

Abstract

A novel, efficient and expedient route to biologically and pharmaceutically important o-hydroxyaryl ketones, xanthones, 4-chromanones and flavones has been developed starting from readily available carboxylic acids and commercially available o-(trimethylsilyl)aryl triflates.
The insertion of arynes into C-N,1 C-C,2 C-Si,3 C-Sn,4 and C-Hal5 bonds and the C=O of aldehydes6 is now a well known reaction. The insertion of a benzene ring into the C-O bond of a carboxylic acid would afford a powerful synthetic approach to a range of complex and biologically important molecules. However, we have previously reported that the reaction of arenecarboxylic acids and arynes in MeCN affords excellent yields of the corresponding aryl esters, i.e., the reaction occurs by insertion into the O-H bond of the carboxylic acid.7 In a closer examination of this chemistry, we have now found reaction conditions which afford good to excellent yields of the corresponding C-O insertion products. Subsequent chemistry of the resulting o-hydroxyaryl ketones affords a novel approach to xanthones, chromanones and flavones.
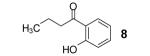
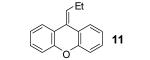
We allowed butyric acid (1) to react with the benzyne precursor 2 in the presence of fluoride ion at an elevated temperature (Scheme 1). To avoid the formation of monoarylation product 5, rather than using relatively acidic acetonitrile as the solvent,8 we turned to far less acidic THF. To our delight, together with the ester 5 the C-O insertion product 8 was formed, as well as an inseparable mixture of bis-aryne addition products 10 and 11.
Scheme 1.
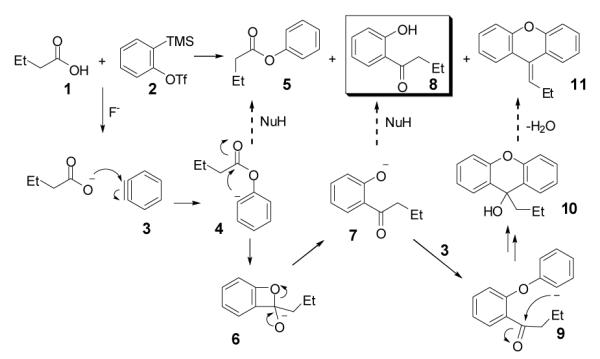
The Proposed Mechanism for the Reaction of Butyric Acid and Benzyne.
The proposed reaction mechanism (Scheme 1) is consistent with a related study of amides reported recently by Greaney’s group.1b The aryl anion intermediate 4 apparently undergoes rearrangement to the phenoxide 7 by a four-membered ring intermediate 6, resulting in formation of the desired o-hydroxyaryl ketone 8 after proton abstraction from the reaction media. Monoarylation product 5 can arise from protonation of the aryl anion 4. By-product 10 is presumably formed from reaction of phenoxide 7 with a second equivalent of benzyne, which, after dehydration, forms the xanthene 11.
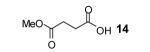
The formation of product 8 encouraged us to pursue this project, since it provides an expeditious route to o-hydroxyaryl ketones, important precursors for the synthesis of biologically useful flavones and chalcones,9 from readily available carboxylic acids. Optimization studies revealed that elevated temperatures (125 °C), dilution of the reaction media (0.017 M) and the addition of 1.5 equiv of the aryne precursor 2 and 4 equiv of CsF allow isolation of the desired product 8 in a 77% yield (Table 1, entry 1). We applied our optimized conditions to other carboxylic acids (Table 1).
Table 1.
Reaction of Carboxylic Acids with Arynesa
Reaction conditions: 0.25 mmol of acid, 1.5 equiv of benzyne precursor and 4.0 equiv of CsF in 15 mL of THF were heated in a closed vial at 125 °C for 24 h.
Isolated yield.
3.0 Equiv of benzyne precursor, 6.0 equiv of CsF and 5 mL of DME were used.
3-Methoxy-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
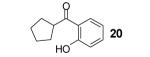
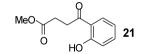
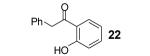
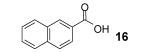
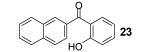
If the benzyne precursor is employed in excess (3 equiv), it is possible to obtain the dehydrated bis-aryne insertion product 11 in a 50% yield.10 This method well tolerates alkene, cycloalkyl, ester and benzyl functional groups; the corresponding products 19-22 have been isolated in 58-72% yields. However, the yields from arenecarboxylic acids are typically low, with inseparable mixtures of products formed. One of the most successful reactions is with β-naphthoic acid (16), which affords the corresponding o-hydroxyaryl ketone 23 in a 44% yield.
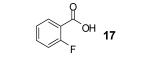
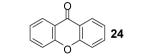
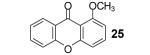
We also envisioned that the in situ formed phenolate anion 7 could potentially undergo a nucleophilic aromatic substitution reaction. This could provide an interesting route to xanthones and analogues, which are very important ring systems in biology and pharmacy.11 Indeed, when o-halobenzoic acids are allowed to react with the benzyne precursor and CsF under our optimized conditions, we were delighted to see formation of the xanthone, with an o-fluoro substituent being the most efficient among the halogens, furnishing xanthone (24) in an 80% yield. The 3-methoxybenzyne precursor 3-methoxy-2-(trimethylsilyl)phenyl triflate, when reacted with the same acid, provided cyclized product 25 in a 71% yield. Only one regioisomer was observed in this case, which suggests that the reaction is controlled electronically rather than sterically.12
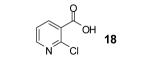
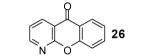
We were also delighted to obtain 4-aza-xanthone (26), albeit in only a 22% yield, starting from 2-chloronicotinic acid (18), since the majority of the reported reactions involving benzynes, even under milder reaction conditions, do not tolerate the nucleophilic nitrogen of a pyridine ring.13
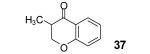
The in situ formed phenolate anion 7 could also potentially undergo a Michael addition reaction. This would provide a novel route to biologically important 4-chromanones and flavones from readily available acrylic and propiolic acids.14 Indeed, when methacrylic acid (27) was allowed to react under the optimized conditions, 4-chromanone 37 was formed, but only in a 58% yield. Optimization studies revealed that addition of the aryne precursor 2 and CsF in two portions favors formation of the product 37, increasing its yield to 84% (Table 2, entry 1). The latter reaction conditions were applied to other 2-alkenoic acids (Table 2).
Table 2.
Reaction of Acrylic and Propiolic Acids with Arynesa
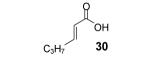
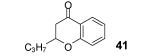
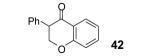
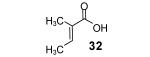
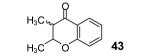
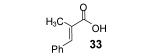
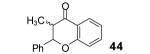
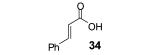
Reaction conditions: 0.25 mmol of acid, 1.5 equiv of aryne precursor and 4.0 equiv of CsF in 15 mL of THF were heated in a closed vial at 125 °C for 18 h. Then an additional 0.5 equiv of aryne precursor and 1.0 equiv of CsF was added and the heating was continued at 125 °C for 6 h.
Isolated yield.
4,5-Dimethoxy-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
The E/Z ratio is ~1.8/1.
The E/Z ratio is ~5.1/1.
The yield also includes the product obtained after base-induced cyclization of the o-hydroxyaryl ketone (see the Supporting Information).
Reaction conditions: 0.25 mmol of acid, 1.5 equiv of aryne precursor and 2.0 equiv of TBAT in 5 mL of toluene were heated at 60 °C for 24 h.
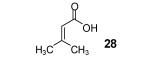
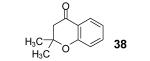
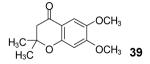
3-Methyl-2-butenoic acid (28) afforded 4-chromanone 38 in a 77% yield (entry 2). The symmetrical dimethoxy-substituted aryne precursor 4,5-dimethoxy-2-(trimethylsilyl)phenyl triflate, when reacted with the same acid, provided cyclized product 39 in a 53% yield.15
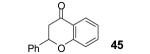
This method well tolerates alkyl and aryl functional groups, with products 40-45 being produced in moderate to good yields (56-82%). In the case of cinnamic acid, additional treatment with base (piperidine/THF/H2O)16 was required to cyclize the hydroxyaryl ketone to the desired flavanone 45 in a 74% overall yield.
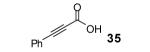
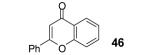
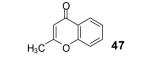
Under our optimized conditions, the major product observed in the case of propiolic acids was the decarboxylated starting material. To decrease this side reaction, we utilized a procedure analogous to that reported by Greaney’s group, which employs triphenyldifluorosilicate (TBAT) in toluene at 60 °C.1b, 17 This afforded flavone (46) and its alkyl analogue 47 in 56 and 64% yields, respectively.
In summary, we have developed a novel, efficient and expedient route to o-hydroxyaryl ketones, xanthones, 4-chromanones, flavones and their analogues by aryne insertion into the C-O bond of readily available carboxylic acids and subsequent rearrangements. The method should prove useful for the preparation of these biologically and pharmaceutically important structures. A variety of functional groups are compatible with the reaction conditions. Further studies on the scope of the method and its applications are in progress.
Supplementary Material
Acknowledgment
We thank the National Science Foundation, the National Institute of General Medical Sciences (GM079593) and the National Institutes of Health Kansas University Center of Excellence in Chemical Methodology and Library Development (P50 GM069663) for their generous financial support. We also thank Dr. Feng Shi at Iowa State University for the preparation of some aryne precursors.
Footnotes
Supporting Information Available: Detailed experimental procedure and characterization data for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1)(a).Liu Z, Larock RC. J. Am. Chem. Soc. 2005;127:13112. doi: 10.1021/ja054079p. [DOI] [PubMed] [Google Scholar]; (b) Pintori DG, Greaney MF. Org. Lett. 2009;12:168. doi: 10.1021/ol902568x. [DOI] [PubMed] [Google Scholar]
- (2).Tambar UK, Stoltz BM. J. Am. Chem. Soc. 2005;127:5340. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- (3).Sato Y, Kobayashi Y, Sugiura M, Shirai H. J. Org. Chem. 1978;43:199. [Google Scholar]
- (4).Yoshida H, Honda Y, Shirakawa E, Hiyama T. Chem. Comm. 2001:1880. doi: 10.1039/b103745p. [DOI] [PubMed] [Google Scholar]
- (5).Yoshida H, Mimura Y, Ohshita J, Kunai A. Chem. Commun. 2007:2405. doi: 10.1039/b701581j. [DOI] [PubMed] [Google Scholar]
- (6).Yoshida H, Watanabe M, Fukushima H, Ohshita J, Kunai A. Org. Lett. 2004;6:4049. doi: 10.1021/ol048298b. [DOI] [PubMed] [Google Scholar]
- (7).Liu Z, Larock RC. Org. Lett. 2004;6:99. doi: 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]
- (8).See reference 7. Acetonitrile leads to almost exclusive formation of monoarylation products.
- (9).Kotali A, Harris PA. Org. Prep. Proc. Int. 1994;26:159. [Google Scholar]
- (10).Okuma K, Nojima A, Matsunaga N, Shioji K. Org. Lett. 2009;11:169. doi: 10.1021/ol802597x. [DOI] [PubMed] [Google Scholar]
- (11).Na Y. J. Pharm. Pharmacol. 2009;61:707. doi: 10.1211/jpp/61.06.0002. [DOI] [PubMed] [Google Scholar]; (b) El-Seedi HR, El-Ghorab DMH, El-Barbary MA, Zayed MF, Goeransson U, Larsson S, Verpoorte R. Curr. Med. Chem. 2009;16:2581. doi: 10.2174/092986709788682056. [DOI] [PubMed] [Google Scholar]; (c) Diderot NT, Silvere N, Etienne T. Adv. Phytomed. 2006;2:273. [Google Scholar]
- (12).For a related observation, see: Dubrovskiy AV, Larock RC. Org. Lett. 2010;12:1180. doi: 10.1021/ol902921s.
- (13).Jeganmohan M, Bhuvaneswari S, Cheng C-H. Chem. Asian J. 2010;5:153. doi: 10.1002/asia.200900324. [DOI] [PubMed] [Google Scholar]
- (14)(a).Brahmachari G. Nat. Prod. Comm. 2008;3:1337. [Google Scholar]; (b) Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; (c) Birt DF, Hendrich S, Wang W. Pharm. Ther. 2001;90:57. doi: 10.1016/s0163-7258(01)00137-1. [DOI] [PubMed] [Google Scholar]; (d) Kim HP, Son KH, Chang HW, Kang SS. J. Pharmacol. Sci. 2004;96:229. doi: 10.1254/jphs.crj04003x. [DOI] [PubMed] [Google Scholar]
- (15).The only detected by-product was the O-monoarylation product.
- (16).Tanaka K, Sugino T. Green Chem. 2001;3:133. [Google Scholar]
- (17).It is noteworthy that this procedure did not work efficiently with substituted acrylic acids.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.