

Abstract
T memory/effector cells (Tmem/eff) isolated from psoriatic patients are chronically activated and poorly suppressed by regulatory T cells (Treg). The proinflammatory cytokine IL-6, which signals through Stat3, allows escape of Tmem/eff cells from Treg-mediated suppression in a murine system. We show here that IL-6 protein is markedly elevated and most highly expressed by CD31+ endothelial cells and CD11c+ dermal dendritic cells (DCs) in lesional psoriatic skin. We hypothesized that exposure to high IL-6 in lesional tissue may lead to the dampened Treg function observed in psoriasis patients. Indeed, we found that IL-6, but not other Stat3-activating cytokines, was necessary and sufficient to reverse human T cell suppression by Treg in an in vitro model using activated DCs as a source of IL-6. IL-6Rα and gp130 expression was significantly elevated in psoriatic effector T cells compared with normal controls. Overall, IL-6Rα expression on Treg exceeded that of effector T cells, and both populations phosphorylated Stat3 in response to IL-6. Phosphorylation of Stat3 in T cells contributes to Th17 differentiation and we identify cells within lesional tissue that coexpress CD3, IL-17, and IL-6, indicating that Th17 cells are present in vivo within the psoriatic Tmem/eff population and contribute to IL-6-mediated resistance to Treg suppression. Taken together, T lymphocytes trafficking into lesional psoriatic skin encounter high IL-6 from endothelial cells, DCs, and Th17 cells, enabling cutaneous T cell escape from Treg suppression and Th17 participation in inflammation. Targeting IL-6 signaling pathways in psoriasis may rebalance Treg/T effector activity and ameliorate disease.
Psoriasis is a prevalent, chronic inflammatory disease of the skin mediated by cross-talk between epidermal keratinocytes, dermal vascular cells, and immunocytes including activated APCs and CD2+CD45RO+ T memory/effector cells (Tmem/eff)3 (1–3). Increased proliferation of keratinocytes in conjunction with inflammation leads to epidermal hyperplasia, characteristic of lesional psoriatic skin. The central, pathogenic role of activated T cells in the development of psoriatic lesions has been demonstrated by the clinical efficacy of drugs that inhibit T cell activation or deplete activated Tmem/eff from psoriatic skin (4–7).
Under conditions of healthy immune homeostasis, the activation of Tmem/eff is controlled by CD4+Foxp3+CD25high, naturally occurring regulatory T cells (Treg), which are indispensable for the maintenance of self-tolerance (8). Depletion of Treg, both in mice and in humans, results in severe autoimmune disease (9) for which there is no compensatory mechanism. The frequency and/or function of Treg is compromised in a number of human autoimmune diseases (reviewed in Ref. 9), and we previously demonstrated that Treg isolated from psoriatic patients are less capable than those from normal donors of suppressing the proliferation of responder T cells from normal and psoriatic donors (10).
Lymphocyte infiltration into superficial perivascular skin is an early critical cellular event in psoriasis. Once in the lesional psoriatic skin, both Th1 and Th17 Tmem/eff are activated by endogenous APCs and produce proinflammatory cytokines and chemokines (11, 12). In turn, these cytokines, particularly in the presence of fibronectin, lead to cell cycle entry of psoriatic keratinocyte stem cells (1), indicating that keratinocytes in the abnormal milieu of psoriatic uninvolved skin are “primed” to respond to T cells (13). Within an altered skin microenvironment rich in EDA-fibronectin (14), dendritic cell (DC)-T cell-keratinocyte cross signaling is thought to lead to epidermal expansion and clinical plaques.
In the present study, we investigated mechanism(s) responsible for the loss of human Treg-mediated suppression of Tmem/eff previously reported in psoriasis and other autoimmune disorders (9, 10). In mice, the proinflammatory cytokine IL-6 was shown to render effector T cells (Teff) refractive to suppression (15). Because IL-6 was previously shown to be associated with pathways active in psoriasis (16, 17), we hypothesized that increased IL-6 protein in psoriatic skin may play a role in dampening Treg function, thus allowing chronic T cell expansion. Indeed, our results indicate that upon entering lesional skin, Teff encounter high levels of IL-6 from endothelial cells, DCs, and Th17 cells. IL-6 exposure leads to diminished Treg cell activity, suggesting that T cells trafficking into psoriatic lesions are modulated by IL-6 to favor activation over suppression. Although the observed loss of functional Treg suppression may be due to IL-6 acting on Teff, Treg, or APC subsets in lesional skin, Stat3 phosphorylation (pStat3) is observed within both T cell subsets and likely contributes to functional alterations in both populations.
Materials and Methods
Isolation and culture of human dermal cells and PBMCs
All studies involving human subjects were approved by the Institutional Review Boards of Case Western Reserve University, University Hospitals Case Medical Center, and the Veterans Affairs Medical Center. Punch biopsies and/or peripheral blood samples were obtained from healthy adult volunteers or patients with moderate plaque psoriasis following informed consent. Primary dermal cells were isolated as previously described (18) and cultured in complete medium (RPMI 1640 containing 10% FBS (Cambrex), L-glutamine, penicillin, streptomycin (all from Cellgro), and 2-ME). PBMCs were prepared from peripheral blood as previously described (10) and adhered to plastic for 1 h to enrich for the nonadherent, lymphocyte-containing fraction. Cell viability was determined by trypan blue exclusion.
Confocal immunofluorescent microscopy
Five-micrometer tissue sections were cut from formalin-fixed, paraffin-embedded blocks. Sections were deparaffinized and rehydrated by sequential 3-min incubations in xylene and ethanol. Ag retrieval was performed to unmask cross-linked epitopes by incubating sections for 20 min in boiling citrate buffer (DakoCytomation) and then cooling them to room temperature. Tissue was labeled overnight at 4°C with goat anti-huIL-6 or goat anti-huIL-17 (both R&D Systems), and mouse anti-huCD45, mouse anti-huCD3, mouse anti-huCD11c, mouse anti-huMac387, mouse anti-huCD31 (all from Abcam), mouse anti-huIL-6 (R&D Systems), or appropriate isotype controls. Alexa Fluor 488- or 594-conjugated chicken anti-goat or chicken anti-mouse secondary Abs (Invitrogen) were used to detect primary Abs and Draq-5 (Axxora) was used as a nuclear marker. Confocal images were acquired using a Zeiss LSM 510 scanning confocal microscope with a ×40 oil immersion Plan-Neofluar objective and a 1.3 numerical aperture. MetaMorph (version 7.1) software (Molecular Devices) was used to calculate the pixel density of labeled cells.
Differentiation and phenotyping of monocyte-derived DCs
CD14+ cells were isolated from PBMCs by negative selection (Stem Cell Technology) and cultured in complete medium supplemented with recombinant human (rh) IL-4 (500 U/ml; Promega) and rhGM-CSF (1000 U/ml; PeproTech) to generate immature DCs. DCs were pulsed for 2 h with 1 μg/ml LPS (Sigma-Aldrich) on day 7 of culture or left untreated. Pulsed cultures were washed thoroughly to remove residual LPS; DCs and conditioned medium were harvested 16 h after the pulse. Conditioned medium was used in T cell functional assays, and DCs were phenotyped with the following mAbs or appropriate isotype controls: anti-CD11c-allophycocyanin, anti-CD14-allophycocyanin, anti-CD11b-Pacific Blue, or anti-CD80-FITC, (all from BD Biosciences). Samples were analyzed using an LSR II flow cytometer (BD Biosciences). Culture supernatants from 2 × 106 DCs or from 2 × 105 dermal cells were assayed for IL-6 and soluble IL-6R (sIL-6R) content by ELISA (Quantikine kit; R&D Systems) according to the manufacturer’s instructions.
Purification of T lymphocyte populations, functional assays, and IL-6R expression
Nonadherent lymphocytes were washed, rested overnight to decrease transient expression of CD25 on activated T cells, and labeled with anti-CD4-allophycocyanin (Invitrogen) and anti-CD25-PE (BD Biosciences). Cells were sorted by high-speed flow cytometry using a FACSAria (BD Biosciences). CD4+CD25− T cells were gated using an isotype control Ab and CD4+CD25high Treg cells were defined as the top 1.5% of CD25-expressing cells within the CD4low gate (19). Two × 104 CD25− T cells were cultured with 1 × 105 allogeneic APCs in 96-well, round-bottom plates. APCs were prepared from total PBMCs by 1-h plastic adherence (to deplete T cells) and the adherent fraction was irradiated with 3000 rad immediately before use in the coculture. Some cocultures were stimulated using microbeads coated with 10 μg/ml anti-CD2/anti-CD3/anti-CD28 (from Miltenyi Biotec) at a 1:4 bead:T cell ratio. When used, CD25high Treg were added at 1:1 or 1:4 ratios with CD25− T cells. Where indicated in Results, either rhIL-6 (1–50 ng/ml) or rhIL-23 (1–100 ng/ml) (both from R&D Systems) were added to the cocultures. Cultures containing rhIL-6 also contained 25 ng/ml sIL-6R (R&D Systems). Some assays were cultured in DC-conditioned medium (DCCM; used undiluted) rather than complete RPMI 1640. All cocultures were pulsed with 1μCi/well [3H]thymidine for the final 16 h of a 7-day culture and harvested using a PhD cell harvester (Cambridge Technology). Proliferation was determined using a Beckman Coulter LS 6000SC scintillation counter. For phenotypic experiments, nonadherent PBMCs were incubated with the following mAbs (or appropriate isotype controls): anti-CD4-Pacific Blue (eBioscience), anti-CD25-allophycocyanin (BD Biosciences), and either anti-IL-6Rα-PE or anti-gp130-PE (both from BD Biosciences). Mean fluorescence intensity (MFI) of the PE channel was calculated for cells within the CD4+CD25− T lymphocyte gate and the CD4+CD25high Treg lymphocyte gate. Change in MFI was calculated by subtracting the MFI of isotype-stained samples from the MFI of Ab-labeled samples.
Immunoblots
Cells were stimulated with rhIL-6 (10 ng/ml) and sIL-6R (25 ng/ml) and immunoblotted as previously described (20) with polyclonal Abs specific for p-Y705-Stat3, total Stat3, or β-actin (all from Cell Signaling Technology), then with goat anti-rabbit HRP (Santa Cruz Biotechnology). Chemiluminescent signals were detected using Kodak BioMax MR film.
Statistical analysis
Statistical analysis was performed using either a two-tailed Student’s paired t test or a two-tailed Wilcoxon rank sum test where indicated. For both tests, values of p ≤ 0.05 were considered significant.
Results
Dermal cells from lesional psoriatic skin, but not nonlesional or normal (healthy) skin, release high levels of IL-6 protein spontaneously in culture
Dermal cell suspensions were prepared from punch biopsy material obtained from lesional and nonlesional psoriatic skin or normal (healthy) skin and cultured for 48 h. Conditioned medium was collected at 0, 12, 24, and 48 h after culture and analyzed for released IL-6 by ELISA. Baseline samples (0 h) were collected immediately following cell culture, and no IL-6 was detected in the medium at this time point (Fig. 1A). Following culture for 48 h, the mean level of spontaneous IL-6 released into the supernatants by lesional psoriasis samples was 2507 ± 263 pg/ml (n = 3), significantly higher than the level of IL-6 secreted by nonlesional psoriasis samples (144.5 ± 83 pg/ml, p = 0.008, n = 3) and normal samples (13 ± 5 pg/ml, p = 0.011, n = 3).
FIGURE 1.

IL-6 is overexpressed in lesional psoriatic skin. A, Dermal single-cell suspensions were prepared from nonlesional and lesional psoriatic skin and normal skin. Cells were cultured for 48 h and IL-6 released to the culture medium was determined by ELISA at the indicated times. *, p ≤ 0.05, n = 3. B and C, Tissue sections from the indicated sources were labeled with anti-IL-6 Ab or isotype control. B, Intensity of IL-6 within IL-6+ cells was calculated using MetaMorph (version 7.1) imaging software and is expressed as raw pixel density (n = 15 fields per tissue source ± SEM). C, Frequency of dermal cells expressing IL-6 (per mm2) was calculated (n = 15 fields per tissue source ± SEM).
Lesional psoriatic skin contains high levels of IL-6bright cells
Normal and psoriatic human skin sections were analyzed for IL-6 expression by immunofluorescence confocal microscopy. IL-6 expression per dermal cell was most intense in lesional psoriatic tissue compared with nonlesional or normal skin (Fig. 1B, p < 0.006, n = 15 fields analyzed in each of 10 patients). Although some IL-6bright cells were also detected in nonlesional psoriatic tissue and normal skin, their frequency (as well as the frequency of total dermal cells) was lower than in lesional psoriatic skin (Fig. 1C, p ≤ 0.02, n = 15 fields analyzed in each of 10 psoriasis patients and 7 normal controls).
IL-6 within the lesional psoriatic dermis localizes to CD45+ leukocytes and CD45− perivascular cells
Most IL-6bright cells in lesional psoriatic skin localized to the papillary tip regions and superficial horizontal vascular plexus (Fig. 2A). Epidermal keratinocytes also produced IL-6 in situ at moderate levels in lesional psoriatic skin (Fig. 2A) and lower levels in nonlesional psoriatic (Fig. 2B) and normal, healthy skin (Fig. 2C).
FIGURE 2.

IL-6 colocalizes with CD45+ cells within lesional psoriatic skin. A–C, Five-micrometer sections of formalin-fixed human skin biopsies were labeled with anti-IL-6 Ab (green) and the nuclear stain Draq-5 (blue). Cytoplasmic IL-6 was detectable by confocal microscopy in dermal infiltrates in lesional (A) and nonlesional psoriatic skin (B) and in normal skin (C). Biopsy specimens obtained from the indicated sources (D–G) were colabeled with Abs specific for CD45 (red, Alexa Fluor 594), IL-6 (green, Alexa Fluor 488), and the nuclear stain Draq-5 (blue). Nearly all CD45+ cells colocalized with IL-6 (white arrows), with some CD45−IL-6+ cells present (yellow arrows). Images are representative of three separate experiments. Scale bar, 50 μm.
To determine the derivation of the IL-6-producing cells, tissue sections were labeled with Abs specific to CD45 and IL-6 (Fig. 2, D–G). CD45+ cells are present in low frequency in both normal (Fig. 2D) and nonlesional psoriatic (Fig. 2E) skin, where they localize primarily to the papillary dermis. Lesional psoriatic skin (Fig. 2, F and G) shows significantly elevated numbers of CD45+ cells, including elongated cells which line the dermal-epidermal junction (Fig. 2F) and organized cellular infiltrates throughout the horizontal plexus (Fig. 2G). The majority of CD45+ cells in normal and psoriatic skin colocalize with IL-6 (white arrows, Fig. 2), although some IL-6+ cells do not express CD45 (yellow arrows, Fig. 2).
IL-6 is produced in highest amounts by DCs and endothelial cells in lesional psoriasis skin
To determine the identity of the IL-6-producing CD45+ and CD45− cells, immunofluorescence staining was performed with Abs specific to CD11c, CD3, Mac387, and CD31. Average IL-6 pixel density for labeled cells was calculated and normalized to the average IL-6 pixel density of keratinocytes from the same sections, generating the fold change in IL-6 intensity for each cell population compared with that of keratinocytes (Table I). This approach allowed us to normalize for varying levels of IL-6 intensity between tissue samples. Among CD45+ leukocytic cells, CD11c+ DCs are the highest producers of IL-6 (1.98 ± 0.4-fold higher than keratinocytes; Table I). These IL-6bright cells are present throughout the papillary dermis and lining the dermal-epidermal junction (Fig. 3A). CD3+ lymphocytes are numerous within psoriatic skin and produce moderate levels of IL-6 (Fig. 3B and Table I), whereas dermal Mac387+ macrophages, found throughout the papillary dermis, produce somewhat less IL-6 (Fig. 3C and Table I). CD31+ endothelial cells, located throughout the papillary tips and horizontal plexus, are uniformly IL-6bright (Fig. 3D), expressing 1.87 ± 0.23-fold higher levels of IL-6 than keratinocytes (Table I).
Table I.
CD11c+ dermal DCs and CD31+ endothelial cells express the highest levels of IL-6 in lesional psoriatic skin
| Average Pixel Density within Indicated Cellsa | Average Pixel Density within Keratinocytesa | pb | Fold Increase over Keratinocytesc | |
|---|---|---|---|---|
| CD11c+ | 966.5 ± 30.3 | 587.1 ± 30.6 | 0.039 | 1.98 ± 0.37 |
| CD3+ | 914.1 ± 35.7 | 676.2 ± 21.3 | 0.0044 | 1.38 ± 0.09 |
| Mac387+ | 1014.0 ± 38.4 | 958.9 ± 28.7 | 0.4056 | 1.08 ± 0.06 |
| CD31+ | 1495.2 ± 81.2 | 832.3 ± 36.3 | 0.0015 | 1.87 ± 0.23 |
n = 9 fields (three fields each among three lesional psoriatic samples).
IL-6 intensity (within indicated cell type) vs IL-6 intensity within keratinocytes in the same field.
Mean IL-6 intensity (within indicated cell type) relative to keratinocytes in the same field.
FIGURE 3.

Immune cell subsets including DCs, as well as CD31+ endothelial cells, represent major sources of IL-6 in lesional psoriatic skin. Double-labeled immunofluorescence shows infiltration of CD11c+ DCs (A), CD3+ T lymphocytes (B), Mac387+ macrophages (C), and CD31+ endothelial cells (D) in lesional psoriasis skin. IL-6-positive cells (yellow arrows), cells positive for the indicated surface marker (green arrows), and colocalized cells (white arrows) are shown. Images are representative of three separate experiments. Scale bar, 50 μm.
IL-17-producing cells are present in lesional psoriasis skin and coexpress CD3 and IL-6
Because many CD3+ lymphocytes in lesional psoriatic skin expressed IL-6 (Fig. 3B) and IL-6 can be a product of pathogenic Th17 cells (21), we determined the microanatomic location of psoriatic Th17 cells and whether they represent a T cell source of IL-6. Numerous IL-17-expressing cells were found throughout the psoriatic papillary dermis (green and white arrows, Fig. 4) and many coexpressed CD3 (white arrows, Fig. 4A), indicating a Th17 phenotype. IL-6-expressing cells (yellow arrows, Fig. 4B) occupy the same microanatomic niche as IL-17-expressing cells, with many double-labeled IL-17+IL-6+ cells present (white arrows, Fig. 4B).
FIGURE 4.
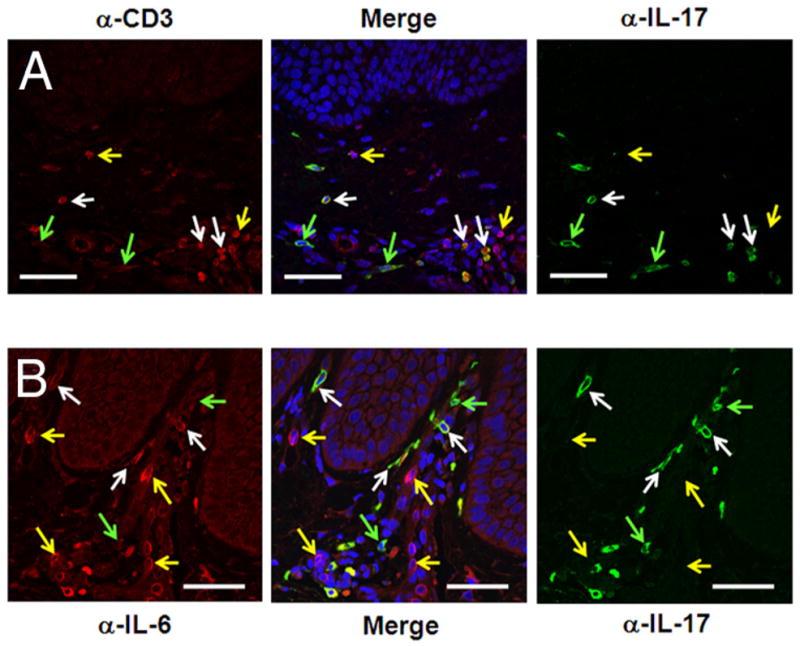
IL-17-producing cells are numerous in lesional psoriatic skin and coexpress CD3 and IL-6. Double-labeled immunofluorescence shows IL-17- producing cells in the dermis of lesional psoriasis tissue (green arrows, A and B), many of which also express CD3 (white arrows, A). CD3+IL-17− cells are denoted by yellow arrows in A. IL-17-expressing cells are found in the micromilieu of IL-6-producing cells (yellow arrows, B) and many colocalized with IL-6 (white arrows, B). Scale bar, 50 μm.
IL-6 reverses the suppressive function of human Treg
We hypothesized that localized, lesional production of IL-6 may modify human Treg function in psoriasis patients. To model the high IL-6 expressed by psoriatic lesional CD11c+ DCs (Table I), we generated mature, LPS-pulsed DCs in vitro from control and psoriatic peripheral blood monocyte precursors. After 8 days of culture, LPS-pulsed, monocyte-derived DCs were found to be CD14− and CD11chigh by flow cytometric staining (data not shown). DC-conditioned medium (DCCM) was collected on day 8 of culture and analyzed for IL-6 and sIL-6R expression by ELISA; IL-6 production by DCs obtained from controls and psoriatic patients following LPS stimulation was high (187 ± 27 and 114 ± 71 ng/ml, respectively, n = 3, p > 0.05; data not shown), similar to the high IL-6 expression observed in tissue-resident DCs (Fig. 3). Unstimulated DCs from normal and psoriatic individuals produced only minimal amounts of IL-6 (<2 ng/ml), and sIL-6R levels were extremely low in all DCCM samples examined (data not shown).
To functionally assess Treg suppression in the presence of DCCM-derived IL-6, flow cytometry-purified responder T cells (CD4+CD25−) and Treg (CD4+CD25high) were cocultured along with irradiated, allogeneic APCs in MLRs and assayed for T cell proliferation by [3H]thymidine incorporation in the presence or absence of DCCM. Treg-mediated suppression of Teff proliferation was reversed upon culture in LPS-pulsed DCCM (31% suppression) compared with non-LPS-pulsed DCCM (75% suppression; Fig. 5A). Responder T cells cultured in non-LPS-pulsed DCCM and those cultured in LPS-pulsed DCCM proliferated to a similar extent in the absence of Treg (Fig. 5A). To determine whether IL-6 in the DCCM was responsible for the reversal of Treg suppression, responder T cells were cocultured with Treg in LPS-pulsed DCCM in the presence of anti-IL-6-neutralizing Ab. In each of three experiments, the addition of anti-IL-6 restored Treg-mediated suppression, either at 20 or 50 ng/ml (Fig. 5B). Use of an isotype control Ab in place of anti-IL-6 did not restore suppression (data not shown). Proliferation of responder T cells was unaffected by the addition of anti-IL-6 in both fresh medium and DCCM (Fig. 5C).
FIGURE 5.

IL-6 is necessary and sufficient for reversal of normal and psoriatic Treg suppressive function. Normal or psoriatic CD4+CD25− lymphocytes were cultured with allogeneic APCs in a MLR or stimulated with anti-CD2/anti-CD3/anti-CD28 microbeads. Where indicated, CD4+CD25− T cells were mixed at 1:1 or 1:4 ratios with CD4+CD25high Treg and cultured for 7 days. Tritiated thymidine ([3H]) was added for the final 16 h of culture. A, Cells were incubated with DCCM, pulsed or not with LPS. B, Proliferation of CD4+CD25− T cells from three separate donors is shown. Cells were cultured in LPS-pulsed DCCM, with or without neutralizing anti-IL-6 mAb. C, CD4+CD25− T cells were cultured with or without neutralizing anti-IL-6 mAb in the indicated medium. D, Normal or psoriatic CD4+CD25− T cells and CD4+CD25high Treg were cocultured with the indicated concentrations of rhIL-6 and sIL-6R. *, p ≤ 0.01, n = 3. E, Normal CD4+CD25− and CD4+CD25high T cells were cocultured with anti-CD2/anti-CD3/anti-CD28 microbeads. F, Normal CD4+CD25− T cells and CD4+CD25high Treg were cocultured with the indicated concentrations of rhIL-23. Results show the mean of three (A–D and F) or four (E) separate experiments ± SEM. N.S., Not significant.
To determine whether IL-6 alone is sufficient to mediate the loss of Treg suppressive capacity, we added rhIL-6 directly to T cell cocultures. Treg did not proliferate, either in the presence or absence of IL-6 (Fig. 5D) or in the presence of anti-IL-6-neutralizing Ab (data not shown). The proliferation of both normal and psoriatic responder T cells was suppressed by the addition of normal Treg in the absence of IL-6 (60 and 80%, respectively, Fig. 5D). The addition of rhIL-6 to normal and psoriatic Teff/Treg cocultures led to a concentration-dependent reversal of Treg function, with a significant loss of Treg suppression observed at both 10 and 50 ng/ml IL-6, concentrations similar to those present in DCCM (p < 0.01 at both concentrations compared with cocultures without IL-6; n = 3). In contrast to allogeneic APC stimulation, IL-6 did not reverse Treg function when cells were stimulated using anti-CD2/anti-CD3/anti-CD28 microbeads in an APC-free system (Fig. 5E; n = 4).
IL-6 signaling leads to Stat3 phosphorylation, an event that also occurs in response to IL-23 signaling through the IL-23R (22). In contrast to IL-6/IL-6R ligation, normal Treg function in cocultures stimulated with allo-APCs remained intact at rhIL-23 concentrations as high as 100 ng/ml (Fig. 5F; n = 3).
CD4+CD25− T cells and CD4+CD25high Treg from normal and psoriatic patients express IL-6R and phosphorylate Stat3 in response to IL-6
CD4+CD25− and CD4+CD25high T cells from normal and psoriatic peripheral blood were labeled with Abs to IL-6Rα and gp130, and expression of these surface receptors was analyzed by flow cytometry. Effector cells and Treg from normal and psoriatic donors expressed both IL-6Rα and gp130 (Fig. 6, A and B). Interestingly, the expression of both IL-6Rα and gp130 was higher in psoriatic CD4+CD25− T cells than in their normal counterparts (Fig. 6A, p = 0.01 and Fig. 6B, p < 0.04, respectively; n = 9). The expression of IL-6Rα in Treg, conversely, was not higher within psoriatic patients compared with normal donors, although psoriatic Treg expressed higher levels of gp130 compared with normal Treg (Fig. 6A, p = 0.4 and Fig. 6B, p = 0.05, respectively; n = 9). A direct comparison between regulatory and effector T cells demonstrated that Treg from both psoriasis patients and normal donors expressed significantly elevated IL-6Rα compared with Teff (Fig. 6A, p < 0.01 for normal cells, n = 9; p < 0.02 for psoriasis cells, n = 9). Indeed, when IL-6Rα expression was analyzed on the combined subjects’ (normal and psoriatic) regulatory vs effector cell populations, Treg showed elevated levels compared with Teff at a strong level of significance (Fig. 6A, p < 0.0001; n = 18). No significant differences in gp130 expression were observed between Treg and Teff from normal donors, psoriatic patients, or when analyzed as a pooled data set (Fig. 6B ;n = 18).
FIGURE 6.

Effector and Treg from normal and psoriatic patients express IL-6R and respond to IL-6 stimulation. A and B, Nonadherent lymphocytes isolated from PBMCs were stained with mAbs specific for CD4, CD25, IL-6Rα, gp130, or appropriate isotype controls. Cells were electronically gated on CD4+CD25− or CD4+CD25high and then analyzed for surface expression of IL-6Rα or gp130. Intensity of IL-6Rα and gp130 expression is expressed as the ΔMFI (MFI of stained samples minus MFI of isotype control samples) (n = 9). Median values are shown by horizontal bars and Wilcoxon rank sum tests were performed with p ≤ 0.05 considered significant. C, Normal and psoriatic CD4+CD25− T cells and CD4+CD25high Treg were isolated from peripheral blood and incubated with or without 10 ng/ml rhIL-6 and 25 ng/ml sIL-6R (to maximize response). Protein lysates were probed with Abs specific for p-Y705 (activated) Stat3, total Stat3, and β-actin. The Western blot shown is a representative experiment from among five individual experiments. N.S., Not significant.
To characterize the functional responsiveness of CD4+CD25− and CD4+CD25high T cells to IL-6, cells from normal and psoriatic donors were stimulated with 10 ng/ml rhIL-6 and analyzed for phosphorylation of Stat3, downstream of IL-6R, by Western blot. Medium was reconstituted with sIL-6Rα, because this soluble receptor is constitutively present in human serum and is necessary for cellular responses to IL-6 (23). Both CD4+ CD25− and CD4+CD25high T cells rapidly phosphorylate Stat3 in response to rhIL-6 in a time-dependent manner (Fig. 6C). CD4+CD25− T cell pStat3 was maximal at 30 min and declined by 60 min (data not shown).
Discussion
Our evidence that human Teff regulation by Treg can be impaired by IL-6 signaling is in agreement with recent work done in mice (15). In this study, we demonstrate high levels of IL-6 in endothelial cells, DCs, macrophages, and IL-17-producing T cells in lesional psoriatic skin. Together with our previous findings of dysfunctional Treg in psoriasis and hyperactivated Teff that are resistant to normal regulation by Treg (10), we now propose that imbalanced Treg/Teff dynamics in psoriasis are mediated by IL-6 signaling events in the lesional skin, acting on T cell subsets which express the IL-6R and which respond functionally through phosphorylation of the transcription factor Stat3.
IL-6 is elevated in psoriasis at both the mRNA (16) and protein (17) levels, and its pleiotropic effects include stimulation of epidermal keratinocyte hyperplasia as well as promoting the differentiation of IL-17-producing T lymphocytes, while inhibiting Treg differentiation (24). We show here that the high levels of IL-6 present within lesional psoriatic skin are derived largely from two populations of cells in intimate contact with T cells migrating into psoriatic lesions, namely, CD11c+ dermal DCs and CD31+ endothelial cells (Fig. 3). The proximity of these cells to T cells entering the skin from blood vessels is likely to result in an initial IL-6 “pulse” as they pass through the endothelium. Furthermore, IL-6 signals are likely delivered during Ag presentation by IL-6high DCs and associated Th17 cells. Additional IL-6 produced by tissue macrophages, T cells, and keratinocytes enhance the IL-6-rich microenvironment in lesional skin, resulting in the strong induction of pStat3 in the memory/effector T cell population. Epidermal overexpression of pStat3 leads to the development of a psoriasis phenotype in mice, suggesting a causal role for activated Stat3 in disease pathogenesis (25). These combined cytokine and cellular alterations in psoriatic skin likely enable the escape of reactive T cells from suppression by Treg.
In addition to high expression in psoriatic lesional skin, IL-6 is also elevated in psoriatic serum (26) and therefore psoriatic T cells are repeatedly exposed to IL-6 in vivo. IL-6 receptor ligand binding activates numerous signaling pathways including Stat3, Stat1, and Ras/ERK (27). Stat3 phosphorylation is the most well-characterized signaling event following IL-6-IL-6R interaction and should occur in all Th1, Th2, Th17, and Treg expressing gp130 (65 ± 4% of CD4+ T cells, n = 18; data not shown). By contrast, reversal of Treg immunosuppression was not mediated by signaling through IL-23R, a Stat3-activating receptor lowly expressed on naive T cells and strongly up-regulated on human Th17 cells (28) (Fig. 5F). Failure to reverse Treg suppression with IL-23 may be due to innate resistance of IL-23R-expressing cells to suppression or the percentage of T cells expressing IL-23R may be too low to allow functional outcomes. Nevertheless, prolonged pStat3 signaling in T cells can promote unrestrained T cell activation (29) and also induce autocrine expression of Stat3 (30), creating a feed-forward mechanism by which IL-6 may promote chronic activation of Teff and loss of Treg control. This concept is consistent with pStat3-induced modification to T cell function following IL-21 signaling, which also disrupts normal Treg-mediated suppression of Teff (31).
Given the high levels of IL-6 in lesional psoriatic skin, dysfunction in Teff-Treg interactions may be mediated by signaling to either Teff or Treg. Stat3 phosphorylation occurs upon IL-6 signaling through a heterodimeric receptor, consisting of IL-6Rα, a ligand-binding domain, and gp130, a signal-transducing domain (27). IL-6 signaling to both CD4+CD25− T cells and CD4+CD25high Treg induces robust Stat3 phosphorylation, indicating that IL-6 may exert its effect by signaling to either T cell subset (Fig. 6). Teff from psoriatic patients expressed elevated IL-6Rα and gp130 levels compared with normal Teff, indicating that T cells from psoriasis patients may be more responsive to low levels of cytokine present in the psoriatic skin microenvironment (Fig. 6, A and B). Interestingly, among Treg, both normal and psoriatic donors expressed higher IL-6Rα levels than Teff (Fig. 6A), suggesting that psoriatic lesional skin may be a potent microenvironment for IL-6 stimulation of both Teff and Treg populations, which may have differential downstream effects on these subtypes and a net effect on inflammation.
T cells stimulated in the absence of accessory cells were resistant to the IL-6-mediated loss of Treg function (Fig. 5E), indicating that allogeneic stimulation by APCs may be required for IL-6 to exert its effects. This suggests that IL-6 may signal directly to APCs or, alternatively, that the trimeric APC/Teff/Treg immune synapse is required for IL-6 to exert its effect on Teff/Treg function. Alternatively, Treg suppression may be restricted to moderate strength of TCR stimulation, such as that observed in MHC mismatch and Ag presentation, while inadequate for stronger, direct TCR stimulation signals.
In addition to promoting a signaling environment in which normal Treg control mechanisms are ineffective, IL-6 contributes directly to the differentiation of pathogenic Th17 cells (32), which are associated with initiation of autoimmunity and inflammation (33). The polarization of naive human T cells to a Th17 phenotype requires Stat3 activation (32), and Stat3 phosphorylation has been demonstrated in both human psoriasis and a murine model of disease (25). Other cytokines that signal through pStat3, including IL-21 and IL-23, are also elevated in psoriasis. Th17 cells, recently identified in cell suspensions from lesional psoriatic skin (34), were reduced following clinical improvement (35) and implicated in disease pathogenesis due to the marked clinical effectiveness of anti-IL-12/23p40, which leads to dramatic reductions of IL-23p19 but not IL-12p35 (5).
Our data place Th17 cells in microanatomic proximity to IL-6-producing cells (Fig. 4), where they also express autocrine IL-6 and may thereby contribute to further Th17 differentiation. Our observations in vivo localize Th17 cells to areas of elevated IL-6 in lesional psoriatic skin, where IL-6 is available and may contribute to Th17 differentiation. Previous reports have suggested that Th17 cells are also resistant to Treg-mediated suppression (36), although our in vitro cocultures using IL-6 did not result in an up-regulation of IL-17 mRNA or protein (data not shown).
Interestingly, activation of Th17 cells in experimental autoimmune encephalitis is associated with escape of Teff from Treg control (37). Given that IL-6 signaling leads to a loss of Treg function (Fig. 5), IL-6-producing cells in the lesion likely contribute to reactivity of tissue Teff by dampening normal Treg control mechanisms. As a critical factor in the homeostatic balance between Th17 cells and Treg (38), the high levels of IL-6 generated by DCs and endothelial cells in lesional skin likely tips the balance in favor of pathogenic Th1 and Th17 cells over Treg, further allowing for unrestrained T cell activation. Thus, targeting IL-6 and its downstream signaling pathways within lesional psoriatic skin holds promise in the restoration of functional Treg control over Tmem/eff activation.
Acknowledgments
We thank W. M. Sramkoski and A. Rodriguez for excellent technical support for the flow cytometry experiments and M. Lam for excellent support for confocal experiments.
Footnotes
This work was funded by National Institutes of Health Grants AR-051498, P30AR39750, and P50AR05508 (to K.D.C.), the Morphology, Flow Cytometry, and Confocal Microscopy core facilities of the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Center and the Case Comprehensive Cancer Center (Grant P30CA43703), and the Murdough Family Center for Psoriasis.
Abbreviations used in this paper: Tmem/eff, T memory effector cell; Treg, regulatory T cell; DC, dendritic cell; rh, recombinant human; sIL-6R, soluble IL-6R; MFI, mean fluorescence intensity; DCCM, DC-conditioned medium.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Bata-Csorgo Z, Cooper KD, Ting KM, Voorhees JJ, Hammerberg C. Fibronectin and α5 integrin regulate keratinocyte cell cycling: a mechanism for increased fibronectin potentiation of T cell lymphokine-driven keratinocyte hyperproliferation in psoriasis. J Clin Invest. 1998;101:1509–1518. doi: 10.1172/JCI171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper KD, Baadsgaard O, Ellis CN, Duell E, Voorhees JJ. Mechanisms of cyclosporine A inhibition of antigen-presenting activity in uninvolved and lesional psoriatic epidermis. J Invest Dermatol. 1990;94:649–656. doi: 10.1111/1523-1747.ep12876222. [DOI] [PubMed] [Google Scholar]
- 3.Morganroth GS, Chan LS, Weinstein GD, Voorhees JJ, Cooper KD. Proliferating cells in psoriatic dermis are comprised primarily of T cells, endothelial cells, and factor XIIIa+ perivascular dendritic cells. J Invest Dermatol. 1991;96:333–340. doi: 10.1111/1523-1747.ep12465237. [DOI] [PubMed] [Google Scholar]
- 4.Ellis CN, Fradin MS, Messana JM, Brown MD, Siegel MT, Hartley AH, Rocher LL, Wheeler S, Hamilton TA, Parish TG, et al. Cyclosporine for plaque-type psoriasis: results of a multidose, double-blind trial. N Engl J Med. 1991;324:277–284. doi: 10.1056/NEJM199101313240501. [DOI] [PubMed] [Google Scholar]
- 5.Toichi E, Torres G, McCormick TS, Chang T, Mascelli MA, Kauffman CL, Aria N, Gottlieb AB, Everitt DE, Frederick B, et al. An anti-IL-12p40 antibody down-regulates type 1 cytokines, chemokines, and IL-12/IL-23 in psoriasis. J Immunol. 2006;177:4917–4926. doi: 10.4049/jimmunol.177.7.4917. [DOI] [PubMed] [Google Scholar]
- 6.Ellis CN, Krueger GG. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N Engl J Med. 2001;345:248–255. doi: 10.1056/NEJM200107263450403. [DOI] [PubMed] [Google Scholar]
- 7.Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, Gottlieb AB, Krueger JG. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1:442–447. doi: 10.1038/nm0595-442. [DOI] [PubMed] [Google Scholar]
- 8.Piccirillo CA, Shevach EM. Naturally-occurring CD4+CD25+ immunoregulatory T cells: central players in the arena of peripheral tolerance. Semin Immunol. 2004;16:81–88. doi: 10.1016/j.smim.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 10.Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, McCormick TS, Cooper KD. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005;174:164–173. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nickoloff BJ. Cracking the cytokine code in psoriasis. Nat Med. 2007;13:242–244. doi: 10.1038/nm0307-242. [DOI] [PubMed] [Google Scholar]
- 12.Somani AK, Yang MF, Cooper KD, McCormick TS. Cytokines and psoriasis: when cytokines become pathokines. G Ital Dermatol Venereol. 2007;142:679–690. [Google Scholar]
- 13.Bata-Csorgo Z, Hammerberg C, Voorhees JJ, Cooper KD. Kinetics and regulation of human keratinocyte stem cell growth in short-term primary ex vivo culture: cooperative growth factors from psoriatic lesional T lymphocytes stimulate proliferation among psoriatic uninvolved, but not normal, stem keratinocytes. J Clin Invest. 1995;95:317–327. doi: 10.1172/JCI117659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ting KM, Rothaupt D, McCormick TS, Hammerberg C, Chen G, Gilliam AC, Stevens S, Culp L, Cooper KD. Overexpression of the oncofetal Fn variant containing the EDA splice-in segment in the dermal-epidermal junction of psoriatic uninvolved skin. J Invest Dermatol. 2000;114:706–711. doi: 10.1046/j.1523-1747.2000.00871.x. [DOI] [PubMed] [Google Scholar]
- 15.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 16.Grossman RM, Krueger J, Yourish D, Granelli-Piperno A, Murphy DP, May LT, Kupper TS, Sehgal PB, Gottlieb AB. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA. 1989;86:6367–6371. doi: 10.1073/pnas.86.16.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuner P, Urbanski A, Trautinger F, Moller A, Kirnbauer R, Kapp A, Schopf E, Schwarz T, Luger TA. Increased IL-6 production by monocytes and keratinocytes in patients with psoriasis. J Invest Dermatol. 1991;97:27–33. doi: 10.1111/1523-1747.ep12477880. [DOI] [PubMed] [Google Scholar]
- 18.Szabo SK, Hammerberg C, Yoshida Y, Bata-Csorgo Z, Cooper KD. Identification and quantitation of interferon-γ producing T cells in psoriatic lesions: localization to both CD4+ and CD8+ subsets. J Invest Dermatol. 1998;111:1072–1078. doi: 10.1046/j.1523-1747.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- 19.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 20.Das L, Levine AD. TGF-β inhibits IL-2 production and promotes cell cycle arrest in TCR-activated effector/memory T cells in the presence of sustained TCR signal transduction. J Immunol. 2008;180:1490–1498. doi: 10.4049/jimmunol.180.3.1490. [DOI] [PubMed] [Google Scholar]
- 21.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 23.Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol. 2006;195:173–183. doi: 10.1007/s00430-006-0019-9. [DOI] [PubMed] [Google Scholar]
- 24.Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci USA. 2007;104:12099–12104. doi: 10.1073/pnas.0705268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, Itami S, Nickoloff BJ, DiGiovanni J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. 2005;11:43–49. doi: 10.1038/nm1162. [DOI] [PubMed] [Google Scholar]
- 26.Szepietowski JC, Bielicka E, Nockowski P, Noworolska A, Wasik F. Increased interleukin-7 levels in the sera of psoriatic patients: lack of correlations with interleukin-6 levels and disease intensity. Clin Exp Dermatol. 2000;25:643–647. doi: 10.1046/j.1365-2230.2000.00727.x. [DOI] [PubMed] [Google Scholar]
- 27.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-β are required for differentiation of human TH17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 31.Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007;178:732–739. doi: 10.4049/jimmunol.178.2.732. [DOI] [PubMed] [Google Scholar]
- 32.Zhou L, I, Ivanov I, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 33.Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- 34.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, Bowman EP, Krueger JG. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 35.Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suarez Farinas M, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183–3194. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stummvoll GH, DiPaolo RJ, Huter EN, Davidson TS, Glass D, Ward JM, Shevach EM. Th1, Th2, and Th17 effector T cell-induced autoimmune gastritis differs in pathological pattern and in susceptibility to suppression by regulatory T cells. J Immunol. 2008;181:1908–1916. doi: 10.4049/jimmunol.181.3.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, Yoshida H, Nishikawa T, Terabe F, Ohkawara T, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2008;105:9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, Riethmacher D, Si-Tahar M, Di Santo JP, Eberl G. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+Foxp3+RORγ+ T cells. J Exp Med. 2008;205:1381–1393. doi: 10.1084/jem.20080034. [DOI] [PMC free article] [PubMed] [Google Scholar]