspase-3, cleaved caspase-3 (c-caspase-3) and total p38 MAP kinase (p38).
D: Western blot showing the positive interaction of bortezomib and radiation in the activation of p38 MAP kinase signaling. TE 12 cells plated in 10cm dishes were treated with control, 2 Gy of radiation alone, bortezomib alone, and bortezomib + radiation (2Gy). Plates were pre-treated for 4 hours with saline or bortezomib (10 nM) before irradiation. Cells were left to grow overnight before the extraction of protein, extracts were then resolved and probed for expression of phospho-p38 MAP kinase, total p38 MAP kinase, and β-actin. Radiation survival. Cells were plated in 96 well plates at the indicated number per well. Cells were then treated with either saline (control) or bortezomib (10 nM) for 4 hrs prior to radiation. Cells were left to grow for two weeks, after this time the total number of colonies were counted. The bars shown are the means obtained from six experiments (*P<0.0001).
It is known that irradiation activates the p38 MAP kinase pathway. Given that pre-operative radiotherapy is a standard treatment for ESCC, we next explored whether there was any positive interaction between bortezomib and irradiation. Treatment of the TE12 cells with either bortezomib (10 nM) or radiation (2 Gy) led to a modest increase in p38 MAP kinase activity (Figure 5D), whereas co-administration of the drug with irradiation significantly increased the level of p38 MAP kinase activity. The enhanced p38 MAP kinase activity also translated into increased cytotoxicity with the administration of bortezomib (4 hrs, 10 nM) prior to irradiation (2 Gy), significantly (P<0.0001) reducing the formation of TE12 colonies over a 14-day period (Figure 5D).
Discussion
In the current study we describe the possible therapeutic utility of the proteasome inhibitor bortezomib in ESCC and elucidate a completely novel mechanism of action for this drug involving p38 MAP kinase-dependent apoptosis. In our initial screen it was noted that only PI1/MG-132/bortezomib had good anti-cancer activity in both the 2D and 3D models. Bortezomib was anti-proliferative and strongly pro-apoptotic in 2D ESCC cell cultures, 3D organotypic cultures and a human ESCC xenograft mouse model. The concentrations of bortezomib required to induce regression of established ESCC xenografts are lower than those reported for other solid tumors (20) and seem to be readily achievable in the clinic (21). Unlike in other squamous cell carcinoma lines, such as PAM212, we did not find any effects upon NFκB signaling following bortezomib treatment (20). At least part of the potential activity of bortezomib against ESCC may be the result of impaired angiogenesis, as bortezomib was found to inhibit tumor-induced vascular network formation in a 3D model of ESCC-induced angiogenesis. It is however, difficult to conclude how bortezomib was exerting its anti-angiogenic effects, particularly as the concentrations of bortezomib used were likely to induce apoptosis of the ESCC cells and may have in fact been the result of impaired ESCC/HMVEC interaction. Any potential anti-angiogenic activity of bortezomib is likely to be of great significance to ESCC as this is known to be a highly angiogenic tumor. A number of studies have shown that angiogenesis, as measured by the extent of tumor microvessel density, as well as VEGF expression, are prognostic factors for ESCC (22–24). It was however difficult to confirm the bortezomib-driven inhibition of angiogenesis within our ESCC xenografts, as the tumors are typically not highly vascularized when grown in this model.
Treatment of human ESCC lines with bortezomib is strongly growth inhibitory, associated with induction of a G2/M phase cell cycle arrest and apoptosis. Induction of G2/M arrest is typically associated with induction of p53 activity and transcription of its down-stream target p21 (25). Previous studies have shown that almost all human ESCC lines have defects in p53 function, arising as a result of mis-sense mutations in the coding sequence (17). In agreement with a lack of functional p53 in the ESCC lines, bortezomib was found to induce cleavage of caspase-3 and PARP without the induction of either p53 or its downstream target p21. In contrast, bortezomib did induce the activation of p53 and p21 in the 1205Lu melanoma cells (which are p53 wild-type). Bortezomib treatment was found to induce the rapid upregulation of the pro-apoptotic protein Noxa in both the ESCC and the melanoma cell lines (10). Although Noxa is known to be a p53-target gene (26), the p53-independent induction of Noxa reported here is consistent with that observed in other studies using p53-mutated cancer cell lines (Jurkat, Sk-MEL-28 and MDA-MB-231) (9, 10, 27).
Previous studies have demonstrated that the induction of reactive oxygen species (ROS) can activate Noxa in response to bortezomib treatment, and that this may be independent of p53 function (27). It has been also shown that ROS generation is associated with bortezomib activity in mantle cell lymphoma and small cell lung carcinoma (27, 28). Here we found that bortezomib treatment was not associated with ROS generation. Instead, we observed an increase in the expression of phospho-H2AX a marker of the DNA damage response. Recent work has shown that H2AX can mediate apoptosis directly where JNK directly phosphorylates H2AX leading to caspase-3 cleavage and caspase-activated DNase activity (18). As bortezomib activates both JNK signaling and induces H2AX phosphorylation, we next asked whether H2AX activity was required for bortezomib induced apoptosis in ESCC lines. Although an siRNA against H2AX yielded good levels of protein knockdown, we did not observe any change in the magnitude of apoptosis induction or DNA laddering following 48 hr bortezomib treatment, suggesting that the interaction of JNK and H2AX was not responsible for the apoptotic effects observed. Another possible explanation for the observed DNA damage response involved the direct role of caspase activation in the cleavage of genomic DNA leading to subsequent H2AX/ATM activation. It was found that inhibition of caspase activity using z-VAD-FMK blocked the phosphorylation of H2AX, suggesting that the DNA damage response occurred following caspase cleavage.
Our initial studies demonstrated activation of the G2/M checkpoint following bortezomib treatment. UV-irradiation is known to induce a G2/M arrest through p38 MAP kinase mediated inhibition of cdc25B activity (19). We next asked whether bortezomib activated the p38 MAP kinase pathway and whether this led to apoptosis in the ESCC cells. Bortezomib treatment was found to induce p38 MAP kinase activity and inhibition of p38 MAP kinase signaling, using an inhibitor or an siRNA, markedly inhibited bortezomib-induced caspase-3 cleavage. p38 MAP kinase activation was also required for the bortezomib-induced increase in Noxa expression and the phosphorylation of H2AX, as this could be also reversed by SB 230580 treatment. Thus, it was demonstrated that activation of p38 MAP kinase is a critical step for bortezomib induced apoptosis in ESCC cells. Thus far, no direct link has been made between the activation of p38 MAP kinase and increased Noxa expression. Previously, it was demonstrated that the p38 MAP kinase activated co-factor p18HAMLET increases Noxa expression following treatment with either UV or cisplatin (29). However, in this instance Noxa is regulated through a p53-dependent mechanism. As our ESCC lines lack any functional p53 activity, this current study provides the first clues that p38 MAP kinase activity may regulate Noxa expression independently of p53.
The essential role for p38 MAP kinase in bortezomib-induced apoptosis in ESCC cells is in stark contrast to findings reported for multiple myeloma cells. Here, bortezomib also activates the p38 MAP kinase pathway, but instead leads to drug resistance through increased expression of Hsp27 (30, 31). In multiple myeloma cells it was demonstrated that p38 MAP kinase pathway inhibition actually enhanced the pro-apoptotic effect of bortezomib through the downregulation of Hsp27, Mcl-1 and Bcl-XL expression whilst upregulating p53 activity (31, 32). Again, in contrast to our findings this study also reported that increased JNK activity lead to enhanced caspase cleavage (31).
Pre-operative radiotherapy is a front-line treatment for ESCC. As irradiation induces p38 MAP kinase, we investigated whether bortezomib-induced p38 activity would enhance responses to radiation treatment. Combined treatment of the ESCC cells with radiation and a single 4 hr treatment with bortezomib enhanced both p38 MAP kinase activity and cytotoxicity in a colony formation assay. We therefore suggest that bortezomib treatment could be a potential radiosensitizing agent for ESCC. In summary, we have demonstrated that bortezomib is a promising potential treatment for ESCC. We also found that bortezomib induces apoptosis in ESCC lines through a novel mechanism involving the p38 MAP kinase-induced Noxa activation, leading to caspase cleavage, which is depicted as a model in Supplemental Figure 4.
Supplementary Material
Acknowledgments
We would like to thank Millenium Pharmaceuticals for supplying the bortezomib (Velcade ®). We would also like to thank all members of the Herlyn lab for their enthusiasm and support.
Grant support: NCI P01CA098101 to M Herlyn.
Abbreviations
- ESCC
esophageal squamous cell carcinoma
- MAPK
mitogen activated protein kinase
- HMVEC
human microvascular endothelial cells
- UV
ultraviolet
Footnotes
Conflict of interest statement: The authors declare no conflict of interest
References
- 1.Enzinger PC, Ilson DH, Kelsen DP. Chemotherapy in esophageal cancer. Semin Oncol. 1999;26:12–20. [PubMed] [Google Scholar]
- 2.Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–7. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 3.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 7.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 8.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–11. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 9.Fribley AM, Evenchik B, Zeng Q, et al. Proteasome inhibitor PS-341 induces apoptosis in cisplatin-resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem. 2006;281:31440–7. doi: 10.1074/jbc.M604356200. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez Y, Verhaegen M, Miller TP, et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65:6294–304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- 11.Yu C, Rahmani M, Dent P, Grant S. The hierarchical relationship between MAPK signaling and ROS generation in human leukemia cells undergoing apoptosis in response to the proteasome inhibitor Bortezomib. Exp Cell Res. 2004;295:555–66. doi: 10.1016/j.yexcr.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Lioni M, Brafford P, Andl C, et al. Dysregulation of claudin-7 leads to loss of e-cadherin expression and the increased invasion of esophageal squamous cell carcinoma cells. Am J Pathol. 2007;170:709–21. doi: 10.2353/ajpath.2007.060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velazquez OC, Snyder R, Liu ZJ, Fairman RM, Herlyn M. Fibroblast-dependent differentiation of human microvascular endothelial cells into capillary-like, three-dimensional networks. Faseb Journal. 2002;16 doi: 10.1096/fj.01-1011fje. [DOI] [PubMed] [Google Scholar]
- 14.Andl CD, Mizushima T, Nakagawa H, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal Keratinocytes in vitro and in vivo. Journal of Biological Chemistry. 2003;278:1824–30. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 15.Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 16.Smalley KS, Contractor R, Haass NK, et al. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007;67:209–17. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 17.Barnas C, Martel-Planche G, Furukawa Y, Hollstein M, Montesano R, Hainaut P. Inactivation of the p53 protein in cell lines derived from human esophageal cancers. Int J Cancer. 1997;71:79–87. doi: 10.1002/(sici)1097-0215(19970328)71:1<79::aid-ijc14>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Lu C, Zhu F, Cho YY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulavin DV, Higashimoto Y, Popoff IJ, et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411:102–7. doi: 10.1038/35075107. [DOI] [PubMed] [Google Scholar]
- 20.Sunwoo JB, Chen Z, Dong G, et al. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001;7:1419–28. [PubMed] [Google Scholar]
- 21.Allen C, Saigal K, Nottingham L, Arun P, Chen Z, Van Waes C. Bortezomib-Induced Apoptosis with Limited Clinical Response Is Accompanied by Inhibition of Canonical but not Alternative Nuclear Factor-{kappa}B Subunits in Head and Neck Cancer. Clin Cancer Res. 2008;14:4175–85. doi: 10.1158/1078-0432.CCR-07-4470. [DOI] [PubMed] [Google Scholar]
- 22.Igarashi M, Dhar DK, Kubota H, Yamamoto A, El-Assal O, Nagasue N. The prognostic significance of microvessel density and thymidine phosphorylase expression in squamous cell carcinoma of the esophagus. Cancer. 1998;82:1225–32. doi: 10.1002/(sici)1097-0142(19980401)82:7<1225::aid-cncr3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 23.Tanigawa N, Matsumura M, Amaya H, et al. Tumor vascularity correlates with the prognosis of patients with esophageal squamous cell carcinoma. Cancer. 1997;79:220–5. [PubMed] [Google Scholar]
- 24.Kitadai Y, Amioka T, Haruma K, et al. Clinicopathological significance of vascular endothelial growth factor (VEGF)-C in human esophageal squamous cell carcinomas. Int J Cancer. 2001;93:662–6. doi: 10.1002/ijc.1379. [DOI] [PubMed] [Google Scholar]
- 25.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 26.Vousden KH, Prives C. P53 and prognosis: new insights and further complexity. Cell. 2005;120:7–10. doi: 10.1016/j.cell.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107:257–64. doi: 10.1182/blood-2005-05-2091. [DOI] [PubMed] [Google Scholar]
- 28.Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–23. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- 29.Cuadrado A, Lafarga V, Cheung PC, et al. A new p38 MAP kinase-regulated transcriptional coactivator that stimulates p53-dependent apoptosis. Embo J. 2007;26:2115–26. doi: 10.1038/sj.emboj.7601657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–6. [PubMed] [Google Scholar]
- 31.Hideshima T, Podar K, Chauhan D, et al. p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene. 2004;23:8766–76. doi: 10.1038/sj.onc.1208118. [DOI] [PubMed] [Google Scholar]
- 32.Navas TA, Nguyen AN, Hideshima T, et al. Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia. 2006;20:1017–27. doi: 10.1038/sj.leu.2404200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.