

Abstract
Objective
Recently, we reported that the forkhead transcription factor, FKHR/FOXO1, is required for vascular endothelial growth factor (VEGF)-mediated upregulation of a number of genes in endothelial cells. Here, we tested the hypothesis that hepatocyte growth factor (HGF), a potent activator of PI3K-Akt in endothelial cells, is capable of depleting the nucleus of FKHR/FOXO1 and thus inhibiting VEGF induction of this class of genes.
Methods and Results
Incubation of human coronary artery endothelial cells with HGF induced prolonged PI3K/Akt-dependent phosphorylation and nuclear exclusion of FKHR/FOXO1. HGF-mediated inhibition of FKHR/FOXO1 activity resulted in secondary attenuation of VEGF-induced expression of FKHR/FOXO1-dependent genes including vascular cell adhesion molecule-1, manganese superoxide dismutase, endothelial specific molecule-1, CBP/p300 interacting transactivator with ED-rich tail-2, bone morphogenetic protein-2, matrix metalloproteinase-10 and MGC5618. At a functional level, pre-incubation of HGF resulted in inhibition of VEGF-induced VCAM-1-mediated monocyte adhesion to endothelial cells. HGF-mediated inhibition of VEGF-inducible VCAM-1 expression and monocyte adhesion was reversed by overexpression of constitutively active phosphorylation-resistant triple mutant (TM)-FKHR.
Conclusion
These findings suggest that physiological agonists of PI3K-Akt signaling pathway may modulate VEGF-FKHR/FOXO1-dependent gene expression in endothelial cells. The data underscore the importance of the “set point” of the endothelial cell when considering mechanisms of signal transduction.
Keywords: HGF, VEGF, Forkhead, Endothelial cells, Gene Expression
Vascular endothelial growth factor (VEGF) plays a critical role in endothelial cell survival, proliferation, migration, and is involved in wound repair, angiogenesis of ischemic tissue, tumor growth, microvascular permeability, vascular protection, and hemostasis 1–8. VEGF has been shown to activate a number of different intracellular signaling pathways, including PKC, PI3K and Akt, MEK1/2, p38 MAPK, and phospholipase Cγ 9–13. VEGF-mediated activation of signaling intermediates, in turn, results in altered activity of transcription factors, including NF-κB, Egr-1, NFAT-1, Ets-1, Stat-3/5, and forkhead transcription factors 14–20.
Hepatocyte growth factor (HGF) (also known as scatter factor) is a high-molecular weight multifunctional polypeptide growth factor. Although HGF originally was described for its ability to stimulate proliferation of liver cells 21, it is now recognized to interact with its receptor, c-MET, in other cell types. For example, in endothelial cells HGF has been shown to promote migration, proliferation, survival and barrier function 22–25. Under in vivo conditions, HGF has been implicated in angiogenesis 23. In cultured endothelial cells, HGF stimulates several signaling pathways and transcription factors including Rac, ERK1/2, p38 MAPK, PI3K, Src, AKT, FKHR/FOXO1, CREB, and ATF 25–28. However, in contrast to VEGF, HGF has not been shown to activate NF-κB 26, 29.
The mammalian members of the winged helix, or forkhead, transcription factors include FKHR (FOXO1), FKHRL1 (FOXO3a), and AFX (FOXO4) 19. Previous studies have demonstrated the presence of FKHR/FOXO1 and AFX in endothelial cells 19. Exposure of endothelial cells to several agonists, including VEGF, angiopoietin or angiotensin II resulted in PI3K-Akt-mediated phosphorylation and nuclear exclusion of FKHR/FOXO1, and subsequent downregulation of pro-apoptotic and anti-proliferative FKHR/FOXO1 target genes, such as GADD45A and p27kip1 19, 30, 31. More recently, we demonstrated that VEGF requires FKHR/FOXO1 activity for the induction of certain genes, including vascular cell adhesion molecule (VCAM)-1, manganese superoxide dismutase (MnSOD), endothelial specific molecule (ESM)-1, CBP/p300 interacting transactivator with ED-rich tail (CITED)-2, bone morphogenetic protein (BMP)-2, matrix metalloproteinase (MMP)-10 and MGC5618 20. These VEGF-inducible, forkhead-responsive genes are referred to as Class II genes, to distinguish them from the classical VEGF-repressible, forkhead-responsive transcripts (Class I) 20. Together with the observation that FKHR/FOXO1 is essential for embryonic vascular development 32, 33, these data suggest that FKHR/FOXO1 is an essential transcription factor that is involved in the coordinated regulation of a distinct set of genes in endothelial cells involved in cell maintenance and health.
The history of signal input, or the “set point” of the endothelial cell, is an important determinant of subsequent signaling. For example, the term “ischemic preconditioning” describes the cytoprotective effect of ischemia-reperfusion or hypoxia on tissues and/or endothelial cells 34. Repeated challenge of endothelial cells with lipopolysaccharide (LPS) leads to endotoxin tolerance characterized by temporary insensitivity to subsequent LPS challenge. Other preconditioning regimens that have been demonstrated to alter endothelial cell signaling include hypoxia 35, heat shock 36, and insulin-like growth factor (IGF)-1 37. As a final example, we recently demonstrated that pre-incubation of endothelial cells with tumor necrosis factor (TNF)-α results in altered thrombin-mediated gene expression by modulating nuclear-cytoplasmic trafficking of p65 NF-κB 38. In the present study, we tested the hypothesis that preconditioning of endothelial cells with the PI3K-AKT agonist, HGF would affect the ability of VEGF to activate Class II genes by limiting the availability of FKHR/FOXO1 in the nucleus (the functional equivalent of FKHR/FOXO1 knockdown). We show that HGF does indeed attenuate VEGF induction of FKHR-dependent genes. These findings add a new level of complexity to forkhead signaling in endothelial cells.
METHODS
Cell culture and reagents
Human coronary artery endothelial cells (HCAEC) were grown in Endothelial Growth Medium-2-MV (EGM-2-MV) BulletKit (Clonetics, San Diego, CA) at 37°C and 5% CO2. Endothelial cells from passage 3 to 6 were used for all experiments. Cells were serum-starved in 0.5% FBS prior to treatment with 50 ng/mL human VEGF-A165 or human 20 ng/mL HGF (PeproTech Inc, Rocky Hill, NJ). Where indicated, cells were pre-incubated for 30 min with 20 ng/mL HGF and then washed with PBS prior to VEGF treatment. Monoclonal anti-hVCAM-1 neutralizing antibody was purchased from Chemicon (Millipore, Tokyo, Japan).
Adenoviruses
HCAEC were transduced with replication-deficient adenoviruses encoding the cDNAs of β-galactosidase (Adv) and triple mutant (TM)-FKHR/FOXO1 as previously described 20, 26. The triple mutant version of FKHR/FOXO1 contains T24A, S256A, and S319A and is resistant to agonist-induced phosphorylation and nuclear export. Adenovirus expressing DN-Akt was previously described 19. Briefly, HCAEC were plated at a density of 1 × 106 cells per 10-cm plate and adenoviruses were added to the cells at five multiplicity of infection (5 MOI) after 5 h of plating. Cells were then allowed to grow for 36 h followed by overnight serum starvation. Expression of adenovirus-based FKHR/FOXO1 and Akt was confirmed using RT-PCR analyses (Supplemental Figure I shows FKHR/FOXO1) and also by Western blots as described previously 19, 20.
siRNA-mediated inhibition of endogenous FKHR/FOXO1
HCAEC were grown to 70–80% confluency in 6-cm plates and transfected with siRNA against the following FKHR/FOXO1 target sequences: siFKHR2 CAG CGC CGA CTT CAT GAG CAA (Qiagen, Valencia, CA) in Opti-MEM containing Lipofectin (10 μg/mL) for 4 h as previously described 20. The cells were then incubated in EGM-2 medium for 24 h and serum-starved in 0.5% serum for 12–16 h before VEGF treatment for the times indicated.
Quantitative real-time PCR. See Supplemental Methods.
Western and Northern blot analyses. See Supplemental Methods.
Immunolocalization studies. See Supplemental Methods.
Nuclear and cytoplasmic cellular fractionation. See Supplemental Methods.
Monocyte adhesion assay. See Supplemental Methods.
Statistical analyses
All values are presented as mean ± SD where appropriate. Statistical significance between two groups was determined by use of a paired t-test, and values of p<0.05 were considered significant.
RESULTS
HGF induces early and prolonged phosphorylation of Akt and FKHR/FOXO1 in primary human endothelial cells
HCAEC were incubated in the absence or presence of VEGF and/or HGF for varying times and assayed for phosphorylation of Akt and FKHR/FOXO1 by Western blot analysis. In response to VEGF, Akt was phosphorylated at 5 min, returning to basal levels by 30 min (Figure 1A, upper panel). VEGF-mediated phosphorylation of FKHR/FOXO1 occurred at 15 min and returned to basal levels by 60 min (Figure 1B). In contrast, HGF-induced phosphorylation of Akt and FKHR/FOXO1 occurred at 5 min and did not return to their basal levels until after 2 h (Figures 1A-B and data not shown). VEGF- and HGF-mediated phosphorylation of FKHR/FOXO1 was inhibited by the PI3K-inhibitor LY294002 (data not shown) and dominant-negative (DN) Akt (Figures 1C–D). Together, these data indicate that while both VEGF- and HGF-mediated phosphorylation of FKHR/FOXO1 occurs through a PI3K-Akt signaling pathway, the effect of HGF on FKHR/FOXO1 phosphorylation in HCAEC occurs earlier and is more sustained.
Figure 1. HGF induces greater and more sustained phosphorylation of endogenous FKHR/FOXO1 in HCAEC compared to VEGF.
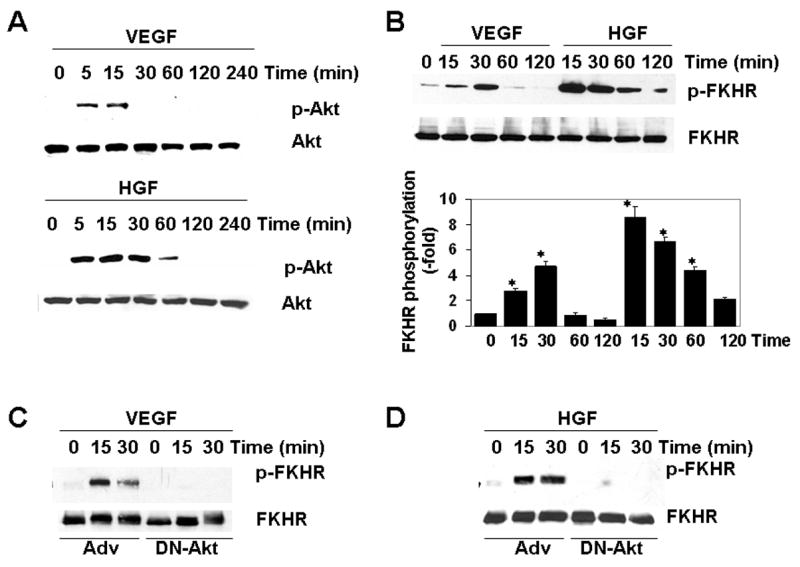
Serum-starved HCAEC were incubated with either VEGF (50 ng/mL) or HGF (20 ng/mL) for the times indicated. HCAEC were harvested for total protein, and Western blots were carried out as described in Methods. A, Membranes were probed for Ser473-Akt (p-Akt) and total Akt. B–D, Membranes were probed for Ser256-FKHR/FOXO1 (p-FKHR/FOXO1) and total FKHR/FOXO1. Bar graph shows quantitation of FKHR/FOXO1 phosphorylation relative to total FKHR/FOXO1 in 3 independent experiments. C and D, HCAEC were transduced with control adenovirus (Adv) or adenovirus expressing dominant-negative Akt (DN-Akt) two days prior to growth factor treatment. Membranes were probed for Ser256-FKHR/FOXO1 (p-FKHR/FOXO1) and total FKHR/FOXO1. Western blots shown are representative of three independent experiments. * p<0.05.
HGF-mediated nuclear exclusion of FKHR/FOXO1 in primary human endothelial cells is prolonged compared to VEGF
Phosphorylation of FKHR/FOXO1 has been shown to promote translocation of the transcription factor from the nucleus to cytoplasm 19, 39. In immunofluorescence assays, incubation of HCAEC with VEGF resulted in cytoplasmic translocation of FKHR/FOXO1 by 15–30 min, an effect that was reversed by 60 min (Supplemental Figure II). HGF-mediated nuclear exclusion of FKHR/FOXO1 occurred by 15 min. However, in contrast to VEGF, the effect of HGF on cytoplasmic translocation persisted at 60 min, returning to basal levels only after 2 h (Figure 2). HCAEC were also processed for cytoplasmic and nuclear fractions and assayed for FKHR/FOXO1 by Western blot. In these assays, nuclear-to-cytoplasmic translocation of FKHR/FOXO1 by VEGF was maximal at 30 min and returned to the basal levels within 60 min, whereas HGF-induced cytoplasmic translocation was maximal from 30 to 60 min but did not return to the basal levels until after 2 h (Supplemental Figure III). In accordance with the phosphorylation data (please see Figs. 1C-D), LY294002 and DN-Akt inhibited VEGF- and HGF-mediated cytoplasmic translocation of FKHR/FOXO1 (Supplemental Figure IV shows the effect of DN-Akt), suggesting that PI3K-Akt-induced phosphorylation is critical for subcellular localization of FKHR/FOXO1 in HCAEC. Consistent with the early and sustained effect of HGF phosphorylation and subcellular localization of FKHR/FOXO1, pre-incubation of HCAEC with HGF for 30 min preempted the effect of VEGF (Supplemental Figure V). Together, these findings suggest that HGF induces sustained nuclear exclusion of FKHR/FOXO1 and that pre-incubation with HGF limits the availability of nuclear FKHR/FOXO1 in VEGF-treated cells.
Figure 2. HGF-mediated nuclear exclusion of FKHR/FOXO1 in HCEAC is more sustained compared to VEGF.
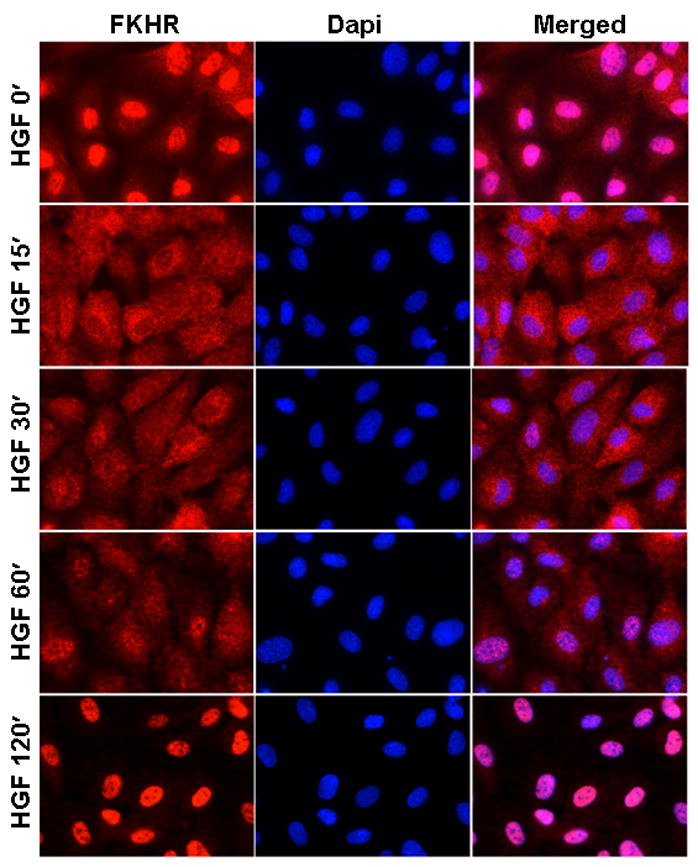
HGF-mediated subcellular localization of FKHR/FOXO1 was assayed as described in Methods. The photomicrographs show FKHR/FOXO1 (red, left), nuclear DAPI (blue, middle), and merged FKHR/FOXO1 and DAPI (right). The bar graph shows the corresponding quantitative analyses of the subcellular localization of FKHR/FOXO1 and was generated by counting nuclear and cytoplasmic localization of FKHR/FOXO1 in 200 HCAEC cells per time point using NIH ImageJ and McMaster Biophotonics as described in Methods; N, nuclear; C, cytoplasmic. The data presented are from three independent experiments. * p<0.05.
Temporal modulation of FKHR/FOXO1 by HGF attenuates VEGF-mediated induction of FKHR/FOXO1-dependent genes in HCAEC
Based on the above findings, we reasoned that HGF-mediated depletion of FKHR/FOXO1 from the nucleus would impair the ability of VEGF to induce those genes that depend on FKHR/FOXO1 as a positive transcription factor (i.e., Class II genes). To test this hypothesis, HCAEC were serum-starved, preincubated with HGF, treated in the absence or presence of VEGF, and harvested for total RNA. In real-time PCR analyses, pretreatment of HCAEC with HGF for 30 min blocked VEGF-mediated induction of FKHR/FOXO1-dependent genes including VCAM-1, MnSOD, BMP2, ESM-1, MMP10, CITED2 and MGC5618 (Table 1). Representative Northern blot analyses for MnSOD and VCAM-1 are shown in Figure 3A. To determine whether this inhibitory effect of HGF was due to unavailability of non-phosphorylated FKHR/FOXO1 in the nucleus, we transduced HCAEC with adenoviruses expressing either β-gal (Adv) or phosphorylation-defective constitutively active triple mutant (TM)-FKHR/FOXO1. As shown in Table 1 and Figure 3B, the ability of HGF to attenuate VEGF-mediated induction of VCAM-1, MnSOD, BMP2, ESM-1, MMP10, CITED2 and MGC5618 expression was reversed by nuclear-localized TM-FKHR/FOXO1. To rule out an effect of HGF preconditioning on VEGFR2 phosphorylation, we carried out immunoprecipitation of VEGFR2 followed by Western blotting with an anti-phospho-tyrosine antibody. Pre-incubation of HCAEC with HGF did not significantly alter VEGF-induced phosphorylation or mRNA expression of VEGFR2 (Supplemental Figure VI). A previous study demonstrated that HGF inhibits VEGF activation of NF-κB 40. However, consistent with our previous results 20, we failed to demonstrate an effect of HGF on VEGF-induced NF-κB signaling. Specifically, HGF had no effect on VEGF-mediated phosphorylation of IκB or NF-κB p65 DNA binding (Supplemental Figure VII). Together, these data suggest that HGF limits the availability of nuclear-localized FKHR/FOXO1, which in turn impairs VEGF-inducible/FKHR/FOXO1-dependent gene expression.
Table 1. HGF-mediated inhibition of VEGF-induced gene expression (HGF+VEGF) is reversed by TM-FKHR (TM-FKHR+HGF+VEGF) in HCAEC as determined by fold induction or reduction using RT-PCR analyses.
The basal levels of expression for each gene in unstimulated, serum-starved (Control) HCAEC were arbitrarily considered as 1 (-fold) per 106 18S mRNA copies. Numbers are expressed as – fold induction (or reduction if less than 1) over the basal level.
| Control | VEGF | HGF | HGF+VEGF | TM-FKHR+HGF+VEGF | FKHRE# | Function | |
|---|---|---|---|---|---|---|---|
| Class I: | |||||||
| BTG-1 | 1 | 0.72±0.08† | 0.5±0.06† | 0.46±0.09 | 2.6±0.4 | 4 | Cell cycle |
| P27kip1 | 1 | 0.57±0.1† | 0.6±0.08† | 0.4±0.05 | 1.7±0.3 | 2 | Cell cycle |
| Class II: | |||||||
| BMP2 | 1 | 4.77±0.64† | 0.87±0.1 | 2.02±0.3* | 6.2±0.74 | 1 | Angiogenesis |
| CITED2 | 1 | 1.6±0.1† | 0.92±0.1 | 0.76±0.1* | 5.4±0.32 | 1 | Cardiac |
| ESM-1 | 1 | 1.78±0.2† | 0.94±0.2 | 1.1±0.2* | 4.2±0.4 | 2 | Angiogenesis |
| MnSOD | 1 | 2.3±0.24† | 0.85±0.1† | 0.81±0.1* | 3.7±0.42 | 1 | Redox regulation |
| MGC5618 | 1 | 2.1±0.29† | 0.73±0.2 | 0.9±0.2* | 4.6±0.36 | 1 | Unknown |
| MMP10 | 1 | 2.6±0.3† | 0.92±0.13 | 1.2±0.3* | 11.2±0.61 | 1 | Matrix remodel |
| VCAM-1 | 1 | 11.4±2.5† | 0.81±0.1† | 2.4±0.4* | 20.8±0.86 | 2 | Cell adhesion |
, number of FKHR consensus elements (FKHRE) in upstream promoter.
p<0.05 relative to Control;
p<0.05 relative to VEGF-treated.
Figure 3. HGF-mediated inhibition of VEGF-induced class II gene expression is reversed by constitutively active TM-FKHR/FOXO1.
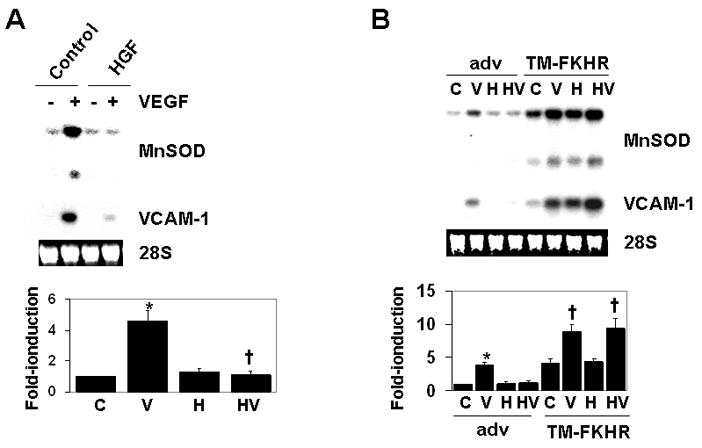
A, HCAEC were serum-starved overnight, and then preincubated in the absence (control) or presence of HGF for 30 min, followed by incubation without (−) or with VEGF (+) for 4 h. Total RNA was extracted and Northern blots analyses were performed using the MnSOD probe; the same membrane was stripped and reprobed for VCAM-1 transcript. Ethidium bromide-stained 28S RNA is shown as a loading control. MnSOD expression is quantitated in the bottom panel (C, control; V, VEGF; H, HGF; HV, pretreatment with HGF followed by VEGF). The data are expressed as mean ± S.D. of three independent experiments. B, HCAEC were transduced with adenoviruses expressing β-galactosidase (Adv) or triple-mutant (TM)-FKHR/FOXO1. Cells were treated in the absence (control, C) or presence of VEGF (V) or HGF (H) for 4 h. Alternatively, cells were preincubated with HGF for 30 min and then treated with VEGF for 4 h (HV). * p<0.05 control versus VEGF-treated; † p<0.05 control adenovirus (Adv)-transduced plus VEGF-treated versus TM-FKHR/FOXO1 adenovirus-transduced plus VEGF, or HGF-pretreated followed by VEGF treatment, as indicated.
FKHR/FOXO1 is required for VEGF-mediated monocyte adhesion to HCAEC
We next wished to determine whether the inhibitory effect of HGF on VEGF signaling was functionally relevant. To that end, we asked whether HGF-mediated attenuation of VCAM-1 expression in VEGF-treated endothelial cells influenced monocyte adhesion. As shown in Figure 4, VEGF resulted in more than 3-fold induction of U937 adhesion to HCAEC. Pre-incubation of HCAEC with HGF resulted in inhibition of VEGF-induced monocyte adhesion (Figure 4, upper panel). This effect was reversed by overexpression of a constitutively active TM-FKHR/FOXO1 (Figure 4, lower panel). In contrast, siRNA-mediated knockdown of siFKHR/FOXO1 in HCAEC resulted in a decrease in the VEGF-induced monocyte adhesion, irrespective of the presence of HGF (Supplemental Figure VIII A). Finally, neutralizing anti-VCAM-1 antibody inhibited VEGF-inducible monocyte adhesion to HCAEC (Supplemental Figure VIII B). These data suggest that HGF-mediated phosphorylation and nuclear exclusion of FKHR/FOXO1 and subsequent attenuation of VEGF-inducible VCAM-1 expression results in reduced monocyte adhesion to endothelial cells.
Figure 4. HGF-mediated inhibition of VEGF-induced monocyte adhesion to HCAEC is reversed by TM-FKHR/FOXO1.

HCAEC were transduced with either Adv or TM-FKHR/FOXO1 as described in Methods. The cells were serum-starved for 12–16h before HGF or VEGF stimulation. Where combined HGF and VEGF treatments are indicated, HCAEC monolayers were treated with HGF for 30 min prior to the addition of VEGF. U937 monocyte adhesion on HCAEC monolayer was determined as described in Methods. Bar graph results shown are mean ± SD (standard deviation). * p<0.05 control versus VEGF-treated; † p<0.05 VEGF-treated versus HGF-pretreated plus VEGF-treated. The results are obtained from three independent experiments. Quantitative bar graphs with p value (<0.05) are shown.
DISCUSSION
VEGF activation of PI3K-Akt results in phosphorylation and nuclear exclusion of the FKHR/FOXO1. Nuclear exclusion, in turn, leads to downregulation of genes that are primarily regulated by forkhead (e.g., p27kip1, BTG-1). However, there is a distinct set of forkhead-responsive genes that are actually upregulated by VEGF 20. VEGF-mediated induction of these genes requires FKHR/FOXO1 and at least one other positive acting factor, such as NF-κB or NFAT 20. The existence of the latter gene class has two implications. First, for VEGF to induce the expression of forkhead-responsive genes there must either be residual FKHR/FOXO1 in the nucleus, or timely reentry of the transcription factor into the nucleus. Second, pre-incubation of endothelial cells with an agonist that selectively alters PI3K-Akt-FKHR/FOXO1 signaling should function as a rheostat for VEGF effect on this gene class. Consistent with this hypothesis, we have demonstrated that HGF, a physiological agonist that activates PI3K-Akt, but not NF-κB 26, blocks the stimulatory effect of VEGF on Class II genes including VCAM-1 and secondary monocyte adhesion.
HGF shares certain properties with VEGF. For example, both growth factors promote endothelial cell proliferation and migration in vitro, and induce angiogenesis in vivo 41. However, HGF and VEGF also exert distinct functions in endothelial cells. For example, VEGF increases endothelial cell permeability, whereas HGF promotes barrier function 28. HGF and VEGF induce largely non-overlapping patterns of gene expression in endothelial cells, suggesting different signal transduction pathways 42. In combination, HGF and VEGF act additively or synergistically to promote endothelial cell survival, proliferation, angiogenesis and downstream gene expression 41. The results of the present study provide evidence for an additional level of cross-talk between HGF and VEGF.
Our data support the hypothesis that HGF inhibits VEGF-mediated gene expression by interfering with the FKHR/FOXO1 arm of the VEGF signaling pathway. Compared with VEGF, HGF induces early, intense and prolonged phosphorylation and nuclear exclusion of FKHR/FOXO1. By depleting the nucleus of a critical threshold of FKHR/FOXO1 and/or delaying nuclear reentry of the transcription factor, HGF may deprive the VEGF signal of a necessary transacting protein required for activation of Class II genes (Figure 5). In further support of our hypothesis is the observation that the effect of HGF on VEGF was reversed by constitutively active TM-FKHR/FOXO1.
Figure 5. Model for HGF inhibition of VEGF-responsive genes that require FKHR/FOXO1 for expression.
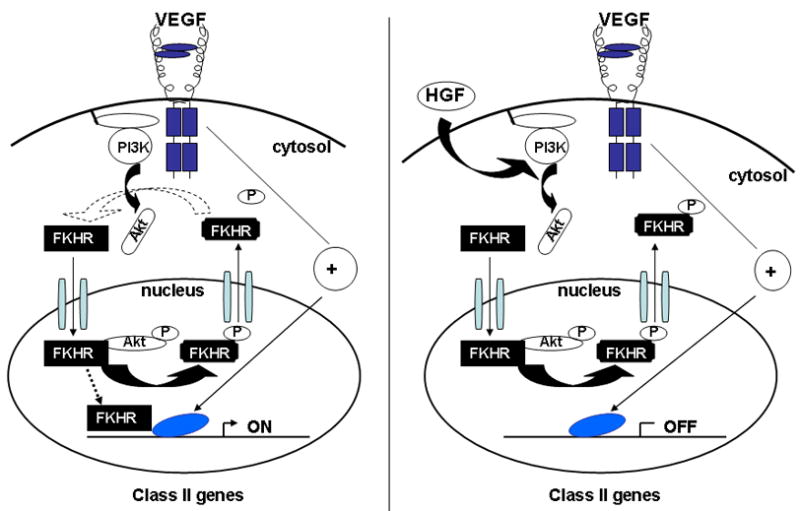
Left panel, VEGF normally induces expression of Class II genes (ON) via a pathway that requires FKHR/FOXO1 and one or more forkhead-independent transcription factor(s) (indicated by blue oval) for optimal expression. Right panel, HGF or other factors that stimulate PI3K-Akt may promote prolonged phosphorylation and nuclear exclusion of FKHR/FOXO1 and thus attenuate subsequent VEGF induction of Class II genes.
Two alternative explanations for an inhibitory effect of HGF on VEGF-mediated gene expression must be considered. First, it is formally possible that HGF-c-Met signaling inhibits VEGFR2 activation. However, our finding that HGF pretreatment failed to alter VEGF-mediated VEGFR2 phosphorylation argues against this mechanism. Second, HGF may inhibit NF-κB, which functions with FKHR/FOXO1 to activate many of the Class II genes. Indeed, a previous study showed that when added together, HGF inhibited VEGF-mediated induction of IκB phosphorylation, p65 nuclear translocation and DNA binding, and VCAM-1/ICAM-1 expression 40. However, we have been unable to demonstrate an effect of HGF on NF-κB activation in the absence or presence of VEGF. The reason for the discrepancy between the two studies is unclear, but may relate to the use of different cell types (HUVEC in the published report, HCAEC in the current study) or the timing of HGF addition (concomitant with VEGF in the published report, prior to VEGF in the current study). Regardless of a potential contribution of NF-κB, our findings point to an important role of the FKHR/FOXO1 pathway in modulating the cross-talk between HGF and VEGF. The functional relevance of this effect is evidenced by the observation that HGF priming reduced VEGF-mediated VCAM-1 dependent adhesion of monocytes. Based on a survey of the HGF-inhibitable Class II genes (Table 2), we predict that HGF signaling may also attenuate VEGF-inducible superoxide dismutase activity, ESM-1 secretion and MMP10 synthesis.
In summary, we have demonstrated that cross-talk between agonists may influence forkhead-dependent signaling in endothelial cells. Stated another way, the capacity of VEGF to promote expression of these Class II genes is dependent on the “set point” of the cell. These findings raise interesting questions about (patho)physiology. HGF is expressed by many cell types, including endothelial cells and vascular smooth muscle cells. HGF levels are increased in certain diseases, such as cancer and atherosclerosis 43, 44. Moreover, recent studies support the therapeutic use of HGF as a means of augmenting angiogenesis. It will be important to consider the potential effects of endogenous and/or exogenous HGF on the VEGF-FKHR/FOXO1 signaling axis in the endothelium.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant HL077348 (to W.C.A. and M.R.A) and American Heart Association Grant SDG 0453284N (to M.R.A.). T.M. was supported by NIBIO, NEDO, the Science and Technology from Ministry of Education, Culture, Sports, Sciences and Technology, and the Takeda Science foundation, Japan.
References
- 1.Dvorak HF. VPF/VEGF and the angiogenic response. Semin Perinatol. 2000;24:75–78. doi: 10.1016/s0146-0005(00)80061-0. [DOI] [PubMed] [Google Scholar]
- 2.Zachary I, Mathur A, Yla-Herttuala S, Martin J. Vascular protection: A novel nonangiogenic cardiovascular role for vascular endothelial growth factor. Arterioscler Thromb Vasc Biol. 2000;20:1512–1520. doi: 10.1161/01.atv.20.6.1512. [DOI] [PubMed] [Google Scholar]
- 3.Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE. 2001;2001:RE21. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 5.Mukhopadhyay D, Zeng H, Bhattacharya R. Complexity in the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF)-receptors signaling. Mol Cell Biochem. 2004;264:51–61. doi: 10.1023/b:mcbi.0000044374.85095.df. [DOI] [PubMed] [Google Scholar]
- 6.Zelzer E, Olsen BR. Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth, and repair. Curr Top Dev Biol. 2005;65:169–187. doi: 10.1016/S0070-2153(04)65006-X. [DOI] [PubMed] [Google Scholar]
- 7.Folkman J. Endogenous angiogenesis inhibitors. Apmis. 2004;112:496–507. doi: 10.1111/j.1600-0463.2004.apm11207-0809.x. [DOI] [PubMed] [Google Scholar]
- 8.Stupack DG, Cheresh DA. Integrins and angiogenesis. Curr Top Dev Biol. 2004;64:207–238. doi: 10.1016/S0070-2153(04)64009-9. [DOI] [PubMed] [Google Scholar]
- 9.Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, Takagi H, Newsome WP, Jirousek MR, King GL. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo D, Jia Q, Song HY, Warren RS, Donner DB. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J Biol Chem. 1995;270:6729–6733. doi: 10.1074/jbc.270.12.6729. [DOI] [PubMed] [Google Scholar]
- 11.Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem. 1997;272:32521–32527. doi: 10.1074/jbc.272.51.32521. [DOI] [PubMed] [Google Scholar]
- 12.D’Angelo G, Struman I, Martial J, Weiner RI. Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N- terminal fragment of prolactin. Proc Natl Acad Sci U S A. 1995;92:6374–6378. doi: 10.1073/pnas.92.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Fisher RJ, Riggs CW, Rhim JS, Lautenberger JA. Inhibition of vascular endothelial growth factor-induced endothelial cell migration by ETS1 antisense oligonucleotides. Cancer Res. 1997;57:2013–2019. [PubMed] [Google Scholar]
- 15.Korpelainen EI, Karkkainen M, Gunji Y, Vikkula M, Alitalo K. Endothelial receptor tyrosine kinases activate the STAT signaling pathway: mutant Tie-2 causing venous malformations signals a distinct STAT activation response. Oncogene. 1999;18:1–8. doi: 10.1038/sj.onc.1202288. [DOI] [PubMed] [Google Scholar]
- 16.Johnson EN, Lee YM, Sander TL, Rabkin E, Schoen FJ, Kaushal S, Bischoff J. NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells. J Biol Chem. 2003;278:1686–1692. doi: 10.1074/jbc.M210250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marumo T, Schini-Kerth VB, Busse R. Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes. 1999;48:1131–1137. doi: 10.2337/diabetes.48.5.1131. [DOI] [PubMed] [Google Scholar]
- 18.Mechtcheriakova D, Wlachos A, Holzmuller H, Binder BR, Hofer E. Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1. Blood. 1999;93:3811–3823. [PubMed] [Google Scholar]
- 19.Abid MR, Guo S, Minami T, Spokes KC, Ueki K, Skurk C, Walsh K, Aird WC. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:294–300. doi: 10.1161/01.ATV.0000110502.10593.06. [DOI] [PubMed] [Google Scholar]
- 20.Abid MR, Shih SC, Otu HH, Spokes KC, Okada Y, Curiel DT, Minami T, Aird WC. A novel class of vascular endothelial growth factor-responsive genes that require forkhead activity for expression. J Biol Chem. 2006;281:35544–35553. doi: 10.1074/jbc.M608620200. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 22.Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant DS, Kleinman HK, Goldberg ID, Bhargava MM, Nickoloff BJ, Kinsella JL, Polverini P, Rosen EM. Scatter factor induces blood vessel formation in vivo. Proc Natl Acad Sci U S A. 1993;90:1937–1941. doi: 10.1073/pnas.90.5.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhlmann CR, Schaefer CA, Fehsecke A, Most AK, Tillmanns H, Erdogan A. A new signaling mechanism of hepatocyte growth factor-induced endothelial proliferation. J Thromb Haemost. 2005;3:2089–2095. doi: 10.1111/j.1538-7836.2005.01541.x. [DOI] [PubMed] [Google Scholar]
- 25.Sengupta S, Gherardi E, Sellers LA, Wood JM, Sasisekharan R, Fan TP. Hepatocyte growth factor/scatter factor can induce angiogenesis independently of vascular endothelial growth factor. Arterioscler Thromb Vasc Biol. 2003;23:69–75. doi: 10.1161/01.atv.0000048701.86621.d0. [DOI] [PubMed] [Google Scholar]
- 26.Abid MR, Schoots IG, Spokes KC, Wu SQ, Mawhinney C, Aird WC. Vascular endothelial growth factor-mediated induction of manganese superoxide dismutase occurs through redox-dependent regulation of forkhead and IkappaB/NF-kappaB. J Biol Chem. 2004;279:44030–44038. doi: 10.1074/jbc.M408285200. [DOI] [PubMed] [Google Scholar]
- 27.Kanda S, Kanetake H, Miyata Y. HGF-induced capillary morphogenesis of endothelial cells is regulated by Src. Biochem Biophys Res Commun. 2006;344:617–622. doi: 10.1016/j.bbrc.2006.03.183. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. Faseb J. 2002;16:950–962. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- 29.Min JK, Kim YM, Kim SW, Kwon MC, Kong YY, Hwang IK, Won MH, Rho J, Kwon YG. TNF-related activation-induced cytokine enhances leukocyte adhesiveness: induction of ICAM-1 and VCAM-1 via TNF receptor-associated factor and protein kinase C-dependent NF-kappaB activation in endothelial cells. J Immunol. 2005;175:531–540. doi: 10.4049/jimmunol.175.1.531. [DOI] [PubMed] [Google Scholar]
- 30.Yu X, Murao K, Imachi H, Cao WM, Li J, Matsumoto K, Nishiuchi T, Ahmed RA, Wong NC, Kosaka H, Unterman TG, Ishida T. Regulation of scavenger receptor class BI gene expression by angiotensin II in vascular endothelial cells. Hypertension. 2007;49:1378–1384. doi: 10.1161/HYPERTENSIONAHA.106.082479. [DOI] [PubMed] [Google Scholar]
- 31.Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, Rudge JS. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes Dev. 2004;18:1060–1071. doi: 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Ikeda K, Motoyama N, Mori N. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 34.Ahmad S, Ahmad A, Gerasimovskaya E, Stenmark KR, Allen CB, White CW. Hypoxia protects human lung microvascular endothelial and epithelial-like cells against oxygen toxicity: role of phosphatidylinositol 3-kinase. Am J Respir Cell Mol Biol. 2003;28:179–187. doi: 10.1165/rcmb.2002-0004OC. [DOI] [PubMed] [Google Scholar]
- 35.Nobata Y, Urakaze M, Temaru R, Sato A, Nakamura N, Yamazaki K, Kishida M, Takata M, Kobayashi M. alpha-Tocopherol Inhibits IL-8 synthesis induced by thrombin and high glucose in endothelial cells. Horm Metab Res. 2002;34:49–54. doi: 10.1055/s-2002-20523. [DOI] [PubMed] [Google Scholar]
- 36.Yasukawa K, Terai M, Shulman ST, Toyozaki T, Yajima S, Kohno Y, Rowley AH. Systemic Production of Vascular Endothelial Growth Factor and fms-Like Tyrosine Kinase-1 Receptor in Acute Kawasaki Disease. Circulation. 2002;105:766–769. doi: 10.1161/hc0602.103396. [DOI] [PubMed] [Google Scholar]
- 37.Che W, Lerner-Marmarosh N, Huang Q, Osawa M, Ohta S, Yoshizumi M, Glassman M, Lee JD, Yan C, Berk BC, Abe J. Insulin-like growth factor-1 enhances inflammatory responses in endothelial cells: role of Gab1 and MEKK3 in TNF-alpha-induced c-Jun and NF-kappaB activation and adhesion molecule expression. Circ Res. 2002;90:1222–1230. doi: 10.1161/01.res.0000021127.83364.7d. [DOI] [PubMed] [Google Scholar]
- 38.Wada Y, Otu H, Wu S, Abid MR, Okada H, Libermann T, Kodama T, Shih SC, Minami T, Aird WC. Preconditioning of primary human endothelial cells with inflammatory mediators alters the “set point” of the cell. Faseb J. 2005;19:1914–1916. doi: 10.1096/fj.05-4037fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 40.Min JK, Lee YM, Kim JH, Kim YM, Kim SW, Lee SY, Gho YS, Oh GT, Kwon YG. Hepatocyte growth factor suppresses vascular endothelial growth factor-induced expression of endothelial ICAM-1 and VCAM-1 by inhibiting the nuclear factor-kappaB pathway. Circ Res. 2005;96:300–307. doi: 10.1161/01.RES.0000155330.07887.EE. [DOI] [PubMed] [Google Scholar]
- 41.Xin X, Yang S, Ingle G, Zlot C, Rangell L, Kowalski J, Schwall R, Ferrara N, Gerritsen ME. Hepatocyte growth factor enhances vascular endothelial growth factor- induced angiogenesis in vitro and in vivo. Am J Pathol. 2001;158:1111–1120. doi: 10.1016/S0002-9440(10)64058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerritsen ME, Tomlinson JE, Zlot C, Ziman M, Hwang S. Using gene expression profiling to identify the molecular basis of the synergistic actions of hepatocyte growth factor and vascular endothelial growth factor in human endothelial cells. Br J Pharmacol. 2003;140:595–610. doi: 10.1038/sj.bjp.0705494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawamoto R, Oka Y, Yoshida O, Takagi Y. Significance of serum circulating hepatocyte growth factor in the development of carotid atherosclerosis. J Atheroscler Thromb. 2003;10:154–159. doi: 10.5551/jat.10.154. [DOI] [PubMed] [Google Scholar]
- 44.Lesko E, Majka M. The biological role of HGF-MET axis in tumor growth and development of metastasis. Front Biosci. 2008;13:1271–1280. doi: 10.2741/2760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.