

Abstract
Iron is deposited in perihematomal tissue after an intracerebral hemorrhage (ICH), and may contribute to oxidative injury. Cell culture studies have demonstrated that enhancing ferritin expression by targeting iron regulatory protein (IRP) binding activity reduces cellular vulnerability to iron and hemoglobin. In order to assess the therapeutic potential of this approach after striatal ICH, the effect of IRP1 or IRP2 gene knockout on ferritin expression and injury was quantified. Striatal ferritin in IRP1 knockout mice was similar to that in wild-type controls three days after stereotactic injection of artificial CSF or autologous blood. Corresponding levels in IRP2 knockouts were increased by 11-fold and 8.4-fold, respectively, compared with wild-type. Protein carbonylation, a sensitive marker of hemoglobin neurotoxicity, was increased by 2.4-fold in blood-injected wild-type striata, was not altered by IRP1 knockout, but was reduced by approximately 60% by IRP2 knockout. Perihematomal cell viability in wild-type mice, assessed by MTT assay, was approximately half of that in contralateral striata at three days, and was significantly increased in IRP2 knockouts but not in IRP1 knockouts. Protection was also observed when hemorrhage was induced by collagenase injection. These results suggest that IRP2 binding activity reduces ferritin expression in the striatum after ICH, preventing an optimal response to elevated local iron concentrations. IRP2 binding activity may be a novel therapeutic target after hemorrhagic CNS injuries.
Keywords: free radical, hemoglobin toxicity, iron, oxidative stress, stroke
Introduction
Iron accumulates within a few days in tissue adjacent to an intracerebral hemorrhage (ICH) (Wu et al., 2003), presumably due at least in part to heme breakdown by the heme oxygenases (Huang et al., 2002; Koeppen et al., 2004). The pathological significance of this phenomenon remains undefined. In experimental studies, deferoxamine, a hydrophilic chelator that detoxifies iron by binding to all six of its coordination sites, has yielded variable results to date (Nakamura et al., 2004; Warkentin et al., 2010). While this discrepancy has been attributed to the predominance of different injury mechanisms in the models used, the limited CNS bioavailability and toxicity of systemically-administered deferoxamine may also have affected outcome (Hanson et al., 2009; Whitten et al., 1965; Whitten et al., 1966). Assessment of an alternate or complementary method to chelate low molecular weight iron in the vicinity of an intracerebral hematoma may therefore be desirable.
Mammalian ferritin is a 24-mer protein with a cage-like configuration that can sequester up to 4000 iron atoms in its mineral core (Arosio and Levi, 2002). Its synthesis is regulated primarily by iron regulatory protein (IRP)-1 and IRP2, which bind to iron responsive elements in the 5’ untranslated regions of both H and L-ferritin subunit mRNA’s and thereby inhibit translation.(Meyron-Holtz et al., 2004). In iron-replete cells, the affinity of IRP1 for ferritin mRNA is markedly reduced, while proteasomal degradation of both IRP1 and IRP2 is enhanced (Guo et al., 1995; Wang et al., 2007a). Attenuating IRP binding activity increases ferritin expression in neurons, astrocytes, and most other cell types, and protects cells from iron-dependent oxidative injury in vitro (Regan et al., 2008a; Santamaria et al., 2004; Wang et al., 2007b). Since perihematomal ferritin levels are rapidly and persistently elevated by experimental ICH per se (Koeppen et al., 2004; Wu et al., 2003), it remains to be determined if targeting IRP binding would further augment expression in vivo or provide any additional benefit. Evaluation of the therapeutic potential of this approach after ICH has been greatly facilitated by the development of IRP knockout mice (LaVaute et al., 2001), which are fertile and healthy for at least the first six months of life. In the present study, we hypothesized that striatal ferritin expression would be increased after ICH by IRP1 or IRP2 gene knockout, and that this increase would be associated with reduced protein oxidation and increased cell viability.
Results
A total of 131 mice (71 males and 60 females) received striatal injections of autologous blood or artificial CSF. Mice were genotyped when 4 to 7 weeks old. Mean weight of wild-type (WT), IRP1 knockout (KO) and IRP2 KO mice prior to striatal blood injection was 25.7±0.3 grams, 25.9±0.5 grams and 25.6±0.4 grams, respectively. At three days after injection, mean weight had decreased to 22.3±0.3 grams in WT, 22.6±0.4 grams in IRP1 KO and 21.7±0.4 grams in IRP2 KO mice. One WT mouse died prior to the end of the three day observation period using the blood injection model.
Mean weight of WT, IRP1 KO and IRP2 KO mice prior to artificial CSF injection was 24.6±0.5 grams, 25.6±0.5 and 24.3±1.1, respectively, and had not changed significantly at 3 days (24.9±0.5 grams in WT mice, 26.5±0.5 grams in IRP1 KO mice and 24.1±1.0 grams in IRP2 KO mice).
Effect of IRP knockout on striatal ferritin expression
Ferritin was weakly expressed in the striata of WT mice that were the product of heterozygous IRP1 knockout matings three days after injection of artificial CSF, and did not differ significantly from that in contralateral striata (Fig. 1). Only one band was consistently detected with the anti-horse spleen ferritin antibody used, perhaps due to the similar molecular weights of murine H and L-ferritin (20.9 and 20.6 kDa, respectively) (Beaumont et al., 1989). Ferritin levels were not altered by homozygous IRP1 gene knockout. Three days after blood injection, mean striatal ferritin was 9.7-fold higher than that in contralateral striata in wild-type mice, and 10.2-fold higher in IRP-1 knockouts (P > 0.05).
Fig. 1.
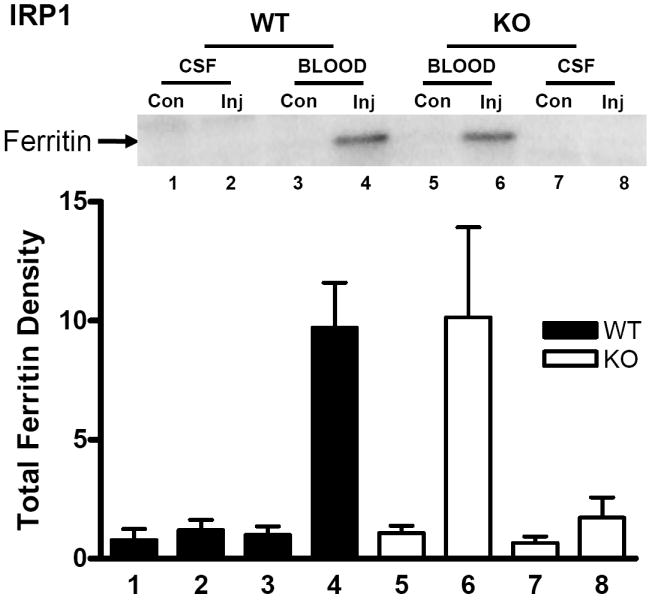
Effect of IRP1 knockout on ferritin expression after ICH. Representative immunoblot of lysates from wild-type (WT) or IRP1 knockout (KO) striata, three days after injection (Inj) with either blood or artificial CSF, or from contralateral (Con) striata, stained with polyclonal anti-horse spleen ferritin. Bars represent mean ferritin band densities (±S.E.M, n = 5/condition for artificial CSF injection and 13/condition for blood injection), normalized to the mean value in WT mice contralateral to blood injection (= 1.0). The same lane order and bar fill pattern are used for Figs. 2-4.
In wild-type mice derived from heterozygous IRP2 knockout matings, striatal ferritin three days after artificial CSF injections was likewise weakly expressed and barely detectable on immunoblots (Fig. 2). However, in homozygous IRP2 knockouts, the mean level in striata contralateral to those injected with artificial CSF was 4.9-fold higher than the corresponding value in wild-type mice. After blood injection, ferritin increased 7.4-fold compared with contralateral mean values in wild-type mice, and 15.7-fold compared with the higher contralateral value in knockouts. At this time point, the mean striatal ferritin in IRP2 knockout blood-injected striata was 8.4-fold higher than in wild-type blood-injected striata.
Fig. 2.
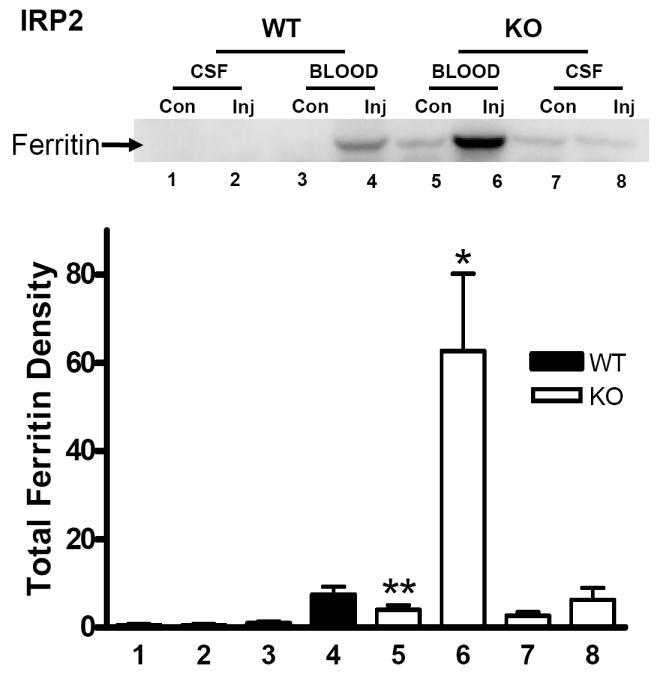
Effect of IRP2 knockout on ferritin expression after ICH. Representative immunoblot of lysates from wild-type (WT) or IRP2 knockout (KO) striata, three days after injection (Inj) with either blood or artificial CSF, or from contralateral (Con) striata, stained with polyclonal anti-horse spleen ferritin. Bars represent mean ferritin band densities (±S.E.M, n=5-7/condition), normalized to the mean value in WT mice contralateral to blood injection (= 1.0). *P< 0.05, **P< 0.01 compared corresponding WT condition, Bonferroni multiple comparisons test.
IRP2 gene knockout reduces protein oxidation after experimental ICH
Prior studies have demonstrated that protein carbonylation is a sensitive marker of hemoglobin and blood toxicity in the mouse striatum (Qu et al., 2005; Qu et al., 2007). In IRP1 wild-type mice, the mean striatal carbonyl signal was increased 2.5-fold in blood-injected striata at three days compared with contralateral striata (Fig. 3). In IRP1 knockouts, carbonyls were increased 2.9-fold (P > 0.05). Injection of an equal volume of artificial CSF had no effect on carbonyl derivative immunoreactivity in either WT or KO mice. In IRP2 wild-type mice, striatal carbonyl levels in blood-injected striata were 2.4-fold higher than in contralateral striata at this time point (Fig. 4). Approximately 60% of this increase was prevented by IRP2 knockout.
Fig. 3.
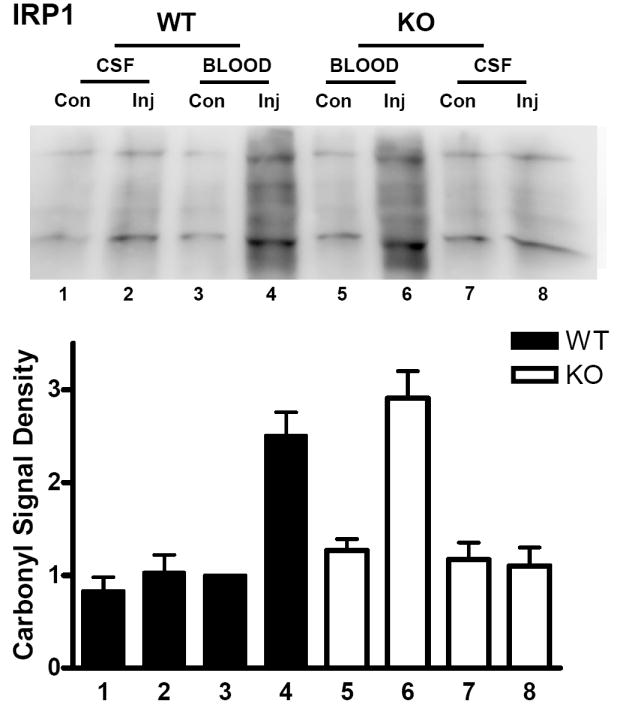
IRP1 knockout has no effect on protein oxidation after ICH. Upper: Immunoblot of striatal lysates from IRP1 knockout (KO) and wild-type (WT) mice 72 hours after injection (Inj) of blood or artificial CSF, or from contralateral (Con) striata, stained with antibody to derivitized protein carbonyls. Lower: Bars represent mean lane densities (±S.E.M, n = 4/condition for artificial CSF injection and 10/condition for blood injection), normalized to the mean value in WT striata contralateral to blood injection (= 1.0).
Fig 4.
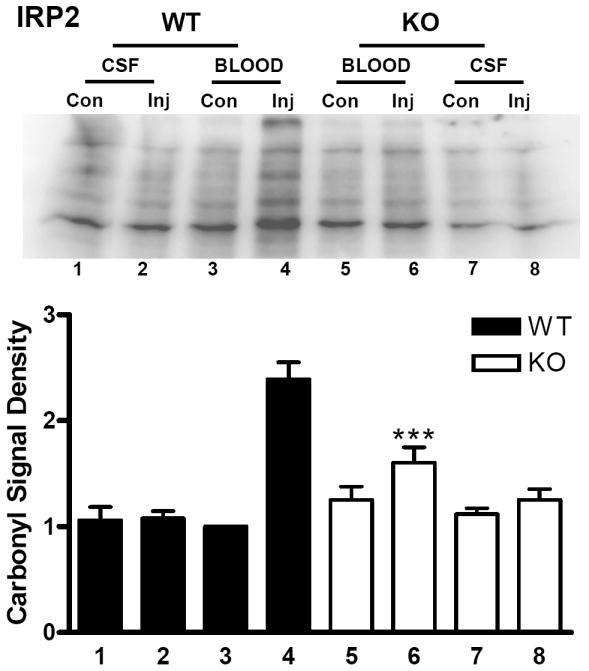
Reduced protein oxidation after ICH in IRP2 knockout mice. Upper: Immunoblot of striatal lysates from IRP2 knockout (KO) and wild-type (WT) mice 72 hours after injection (Inj) of blood or artificial CSF, or from contralateral (Con) striata, stained with antibody to derivitized protein carbonyls. Lower: Bars represent mean lane densities (±S.E.M, 5-7/condition), normalized to the mean value in WT striata contralateral to blood injection (= 1.0). ***P< 0.001 compared with the mean signal in WT striata injected with blood, Bonferroni multiple comparisons test.
Effect of IRP knockout on striatal cell viability
After striatal cell dissociation and suspension, cell viability three days after blood injection in wild-type mice produced by heterozygous IRP1 or IRP2 knockout breeders was approximately half of that in contralateral striata, as assessed by the MTT assay (Figs. 5,6). This ratio was similar in IRP1 knockouts (Fig. 5), but was significantly increased in IRP2 knockouts (Fig 6).
Fig 5.

IRP1 knockout has no effect on striatal cell viability after ICH. Representative photomicrographs of sections from wild-type (WT) and IRP1 knockout (KO) striata 72 h after striatal blood injection, stained with hematoxylin and eosin. The left border of each photo is 350 μm from the injection site. Injured neurons have condensed, darkly-stained nuclei. Scale bar = 50 μm. Bar graph represents mean striatal cell viability at this time point (±S.E.M, 4-6/condition), as assessed by cellular conversion of MTT to formazan. All values are normalized to those in contralateral striata (= 1.0). WT and KO mice injected with artificial CSF had similar values, so results were combined.
Fig 6.

IRP2 knockout increases striatal cell viability after ICH. Representative photomicrographs of sections from wild-type (WT) and IRP2 knockout (KO) striata 72 h after striatal blood injection, stained with hematoxylin and eosin. The left border of each photo is 350 μm from the injection site. Scale bar = 50 μm. Bar graph represents mean striatal cell viability three days after striatal blood or collagenase injection (±S.E.M, 5-11/condition), as assessed by cellular conversion of MTT to formazan. All values are normalized to those in contralateral striata (= 1.0) to yield MTT signal ratio. WT and KO mice injected with artificial CSF had similar values, so results were combined. ***P< 0.001, **P< 0.01 compared with the ratio in WT mice injected with blood or collagenase.
The iron chelator deferoxamine is protective after striatal blood injection in rodents and piglets (Gu et al., 2009; Nakamura et al., 2004), but has recently been reported to be ineffective when ICH was induced by collagenase injection (Warkentin et al., 2010). The effect of IRP2 knockout was therefore also assessed using the latter model. Thirty-seven mice (23 wild-type, 14 knockouts) received striatal injections of 0.015 units collagenase rather than autologous blood. Twelve wild-type and three IRP2 knockout mice died during the three day observation period (relative risk for death in wild-type = 2.5, two sided P value = 0.0905). The mean pre-injection weight in the 11 surviving mice in each group was 28.1 grams (wild-type) and 27.1 grams (knockout), and decreased to 24.9 grams and 24.1 grams at three days, respectively. Striatal cell viability in wild-type mice at this time point was 46.1±6.1% of contralateral, and increased to 71.9±4.1% in IRP2 knockouts (P = 0.002).
Discussion
The present study accomplishes two aims. First, we have identified IRP2 as a key inhibitor of ferritin upregulation after ICH in the mouse striatum. Although ferritin was induced in wild-type mice three days after blood injection, IRP2 apparently attenuated this effect, since an additional 8.4-fold increase was observed in IRP2 knockouts. Second, we have presented evidence suggesting that this ferritin hyperexpression is protective. Protein carbonyls, which are sensitive markers of iron-dependent oxidative injury (Levine et al., 1994), were significantly reduced in the hemorrhagic striata of IRP2 knockout mice, compared with their wild-type counterparts. Cell viability at three days was increased, whether the hematoma was produced by blood or collagenase injection. These results are consistent with our observations in primary cortical cell cultures that IRP2 knockout neurons are protected from the iron-dependent injuries produced by hemoglobin and hydrogen peroxide (Regan et al., 2008a; Regan et al., 2008b), and provide the first in vivo evidence that ferritin induction may be beneficial after ICH.
Prior studies in vitro have demonstrated that both IRP1 and IRP2 contribute to basal regulation of ferritin synthesis. Wang et al. observed that IRP1 knockdown in HeLa cells had no effect on ferritin levels, IRP2 knockdown resulted in a moderate increase, while knockdown of both produced synergistic hyperexpression and protected cells from hydrogen peroxide toxicity (Wang et al., 2007b). In primary neuron-glia cultures, IRP1 knockout increased ferritin expression 1.9-fold over wild type cells after hydrogen peroxide exposure, compared with a 6.9-fold increase in IRP2 knockouts (Regan et al., 2008b), with increased expression in both neurons and glial cells (Regan et al., 2008a). The present observation that striatal ferritin was not increased whatsoever at baseline or after blood injection in IRP1 knockout mice is consistent with results of Meyron-Holtz et al. (Meyron-Holtz et al., 2004) that IRP2 can fully compensate for loss of IRP1 in most tissues in vivo, and suggests that it is also sufficient after brain hemorrhage. Although IRP1 cannot fully compensate for loss of IRP2, it does appear to have a regulatory role, since ferritin expression was not maximized by IRP2 knockout per se, but rather increased significantly after blood injection. Mice lacking both IRP1 and IRP2 do not survive gestation (Smith et al., 2006), so the precise effect of IRP1 in the absence of IRP2 cannot be evaluated using a double knockout approach. However, the present results demonstrate that IRP2 binding activity is the more promising therapeutic target after ICH.
The hypothesis that iron toxicity contributes to the pathogenesis of ICH has been supported by the results of a series of studies in which bolus dosing of the iron chelator deferoxamine (DFO) attenuated edema, oxidative injury markers, and perihematomal cell death after striatal blood injection, and improved neurological outcome (Nakamura et al., 2004; Okauchi et al., 2009; Okauchi et al., 2010). Conversely, Warkentin et al. (2010) have recently reported that DFO had no effect when ICH was induced by collagenase injection. Although model-specific and laboratory-specific variables may account for this discrepancy, the limitations of DFO as an acute therapeutic agent should also be considered. When administered by high dose bolus intravenous injection, it often produces hypotension, and then is rapidly eliminated via renal filtration, with a plasma half life of 5.5 minutes in male Swiss-Webster mice (Hallaway et al., 1989; Whitten et al., 1965; Whitten et al., 1966). For these reasons DFO is administered only by continuous infusion in clinical settings. In aqueous solutions DFO is a positively charged amine that penetrates biological membranes slowly (Cable and Lloyd, 1999), and may have limited CNS bioavailability. Hanson et al.(2009) recently reported that the mean DFO concentration in the rat striatum 25 minutes after intravenous administration of a 6 mg bolus was 0.14 μM, compared with 44 μM in blood and 156 μM in the kidney. Although delivery to striatal tissue at risk may increase with breakdown of the blood-brain barrier after ICH, data to that effect have not yet been published. IRP2 knockout mice may provide an alternate model to further test the iron toxicity hypothesis of hemorrhagic brain injury, without concerns about confounding hemodynamic and pharmacokinetic variables.
Transition metals such as iron and copper catalyze the oxidation of the side chains of lysine, arginine, proline and threonine residues to carbonyl derivatives (Stadtman and Levine, 2003), which can be readily detected on immunoblots with specific antibodies. It has been previously reported that hemoglobin increases protein carbonylation in cultured cells, consistent its iron-dependent pro-oxidant effect (Sadrzadeh et al., 1987), and that carbonyl levels are significantly reduced by exogenous membrane-permeable iron chelators or IRP2 knockout (Comporti et al., 2002; Regan et al., 2008a). The present study extends the latter observation to the intact mouse striatum, and provides novel evidence that augmentation of the iron binding capacity of perihematomal cells may attenuate tissue oxidation after experimental ICH.
Cellular reduction of tetrazolium salts such as TTC and MTT to colored formazan products is a frequently used marker of cell viability in cell culture and in stroke models. Since the area of reduced staining in tissue slices of hemorrhagic mouse striata is often not clearly defined (Qu et al., 2005; Qu et al., 2007), the reducing capacity of the entire striatum was quantified after cell dissociation by trituration. This method allows delivery of the same concentration of tetrazolium to all cells, and in this model quantifies striatal injury in a manner that correlates well with more laborious cell counts (Qu et al., 2005; Qu et al., 2007). The mouse striatum is particularly well-suited for this approach for three reasons. First, it is architecturally-distinct in fresh tissue preparations and therefore easily dissected free of surrounding tissue. Second, its small size (mean volume 23.6 ± 0.6 mm3, Rosen and Williams, 2001) allows complete dissociation of the entire structure, eliminating tissue sampling bias. Third, it has a relatively homogeneous cellular composition, containing predominantly medium spiny neurons and few astrocytes, with a neuron:astrocyte ratio ~ 5:1 in 6 month old mice (Sturrock, 1980). The 50% decrease in MTT reduction three days after blood injection was similar to prior observations in wild-type mice using this model (Qu et al., 2007), and was also approximated by injection of 0.015 units collagenase. The attenuation of this effect in IRP2 knockout mice when ICH was induced by either blood or collagenase injection suggests that ferritin hyperexpression may be beneficial in both models.
The three day mortality rate of wild-type mice after collagenase injection (52%) is comparable to that reported by Tang et al. (Tang et al., 2004) and by Titova et al. (Titova et al., 2008), and contrasts with the > 99% survival after striatal blood injection. Since surviving collagenase-injected mice and blood-injected mice sustained a similar reduction in cell viability, as assessed by MTT assay, these results suggest greater variability in the former model when using this species. A trend toward increased survival was identified in IRP2 knockouts after collagenase injection (21% mortality), but this difference did not quite reach statistical significance.
Although young IRP2 knockouts have a normal phenotype and are fertile, Lavaute et al. (2001) described a late-onset movement disorder in the same line of IRP2 knockouts as those used in the present study. The clinical manifestations of this syndrome included ataxia, bradykinesia, and tremor; ferritin-bound iron deposits in neurons and oligodendrocytes were noted on necropsy. Conversely, using independently-generated IRP2 knockouts, Galy et al. (2006) observed no iron deposition or neurodegeneration whatsoever in aged (13-14 months-old) mice, and only mild alterations in grooming behavior and balance. All mice receiving striatal blood or collagenase injections in the present study were only 3-5 months old, and these young IRP2 knockouts not surprisingly had a phenotype that was grossly indistinguishable from that of their wild-type littermates. Furthermore, baseline levels of protein carbonyls in the two groups were similar, indicating that the knockouts did not have a higher background level of iron-mediated oxidative stress. These observations suggest that complete lack of IRP2 binding activity may be tolerated for several months by young or middle-aged rodents. The nontoxic interval in older animals remains to be determined.
Since ferritin sequesters iron but not heme, the protective effect of ferritin hyper-expression is consistent with our prior observations that hemoglobin neurotoxicity is mediated by heme breakdown and iron release in murine cells (Rogers et al., 2003). However, the benefit of inhibiting heme breakdown may be limited by the toxicity of hemin, the oxidized form of heme that accumulates in intracranial hematomas (Robinson et al., 2009). In contrast to free iron or hemoglobin, hemin is highly lipid soluble, accumulates in cell membranes, and diffuses into perihematomal tissue within 2-3 days of experimental ICH (Gu et al., 2009; Koeppen et al., 2004). In the CNS, its cellular uptake may also be facilitated by expression of heme carrier protein-1 in astrocytes and perhaps other cell populations (Dang et al., 2010). Experimental evidence suggests that intact hemin initiates lipid peroxidation via breakdown of preformed lipid hydroperoxides (Van den Berg et al., 1988). It may also disrupt cellular ionic gradients via a non-oxidative mechanism, resulting in swelling and lysis (Chou and Fitch, 1981). Since hemin and iron are potent neurotoxins, combination therapy that targets both of these hemoglobin degradation products may be preferable and seems worthy of further investigation.
The present data identify IRP2 binding to ferritin mRNA as a possible therapeutic target after ICH. In cell culture models, both genetic and pharmacologic approaches have been successfully used to reduce IRP2 binding activity. Wang et al. (2007b) reported IRP2 knockdown with small interfering RNA in HeLa cells which was sufficient to increase ferritin expression and reduce cell death after hydrogen peroxide treatment. However, the efficacy of this approach in vitro, where it produced nearly-complete IRP2 knockdown, may be difficult to replicate in vivo. Pharmacologic antagonism of IRP binding has not been intensively investigated, although evidence to date suggests that it may be feasible. In vitro, a high concentration (5 mM) of the aconitase inhibitor oxalomalate reduced both IRP1 and IRP2 binding activity (Festa et al., 2000), increased cell ferritin, and attenuated the oxidative injury produced by ferric ammonium citrate (Santamaria et al., 2004). In vivo, administration of the oxalomalate precursors oxaloacetate and glyoxalate to mice reduced liver IRP binding activity, although ferritin levels were not measured. A similar effect was produced in vivo by all-trans-retinoic acid, although only in iron-deficient animals (Schroeder et al., 2007). These studies provide support for the concept that IRP2 binding may be inhibited by low molecular weight compounds that have access to the CNS. However, further testing of this approach may require the identification of more potent and selective antagonists, which should be facilitated by further characterization of the iron regulatory element binding sites of IRP2.
Experimental Procedures
Experimental Animals
Breeding and housing of mice was conducted exclusively at our animal care facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal care and treatments were in compliance with the guidelines described in Principles of Laboratory Animal Care, and were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. Animals were provided with food and water ad libitum and a 12-hour light/dark cycle. IRP1 and IRP2 knockout mice (Aco1 -/- and Ireb2 -/-, respectively) were descendants of those generated by Rouault and colleagues (Meyron-Holtz et al., 2004) and were crossbred with wild-type B6;129 mice that are maintained in our colony. Heterozygous mice were then used for breeding. Genotype was determined by PCR of genomic DNA extracted from proteinase K-treated tail clippings, using the following primer sets:
IRP1 wild-type: forward: 5’-GAGAGGTCCTCCCTCTTGCT-3’ reverse: 5’-CCACTCTCTCGAAGGTAGTAG-3’.
IRP2 wild-type forward: 5’ - TGT TCC TGT CAG TCC TCG TG - 3’; reverse: 5’ - GGC CAG ACT GGT CTT CAG AG - 3’.
NeoR insert forward: 5’ - GAT CTC CTG TCA TCT CAC CT-3’; reverse: 5’ - TCA GAA GAA CTC GTC AAG AA-3’.
NeoR insert primers were the same for IRP1 and IRP2 knockouts. Wild-type and knockout primer reactions were run separately. Absence of wild-type IRP gene expression in mice identified as homozygous knockouts by this method was confirmed by RT-PCR, as previously described (Regan et al., 2008b), using primers specific for the IRP1 and IRP2 knockout insertion sites.
Striatal Blood or Collagenase Injections
Mice were anesthetized with 2% isoflurane in oxygen, which was delivered with a tightly-fitted mask. Blood was collected from a tail vein into a syringe that had been flushed once with sterile 3.5% trisodium citrate (Acros Organics, Cat. # 227130010). After mice were secured into a stereotactic frame (David Kopf Instruments, Tujunga, CA), a 30-gauge blunt needle was introduced through a burr hole into the right striatum at the following coordinates relative to the bregma: 2 mm lateral, 1 mm posterior, and 3.5 mm below the surface of the skull. Each mouse was then injected with 25 μl blood (1 μl /minute) using a minipump (Nanomite Injector Syringe Pump, Harvard Apparatus, Holliston, MA). Additional mice were injected with an equal volume of artificial CSF (NaCl 148 mmol/L, KCl 3 mmol/L, CaCL2 1 mmol/L, MgCL2 0.8 mmol/L, Na2HPO4 0.8 mmol/L, NaH2PO4 0.2 mmol/L) as a surgical control, also using a syringe that had been flushed once with sterile 3.5% trisodium citrate. Alternatively, hemorrhage was induced by injection of 0.015 units sterile collagenase (Sigma-Aldrich, St. Louis, MO, Cat. # C2399) in 2 μl artificial CSF, without citrate. The injection needle was slowly withdrawn 10 minutes later and the wound was sutured.
At 72 hours after injection, mice were deeply anesthetized with isoflurane and then were euthanized by cervical dislocation. Brains were rapidly removed and placed into a 60-mm culture dish containing 3 ml of dissecting medium consisting of Hanks Balanced Salt Solution (Gibco/Invitrogen, Carlsbad, CA, Cat. #14185) supplemented with 27.8 mM glucose, 20.5 mM sucrose, and 4.2 mM sodium bicarbonate (Rose et al., 1993). Both striata were then excised under a dissecting microscope, and tissue samples were collected for analysis.
Immunoblotting
Striata were dissociated in cold lysis buffer (210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L HEPES, 1 mmol/L ethylenediaminetetraacetic acid, and 0.1% sodium dodecyl sulfate) by repeated passage through a Pasteur pipette, and were then sonicated. After centrifugation (16,000 × g, 5 minutes), the supernatant was collected and its protein concentration was assayed (BCA method, Pierce, Rockford, IL). Protein samples (40 μg in 18 μl) were then diluted with 6μl 4× loading buffer (Tris-Cl 240 mmol/L, β-mercaptoethanol 20%, SDS, 8%, glycerol 40%, bromphenol blue 0.2%) and were boiled for 3 minutes. Proteins were then separated on 15% or 7.5% polyacrylamide gels (Bio-Rad, Hercules, CA); equal loading was confirmed with Coomassie blue staining. After protein transfer to a polyvinylidene difluoride membrane filter, nonspecific sites were blocked by incubating for 1 hour at 37°C with 5% nonfat dry milk in a buffer containing 20 mmol/L Tris, 500 mmol/L NaCl, and 0.1% Tween 20 (pH 7.5). Membranes were incubated with rabbit anti-horse spleen ferritin (Sigma Aldrich, Product No. F5762, 1:2000 dilution) at 4°C overnight, with gentle shaking. This antibody was more sensitive than the commercially available H- or L-ferritin-specific antibodies that were tested, and consistently detected ferritin expression in CSF-injected and contralateral striata. Membranes were then incubated with horseradish peroxidase conjugated goat anti rabbit immunoglobulin G (1:3000 dilution, Pierce, Rockford, IL, Cat # 1858415) at 37°C for 1 hour. Immunoreactive proteins were visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) and Kodak Gel Logic 2200.
Protein Oxidation Assay
Striatal samples were collected and dissociated as described above for immunoblotting. After sonication, centrifugation (16,000 × g, 5 minutes), and protein assay, samples were denatured in 4.5% sodium dodecyl sulfate. Carbonyl groups were then converted to dinitrophenyl (DNP) derivatives by treatment with 2,4-dinitrophenylhydrazine using the Oxyblot kit (Millipore, Billerica, MA) and following the manufacturer’s instructions. Proteins were then separated on a 12% polyacrylamide gel followed by membrane transfer as described above. Protein carbonyl groups were detected with rabbit anti DNP primary antibody (1:300 dilution; Millipore) followed by horseradish peroxidase conjugated goat anti rabbit immunoglobulin G secondary antibody (1:150 dilution). Immunoreactive proteins were visualized as described above.
Cell Viability Assay
Striatal cell viability was assessed using the MTT assay, modified for use on mouse striatal tissue as previously described (Qu et al., 2005; Qu et al., 2007). MTT is reduced by viable cells to formazan; the purple product is then readily extracted from cells and quantified by spectrophotometry. The in vivo modification makes use of the fact that CNS tissue can be dissociated into individual cells or small groups of cells by gentle trituration without loss of cell viability, as determined by trypan blue exclusion or the ability to reduce tetrazolium salts to formazan. This dissociation technique, which is used extensively in our laboratory to prepare primary cell cultures, ensures that a uniform concentration of tetrazolium is delivered to all cells. Prior studies have demonstrated that this method of measuring cell viability correlates well with cell counts (Qu et al., 2005; Qu et al., 2007).
Immediately after dissection, each striatum was minced with forceps and placed into separate 15-ml centrifuge tubes containing 1 ml dissecting medium (described in Injections section). Tissue was then dissociated by gentle trituration through a Pasteur pipette, followed by repeated passage through another pipette with a narrowed tip. One milliliter of 0.6 mmol/L MTT in Dulbecco’s modified Eagle medium (Invitrogen) was added to the cell suspension, which was then mixed by brief vortexing. After incubation in a 37°C water bath for 4 minutes, cells were collected by low speed centrifugation (1380 × g, 1 minute), and the supernatant was completely removed and discarded. The formazan reaction product was then extracted from the cells by adding 2 ml isopropanol and vortexing. After centrifugation (1380 x g, 5 minutes), the absorbance of the supernatant was quantified at 562 nm with a reference wavelength of 650 nm. The formazan signal in the injected striatum was divided by that in the contralateral striatum and was expressed as this ratio.
Hematoxylin and Eosin Staining
After mice were deeply anesthetized with pentobarbital (75 mg/kg), they were perfused with 4% paraformaldehyde in 0.1 mol/L PBS (pH 7.4). Brains were removed and kept in the same paraformaldehyde solution at 4°C for 48 hours, and were then embedded in paraffin. They were then cut coronally through the needle entry site (identifiable on the brain surface), as well as 2mm anterior and 2mm posterior to that plane. Sections (5 mm) were deparaffinized with xylene and graded alcohol and stained with hematoxylin and eosin. Sections were examined with an Olympus BX 51 microscope. The needle track and injection site were visualized, and 200X images were captured.
Statistical Analysis
Immunoblot and assay data were analyzed with one-way analysis of variance followed by the Bonferroni multiple comparisons test. Mortality data were analyzed with Fisher’s exact test. Significance was assigned to a P value less than 0.05.
Acknowledgments
This study was supported by grants from the National Institutes of Health (NS42273) to RFR.
Abbreviations
- ICH
intracerebral hemorrhage
- IRP
iron regulatory protein
- KO
knockout
- MTT
Methylthiazolyldiphenyl-tetrazolium bromide
- TTC
2,3,5-Triphenyl-tetrazolium chloride
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- Arosio P, Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med. 2002;33:457–63. doi: 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- Beaumont C, Dugast I, Renaudie F, Souroujon M, Grandchamp B. Transcriptional regulation of ferritin H and L subunits in adult erythroid and liver cells from the mouse. Unambiguous identification of mouse ferritin subunits and in vitro formation of ferritin shells. J Biol Chem. 1989;264:7498–504. [PubMed] [Google Scholar]
- Cable H, Lloyd JB. Cellular uptake and release of two contrasting iron chelators. J Pharm Pharmacol. 1999;51:131–4. doi: 10.1211/0022357991772231. [DOI] [PubMed] [Google Scholar]
- Chou AC, Fitch CD. Mechanism of hemolysis induced by ferriprotoporphyrin IX. J Clin Invest. 1981;68:672–677. doi: 10.1172/JCI110302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comporti M, Signorini C, Buonocore G, Ciccoli L. Iron release, oxidative stress and erythrocyte ageing. Free Radic Biol Med. 2002;32:568–76. doi: 10.1016/s0891-5849(02)00759-1. [DOI] [PubMed] [Google Scholar]
- Dang TN, Bishop GM, Dringen R, Robinson SR. The putative heme transporter HCP1 is expressed in cultured astrocytes and contributes to the uptake of hemin. Glia. 2010;58:55–65. doi: 10.1002/glia.20901. [DOI] [PubMed] [Google Scholar]
- Festa M, Colonna A, Pietropaolo C, Ruffo A. Oxalomalate, a competitive inhibitor of aconitase, modulates the RNA-binding activity of iron-regulatory proteins. Biochem J. 2000;348(Pt 2):315–20. [PMC free article] [PubMed] [Google Scholar]
- Galy B, Holter SM, Klopstock T, Ferring D, Becker L, Kaden S, Wurst W, Grone HJ, Hentze MW. Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse. Nat Genet. 2006;38:967–9. doi: 10.1038/ng0906-967. discussion 969-70. [DOI] [PubMed] [Google Scholar]
- Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke. 2009;40:2241–3. doi: 10.1161/STROKEAHA.108.539536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Phillips JD, Yu Y, Leibold EA. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem. 1995;270:21645–51. doi: 10.1074/jbc.270.37.21645. [DOI] [PubMed] [Google Scholar]
- Hallaway PE, Eaton JW, Panter SS, Hedlund BE. Modulation of deferoxamine toxicity and clearance by covalent attachment to biocompatible polymers. Proc Natl Acad Sci U S A. 1989;86:10108–12. doi: 10.1073/pnas.86.24.10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson LR, Roeytenberg A, Martinez PM, Coppes VG, Sweet DC, Rao RJ, Marti DL, Hoekman JD, Matthews RB, Frey WH, 2nd, Panter SS. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J Pharmacol Exp Ther. 2009;330:679–86. doi: 10.1124/jpet.108.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–597. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, 3rd, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–14. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–57. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, Rouault TA. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. Embo J. 2004;23:386–95. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–8. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- Okauchi M, Hua Y, Keep RF, Morgenstern LB, Xi G. Effects of deferoxamine on intracerebral hemorrhage-induced brain injury in aged rats. Stroke. 2009;40:1858–63. doi: 10.1161/STROKEAHA.108.535765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okauchi M, Hua Y, Keep RF, Morgenstern LB, Schallert T, Xi G. Deferoxamine Treatment for Intracerebral Hemorrhage in Aged Rats. Therapeutic Time Window and Optimal Duration. Stroke. 2010;41:375–82. doi: 10.1161/STROKEAHA.109.569830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Chen J, Benvenisti-Zarom L, Ma X, Regan RF. Effect of targeted deletion of the heme oxygenase-2 gene on hemoglobin toxicity in the striatum. J Cereb Blood Flow Metab. 2005;25:1466–1475. doi: 10.1038/sj.jcbfm.9600143. [DOI] [PubMed] [Google Scholar]
- Qu Y, Chen-Roetling J, Benvenisti-Zarom L, Regan RF. Attenuation of oxidative injury after induction of experimental intracerebral hemorrhage in heme oxygenase-2 knockout mice. J Neurosurg. 2007;106:428–35. doi: 10.3171/jns.2007.106.3.428. [DOI] [PubMed] [Google Scholar]
- Regan RF, Chen M, Li Z, Zhang X, Benvenisti-Zarom L, Chen-Roetling J. Neurons lacking iron regulatory protein-2 are highly resistant to the toxicity of hemoglobin. Neurobiol Dis. 2008a;31:242–249. doi: 10.1016/j.nbd.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan RF, Li Z, Chen M, Zhang X, Chen-Roetling J. Iron regulatory proteins increase neuronal vulnerability to hydrogen peroxide. Biochem Biophys Res Commun. 2008b;375:6–10. doi: 10.1016/j.bbrc.2008.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SR, Dang TN, Dringen R, Bishop GM. Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14:228–35. doi: 10.1179/135100009X12525712409931. [DOI] [PubMed] [Google Scholar]
- Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Rad Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine neocortical cell culture. In: Tyson CA, Frazier JM, editors. Methods in Toxicology; Part A: In Vitro Biological Systems. Vol. 1. Academic Press; San Diego: 1993. pp. 46–60. [Google Scholar]
- Rosen GD, Williams RW. Complex trait analysis of the mouse striatum: independent QTLs modulate volume and neuron number. BMC Neurosci. 2001;2:5. doi: 10.1186/1471-2202-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrzadeh SMH, Anderson DK, Panter SS, Hallaway PE, Eaton JW. Hemoglobin potentiates central nervous system damage. J Clin Invest. 1987;79:662–664. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria R, Irace C, Festa M, Maffettone C, Colonna A. Induction of ferritin expression by oxalomalate. Biochim Biophys Acta. 2004;1691:151–9. doi: 10.1016/j.bbamcr.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Schroeder SE, Reddy MB, Schalinske KL. Retinoic Acid Modulates Hepatic Iron Homeostasis in Rats by Attenuating the RNA-Binding Activity of Iron Regulatory Proteins. J Nutr. 2007;137:2686–2690. doi: 10.1093/jn/137.12.2686. [DOI] [PubMed] [Google Scholar]
- Smith SR, Ghosh MC, Ollivierre-Wilson H, Hang Tong W, Rouault TA. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol Dis. 2006;36:283–7. doi: 10.1016/j.bcmd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–18. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- Sturrock RR. A comparative quantitative and morphological study of ageing in the mouse neostriatum, indusium griseum and anterior commissure. Neuropathol Appl Neurobiol. 1980;6:51–68. doi: 10.1111/j.1365-2990.1980.tb00204.x. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–45. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- Titova E, Ostrowski RP, Kevil CG, Tong W, Rojas H, Sowers LC, Zhang JH, Tang J. Reduced brain injury in CD18-deficient mice after experimental intracerebral hemorrhage. J Neurosci Res. 2008;86:3240–5. doi: 10.1002/jnr.21762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg JJ, Kuypers FA, Qju JH, Chiu D, Lubin B, Roelofsen B, Op den Kamp JA. The use of cis-parinaric acid to determine lipid peroxidation in human erythrocyte membranes:comparison of normal and sickle erythrocyte membranes. Biochem Biophys Acta. 1988;944:29–39. doi: 10.1016/0005-2736(88)90313-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Fillebeen C, Chen G, Biederbick A, Lill R, Pantopoulos K. Iron-dependent degradation of apo-IRP1 by the ubiquitin-proteasome pathway. Mol Cell Biol. 2007a;27:2423–30. doi: 10.1128/MCB.01111-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Di X, D’Agostino RB, Jr, Torti SV, Torti FM. Excess capacity of the iron regulatory protein system. J Biol Chem. 2007b;282:24650–9. doi: 10.1074/jbc.M703167200. [DOI] [PubMed] [Google Scholar]
- Warkentin LM, Auriat AM, Wowk S, Colbourne F. Failure of deferoxamine, an iron chelator, to improve outcome after collagenase-induced intracerebral hemorrhage in rats. Brain Res. 2010;1309:95–103. doi: 10.1016/j.brainres.2009.10.058. [DOI] [PubMed] [Google Scholar]
- Whitten CF, Gibson GW, Good MH, Goodwin JF, Brough AJ. Studies in acute iron poisoning. I. Desferrioxamine in the treatment of acute iron poisoning: clinical observations, experimental studies, and theoretical considerations. Pediatrics. 1965;36:322–35. [PubMed] [Google Scholar]
- Whitten CF, Chen YC, Gibson GW. Studies in acute iron poisoning. II. Further observations on desferrioxamine in the treatment of acute experimental iron poisoning. Pediatrics. 1966;38:102–10. [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakemura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]